
Abstract
Dimensionally Stable Anodes (Sb2O5-doped Ti/RuO2-ZrO2) are synthesized at four different Zr/Ru molar ratios using the Pechini method to account for the effects of ZrO2 content on the active chlorine formation. X-ray diffraction for ternary catalysts reveal co-deposited phases consisting of a tetragonal crystalline structure (P42/mnm) for RuO2, and two phases corresponding to monoclinic (P2/m) and tetragonal (P42/mnm) crystalline structures for ZrO2. SEM micrographs show that ZrO2 alters grain growth and film morphology as its content is increased. Electrochemical tests performed in 1 mol L−1 H2SO4 and 0.1 mol L−1 NaCl demonstrate that the kinetic rate to perform the oxygen evolution reaction (OER) decreases with the simultaneous augment of ZrO2 and Sb2O5 compositions in the ternary anode, while in the ZrO2 absence, Sb2O5 catalyzes the OER (Ti/SbRu) compared to the Ti/Ru and ternary electrodes. Although the chlorine evolution reaction (CER) preferentially occurs by the suppression of OER in chloride media, the CER rate mainly increases due to the rise of the ZrO2 content. This finding is corroborated with an iodometric method using UV-Vis spectroscopy, which shows that the electro-generation capacity to form active chlorine species is significantly enhanced when ZrO2 is added, compared to Ti/RuO2 and Sb2O5 doped Ti/RuO2 materials. Accelerated life tests conducted with all catalysts indicate that all ternary anodes present failure times considerably longer than Ti/Ru, Ti/SbRu and Ti/RuZr electrodes, suggesting a stability enhancement due to the combined presence of Sb and Zr species.
Export citation and abstract BibTeX RIS
RuO2 and valve metal oxides are typically deposited on Ti for multiple electro-catalytic uses,1,2 as a result of their chemical and mechanical properties (e.g. high surface area, high catalytic activity, high stability to anodic corrosion), whence theses electrodes have become recognized as Dimensionally Stable Anodes (DSA).3,4 The most important reactions occurring on DSAs are anodic Oxygen Evolution (OER) and Chlorine Evolution (CER) in chloride-free and chloride containing electrolytes, respectively.
In recent years, there are growing interests to perform direct electrolysis of water with chloride content for generation of active chlorine species (Cl2-active, i.e. Cl2 E° = 1.36 V/SHE, HClO E° = 1.49 V/SHE and/or ClO− E° = 0.89 V/SHE), since they have been successfully utilized for electrochemical water disinfection5,6 and to eliminate organic micro-pollutants7 from wastewater. Cl2-active production involves a complex reaction mechanism related to chloride oxidation, and the chemical/electrochemical reactivity of some of its oxidation products. The occurrence of this mechanism on oxide coated catalysts (MOx) is described as follows:8,9
Anode
![Equation ([1])](https://content.cld.iop.org/journals/1945-7111/163/9/H818/revision1/d0001.gif)
![Equation ([2])](https://content.cld.iop.org/journals/1945-7111/163/9/H818/revision1/d0002.gif)
![Equation ([3])](https://content.cld.iop.org/journals/1945-7111/163/9/H818/revision1/d0003.gif)
where MOx and MOx − OH•ads are the non-oxidized and oxidized surface sites, respectively (reaction 1); while chloride oxidation on the anode leads to the formation of adsorbed hypochlorous acid (reaction 2) along with the concomitant oxygen evolution reaction (not shown). Hypochlorous species can further interact with chloride ion and yields chlorine in solution, according to reaction 3. On the other hand, electro-generated chlorine can react with water depending on pH to form hypoclorous acid and hypochlorite ions in bulk solution. As suggested in reactions 1–3, the capacity of electrode surface for charge transfer and surface active sites to adsorb/desorb chloride ions determine the efficiency of chlorine evolution. These effects could be associated with the composition of valve metal oxides and the surface charge of anode, as evidenced in reports establishing that alloying, mixing and doping with these agents remarkably improve electro-catalysis.10–13 Therefore, a binary or ternary electro-catalyst containing RuO2 at a determined composition can be synthesized for these purposes.
The incorporation of Sb2O5 as dopant to DSAs has been typically oriented to improve electronic conductivity and the stability of DSAs.14,15 Chen et al. analyzed the electrochemical behavior of Ti/RuO2-Sb2O5-SnO2 (15:15:70, Ru:Sb:Sn), concluding that Sb improved the stability of RuO2 electrode, and its service life time.16 Cao et al. demonstrated that Sb incorporation into nanostructured RuO2 composites comprised of TiO2 nanotube arrays (TNTs) increased the anode activity in chloride media.17
On the other hand, ZrO2 is an inert oxide that despite its poor conductivity presents high corrosion resistance, good thermostability, and it has been widely used in catalysis as additive. Likewise, it has been proposed as "diluent" for RuO2,18 since it improves its dispersion and increases the number of catalytic active sites,19,20 although it has been seldom studied in a different application as diluent. Burke et al. demonstrated that the presence of ZrO2 in the active RuO2 layer substantially increases its life time during oxygen gas evolution.21 Comninellis et al. examined catalytic properties toward OER of binary coatings of RuO2 (conducting component) and ZrO2 (inert oxide).22 In spite of these important contributions, the effect of ZrO2 content to improve CER electro-catalysis on RuO2 materials has not been discussed to our current state of knowledge. Additionally, none study has accounted for the synergic effect that Sb doping and the addition of an agent dispersant (ZrO2) could stem in the morphology and capacity to form active chlorine species on the surface of a RuO2 catalyst.
In a previous work conducted by our research group, a stable Sb2O5 doped Ti/RuO2-ZrO2 catalyst (1:0.3:0.004 Ru: Zr: Sb) was designed for chlorine generation (oxidizing agent for indirect oxidation) to successfully degrade Indigo Carmine in a FM01-LC reactor and reactive black 5 at micro-electrolysis level.23,24 However, a fixed ZrO2 composition was used for this macroscopic analysis, whence an optimum ratio of this component in the ternary oxide and the electro-catalytic effects of varying its proportion toward CER remain unknown. This has spurred the present fundamental evaluation aiming to investigate the effect of ZrO2 content within Sb2O5 doped Ti/RuO2 anodes. The materials are prepared using Pechini method and their microstructural properties are characterized by X-ray diffraction (XRD) and scanning electronic microscopy (SEM-EDS). The efficiency to form active chlorine species is electrochemically (cyclic voltammetry) assessed against the competing oxygen evolution reaction occurring in NaCl and H2SO4 solutions; and the capacity of each catalyst to generate active chlorine species is iodometrically corroborated through UV-vis spectroscopy. The controlling effects of these mechanisms are discussed as a function of ZrO2 variation within Sb2O5 doped Ti/RuO2 catalysts.
Experimental
Preparation of electrodes
Sb2O5 doped-Ti/RuO2-ZrO2 films (Ti/SbRuZr 1, Ti/SbRuZr 2, Ti/SbRuZr 3 and Ti/SbRuZr 4, refer to Table I for further details of molar ratios) as well as the comparison electrodes (Ti/Ru, Ti/RuZr and Ti/SbRu) were impregnated on titanium plates using the Pechini method according to Palma-Goyes et al.23 Analytic grade reagents of RuCl3 (Sigma-Aldrich), ZrO(NO3)2·xH2O (Sigma-Aldrich) and SbCl3 (Sigma-Aldrich) were utilized as metallic precursors in a polymeric mixture prepared with a different molar ratio of Ru, Zr and Sb (Table I), with 2.4 mol L−1 citric acid as chelating agent and 18 mol L−1 ethylene glycol (EG) as reaction solvent. The polymeric mixture was kept stirred for 30 minutes at 80°C and subsequently deposited on Ti plates (2 × 2 cm), which were previously treated in oxalic acid (Merck) at 75°C for 30 min. These plates were dried at 180°C and calcined at 550°C for 1 h in a furnace. Both temperatures were selected from thermogravimetric analysis (TGA) and differential thermal analysis (DTA) of the raw polymeric mixture. The coating procedure was repeated eight times to thicken the coating.
Table I. Precursors and nominal molar ratios employed to prepare DSAs.
| Electrode | Metal precursor 1 | Metal precursor 2 | Metal precursor 3 | Molar ratio of metal 1 | Molar ratio of metal 2 | Molar ratio of metal 3 |
|---|---|---|---|---|---|---|
| Ti/Ru | RuCl3 | —– | —– | 1 | 0 | 0 |
| Ti/RuZr | RuCl3 | ZrO(NO3)2·xH2O | —– | 1 | 1 | 0 |
| Ti/SbRu | RuCl3 | —— | SbCl3 | 1 | 0 | 0.004 |
| Ti/SbRuZr 1 | RuCl3 | ZrO(NO3)2•H2O | SbCl3 | 1 | 1 | 0.004 |
| Ti/SbRuZr 2 | RuCl3 | ZrO(NO3)2•H2O | SbCl3 | 1 | 0.8 | 0.004 |
| Ti/SbRuZr 3 | RuCl3 | ZrO(NO3)2•H2O | SbCl3 | 1 | 0.5 | 0.004 |
| Ti/SbRuZr 4 | RuCl3 | ZrO(NO3)2•H2O | SbCl3 | 1 | 0.3 | 0.004 |
Microstructural characterization of electrodes
SEM images were collected to determine the morphology of deposited oxide films with a high-field emission microscope SM-7600F, using an accelerating voltage of 10.0 kV. Chemical composition was evaluated using energy-dispersive X-ray spectroscopy (EDS). X-ray diffraction (XRD) data were collected with a PANalytical Empyrian diffractometer in a Bragg-Brentano (θ-2θ) configuration operated with a Cu Kα1/Kα2 radiation (λ = 1.5418 Å) at 45 kV and 40 mA from 25 to 45° 2θ using a PIXcel 3D detector. The crystalline phases were identified using the JCPDS-ICDD cards. The full width at half maximum (FWHM) value for average crystallite size calculation using the Scherrer equation was obtained from a profile refinement using the FULLPROF program.
Electrochemical analysis
Cyclic voltammetry (CV) was conducted in an Autolab PGSTAT 302N potentiostat/galvanostat at a scan rate of 20 mV s−1, in 0.1 mol L−1 NaCl and 1.0 mol L−1 H2SO4 electrolytes at room temperature. A three-electrode cell (60 mL) was used to carry out these electrochemical experiments. DSAs (Ti/Ru Ti/SbRu, Ti/RuZr, Ti/SbRuZr 1, Ti/SbRuZr 2, Ti/SbRuZr 3, and Ti/SbRuZr 4) and Ag/AgCl(s)/KCl(3M) (Ag/AgCl) were used as working (3 cm2 of geometric area) and reference electrodes, respectively; and a pure graphite rod (0.5 mm diameter × 16 mm long, Alfa AESAR, 99.999%) as counter electrode.
Accelerated Life Test (ALT) was conducted under electrolysis at a constant current density of 1 A cm−2 in 1 mol L−1 H2SO4, using each DSA as working electrode (anode) and a titanium plate as counter (cathode). Failure of DSA was considered when the cell voltage reached 7 V vs Ag/AgCl from an initial value around 3 V.
Electroactive areas were estimated employing the facile kinetics of the Fe(CN)64−/Fe(CN)63− couple occurring on all DSAs. Randles-Sevcik equation as described for linear sweep voltammetry was used to evaluate these areas:25
![Equation ([4])](https://content.cld.iop.org/journals/1945-7111/163/9/H818/revision1/d0004.gif)
where n is the number of electrons participating in the oxidation of Fe(CN)64−/Fe(CN)63−, A is the electroactive area of the electrode (cm2), D is the diffusion coefficient of Fe(CN)64− species (cm2 s−1), C is its bulk concentration (mol cm−3), and v is the scan rate of the potential perturbation (V s−1). Peak current (ip) was sampled from multiple voltammograms conducted at different scan rates, and a plot of ip vs  was constructed. Note that for a reversible system, the peak current will linearly increase with
was constructed. Note that for a reversible system, the peak current will linearly increase with  . The slope of the resulting line is proportional to the electroactive area of the DSA (refer to Eq. 4). Table II presents electroactive areas evaluated for each DSA (Ti/Ru, Ti/SbRu, Ti/RuZr, Ti/SbRuZr 1, Ti/SbRuZr 2, Ti/SbRuZr 3, and Ti/SbRuZr 4).
. The slope of the resulting line is proportional to the electroactive area of the DSA (refer to Eq. 4). Table II presents electroactive areas evaluated for each DSA (Ti/Ru, Ti/SbRu, Ti/RuZr, Ti/SbRuZr 1, Ti/SbRuZr 2, Ti/SbRuZr 3, and Ti/SbRuZr 4).
Table II. Electroactive areas obtained from the parameters involved during 4 mmol L−1 Fe(CN)63−/Fe(CN)64− reduction on different electrode materials, and estimated using Randles-Sevcik equation (Eq. 4).
| Electrode | Electroactive area/cm2 |
|---|---|
| Ti/Ru | 4.0 |
| Ti/SbRu | 3.9 |
| Ti/RuZr | 7.2 |
| Ti/SbRuZr 1 | 7.6 |
| Ti/SbRuZr 2 | 6.9 |
| Ti/SbRuZr 3 | 5.0 |
| Ti/SbRuZr 4 | 8.0 |
Chemical analysis
Evolution of chlorinated oxidizing species was determined iodometrically.23,26 Aliquots were sampled from the reactor and added to a quartz cell containing potassium iodide (0.1 mol L−1) and ammonium heptamolybdate (0.01 mol L−1). The absorbance was recorded at 350 nm after 2 minutes of reaction. The formation rate of Cl2-active (μ mol L−1 min−1) was evaluated by linearizing the graph of evolution of Cl2-active (μ mol L−1) vs electrolysis time (min). This rate is an indicative of the amount of oxidizing agent formed per unit of time on the anode surface.
Results and Discussion
Microstructural characterization
Fig. 1a shows SEM images collected for the coated surfaces varying the ZrO2 content at three different magnifications (×1000, 3000 and 10.000). As observed, the anode morphologies are significantly affected by the presence and concentration of ZrO2 oxide, and they consist of mud-cracks structures combined with flat areas. Additionally, the ZrO2 presence changes the compact appearance of the film by a heterogeneous morphological aspect. This provides an indirect evidence of a homogeneous distribution of a visible porosity across the film surface which alters grain growth, and increase the number of cracks and surface area of electrode. Although the Sb2O5 content does not significantly modify the morphology of the Ti/RuO2 coating, there is an effect when it coexists with ZrO2 in the ternary oxide (Ti/SbRuZr 1, Ti/SbRuZr 2, Ti/SbRuZr 3 and Ti/SbRuZr 4). This effect consists of increasing the visible porosity level as the ZrO2 content is augmented. On the other hand, the EDS mapping of the element distributions on Ti/RuZr, Ti/SbRu and Ti/SbRuZr 1-Ti/SbRuZr 4 coatings provides clear evidence of an uniform element distribution on the electrode surface that remains homogeneous even with the ZrO2 increment, and the dispersion of RuO2, ZrO2 and Sb2O5 phases. Table III presents SEM-EDS analysis for each anode, making evident that the trend of nominal composition (Table I) is kept for Ru, Zr and Sb distributions.

Figure 1. SEM images obtained for different DSAs deposited on Ti using Pechini method. Molar ratios used to prepare the anodes are presented in Table I.
Table III. Atomic metal composition obtained from SEM-EDS analyses for each DSA.
| Electrode | % Ru | % Zr | % Sb | % O | % Ti |
|---|---|---|---|---|---|
| Ti/Ru | 12.1 | 0 | 0 | 66.9 | 20.8 |
| Ti/RuZr | 13.8 | 12.48 | 0 | 73.0 | 3.68 |
| Ti/SbRu | 14.1 | 0 | 1.06 | 71.1 | 13.0 |
| Ti/SbRuZr 1 | 9.26 | 14.95 | 1.26 | 73.2 | 1.28 |
| Ti/SbRuZr 2 | 10.2 | 10.7 | 1.55 | 74.0 | 3.34 |
| Ti/SbRuZr 3 | 16.7 | 9.49 | 1.60 | 70.7 | 1.39 |
| Ti/SbRuZr 4 | 12.7 | 2.76 | 1.07 | 70.6 | 12.9 |
Fig. 2 displays the XRD analysis conducted with the electrode films supported on Ti. Fig. 2a shows X-ray patterns for Ti/Ru, Ti/SbRu and Ti/RuZr, and Fig. 2b shows the corresponding experiments for Ti/SbRuZr 1-Ti/SbRuZr 4. The identification of the diffraction peaks corresponds to the files reported in the JCPDS database. Peaks in Fig. 2a and 2b at 2θ = 28 and 36 are attributed to rutile-type phase (RuO2 tetragonal, P42/mnm), while peaks at 31, and 39 along with 41 are for ZrO2 tetragonal (P42/mnm) and Ti hexagonal (P63/mmc), respectively. Signals related to Sb2O5 are not detected, most likely due to the lowest molar content used to fabricate the anodes. The presence of defined signals associated to RuO2 and ZrO2 confirm co-deposited phases in agreement with SEM-EDS results, and studies described by Hrovat et al.27 and Comninellis et al.22 for Ti/RuO2 with ZrO2 as metal valve. An additional feature from Fig. 2b reveals that the ZrO2 tetragonal phase becomes evident as the ZrO2 content is increased from 0.5 Zr/Ru metal molar ratio. This information discards the formation of solid solution between both oxides, which can result from a favored preferential crystal orientation in the [−1 1 0] direction due to the natural layer of TiO2 grown on Ti substrate. According to the lattice parameters (a, b, c) obtained by a matching profile refinement, the planes (1 1 0) for TiO2 and RuO2 present a 98% matching in the [h k 0] direction. Concerning to the main crystalline peak (0 1 1) of the tetragonal structure for the ZrO2, this plane presents 87% matching in the [h 0 0] direction. However, the monoclinic phase of ZrO2 is not excluded since its main peak is found in the same 2θ region as the main peaks of TiO2 and RuO2 phases (27-28 degrees). This behavior was also reported by Srinivasan et al., analyzing the effect of sulfate on the crystal structure of zirconia.28
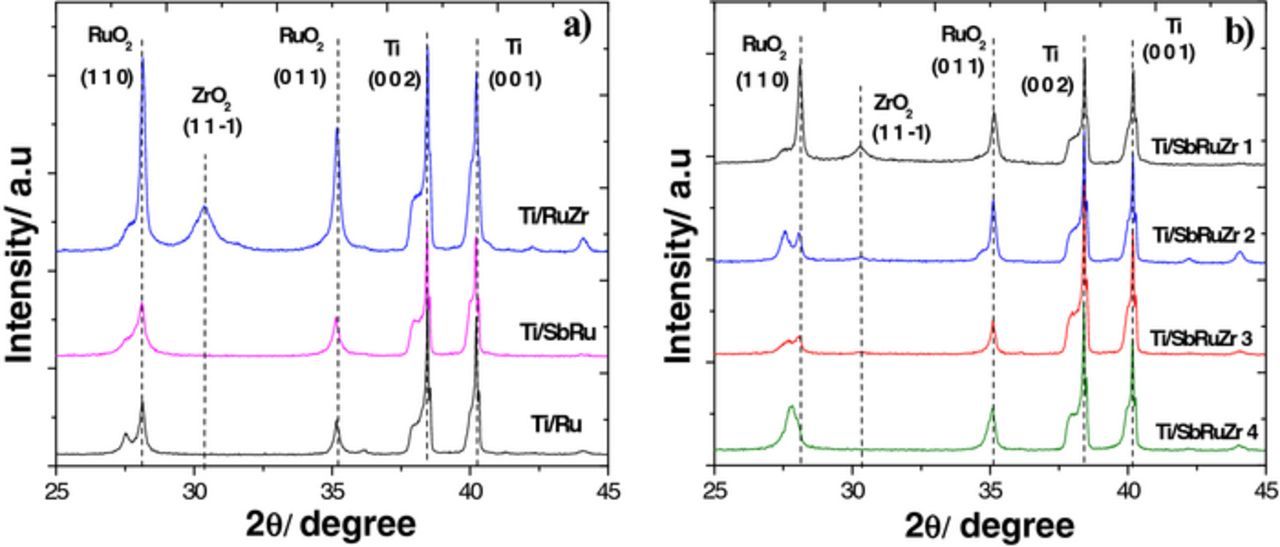
Figure 2. XRD patterns collected from the following DSAs: a) Ti/Ru, Ti/RuZr and Ti/SbRu, b) Ti/SbRuZr 1, Ti/SbRuZr 2, Ti/SbRuZr 3, Ti/SbRuZr 4.
On the other hand, a semiquantitative calculation of average crystallite size on the surface was estimated using the Caglioti formula and the instrumental contribution was subtracted using a LaB6 NIST standard reference in order to obtain a more accurate description of its microstructure. The average crystallite size of both anodes was then calculated using the Scherrer equation (Eq. 5), using the principal plane (1 1 0) in the tetragonal phase, and determining the half maximum intensity.
![Equation ([5])](https://content.cld.iop.org/journals/1945-7111/163/9/H818/revision1/d0005.gif)
where τ is the mean grain size; λ is the X-ray wavelength; β is the line broadening at half maximum intensity (FWHM), and θ is the Bragg angle. Results obtained from Scherrer equation (Table IV) reveal that crystallite size is considerably decreased with ZrO2 incorporation. Nucleation and growth processes could account for this behavior. The [Ru(H2O)6]3+ complex is coordinated to hydroxyl and carboxyl groups from the citric acid, and subsequently dispersed via ethylene glycol solvent. A random metal distribution occurs as ZrO(OH)+ ion complex is incorporated into the polymeric matrix. After that, the nucleation starts during the thermal oxidation process for both amorphous oxides, and the formation of a crystalline pattern is revealed by XRD, as the temperature rises above 500°C. RuO2 crystal growth relies on the chemical homogeneity of the medium, and the presence of ZrO2 directly affects the limit of crystal growth (size of RuO2) since these phases cannot form a solid solution. Note that most likely this effect could increase the surface area of the coatings, significantly affecting the electro-catalytic activity of the synthesized materials.
Table IV. Average crystallite size of electro-catalysts obtained from Scherrer equation.
| Electrode | Average crystallite size/nm |
|---|---|
| Ti/Ru | 108.87 |
| Ti/RuZr | 79.52 |
| Ti/SbRu | 108.6 |
| Ti/SbRuZr 1 | 61.17 |
| Ti/SbRuZr 2 | 63.30 |
| Ti/SbRuZr 3 | 61.24 |
| Ti/SbRuZr 4 | 28.78 |
Electrochemical analysis to evaluate ZrO2 and Sb2O5 effects on Cl2 active rate formation
DSAs are known to be good catalysts to yield oxygen and chlorine species. However, the electro-catalysts synthesized in the present study are intended to form active chlorine, whereby it is overriding to increase the overpotential (i.e. energy barriers) of OER, since it can efficiently compete against CER due to the water presence in chloride media. In order to evaluate the occurrence of oxygen evolution and chorine evolution reactions for Ti/Ru, Ti/RuZr, Ti/SbRu, Ti/SbRuZr 1, Ti/SbRuZr 2, Ti/SbRuZr 3 and Ti/SbRuZr 4 catalysts, CV experiments were initially conducted in electrolytes containing 1.0 mol L−1 H2SO4 (Figure 3), where it is only expected the occurrence of OER due to the absence of chlorides. The selection of a low pH is mainly dictated by the requirement of a stable and active material for OER under acidic conditions.29 Fig. 3a shows CVs conducted on Ti/Ru, Ti/RuZr, Ti/SbRu and Ti/SbRuZr 1 coatings (refer to Table III for further details of composition). The inset of this figure presents the typical behavior of the Ti/Ru catalyst, describing changes in the Ru oxidation state during the potential scan (OCP to 1.0 V vs Ag/AgCl), which agrees with characteristic shapes reported in the literature for this metal.30,31 The curve shows broad peaks around 0.5 V vs Ag/AgCl, which are attributed to the surface redox transition from Ru (III)/Ru (IV) and Ru (IV)/Ru (VI) oxidation states. A similar behavior was obtained for the other coatings (data not shown). Sampling of OCP and polarization values under the occurrence of OER at 3.3 mA cm−2 are summarized in Table V for each electrode, in order to establish a quantitative comparison between catalysts for this contribution. Note that this current has been selected since it generates an important occurrence of OER for all CV experiments (i.e. significant rise in current density). An additional feature in Fig. 3a reveals that the OER potential (around 1.37 V) on Ti/Ru is significantly shifted to more positive potentials around 1.47 V and the current density significantly lowered down, when ZrO2 is incorporated into the electrode (i.e. Ti/RuZr). This is qualitatively corroborated in the polarization values shown in Table V. These observations could be associated with a lower conductivity of ZrO2, which also inhibits this reaction. On the other hand, the addition of Sb2O5 (Ti/SbRu) displaces the onset of OER to less positive potentials (1.29 V), and the current becomes higher with respect to the sole Ti/Ru catalyst, confirming the enhancement of electronic conductivity due to Sb addition. Note that for Ti/SbRuZr 1 electrode, the potential to start OER is slightly less positive compared to Ti/Ru, which implies that the Sb incorporation into the Ti/RuZr anode removes the OER inhibition occurring on the binary catalyst (Ti/RuZr). Thus, the new question arising due to this finding is whether the presence of Sb in the ternary electrode catalysts the OER.
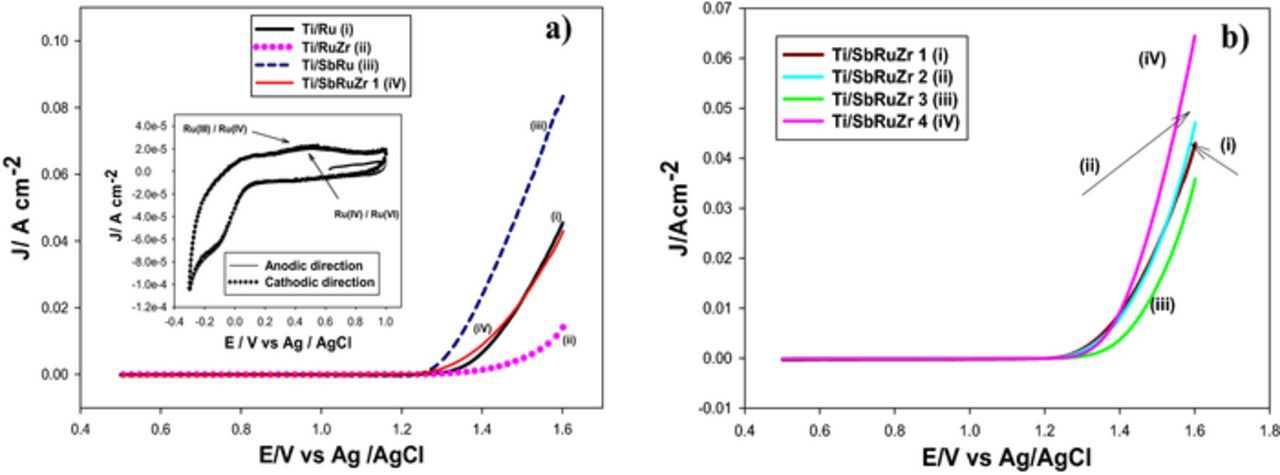
Figure 3. Cyclic voltammetry experiments conducted in electrolytes containing 1.0 mol L−1 H2SO4 using a scan rate of 20 mV s−1 on the following DSAs: a) Ti/Ru, Ti/RuZr and Ti/SbRu 1, b) ternary oxides Ti/SbRuZr 1, Ti/SbRuZr 2, Ti/SbRuZr 3 and Ti/SbRuZr 4. The figure inset in a) displays a magnification in the range from −0.4 to 1.0 V for the Ti/Ru catalyst, where the scan potential was initiated in anodic and cathodic directions.
Table V. Open circuit potentials and polarization values for oxygen evolution reaction calculated from voltammetry analyses at 3.3 mA cm−2 for DSAs.
| Electrode | OCP/V vs Ag/AgCl | Polarization*/V |
|---|---|---|
| Ti/Ru | 0.66 | 0.71 |
| Ti/RuZr | 0.59 | 0.88 |
| Ti/SbRu | 0.57 | 0.72 |
| Ti/SbRuZr 1 | 0.62 | 0.71 |
| Ti/SbRuZr 2 | 0.62 | 0.72 |
| Ti/SbRuZr 3 | 0.71 | 0.68 |
| Ti/SbRuZr 4 | 0.63 | 0.71 |
*Calculated from the difference between applied potential and OCP at a rate of 3.3 mA cm−2.
In order to clarify the above statement, CV experiments were carried out in 1.0 mol L−1 H2SO4 electrolytes to compare the OER electro-activity of ternary electrodes. Fig. 3b displays this comparison on ternary catalysts (Ti/SbRuZr 1, Ti/SbRuZr 2, Ti/SbRuZr 3, Ti/SbRuZr 4). Note that SbRuZr 1 presents the highest content of ZrO2 in the catalyst, while SbRuZr 4 possesses the lowest (Table III). For Ti/SbRuZr 1 and Ti/SbRuZr 2 electrodes, the onset potential to perform the OER is virtually the same, and even similar current densities are obtained, which suggests that for the range of Zr composition in these catalysts, the kinetics of this contribution does not considerably vary. Note that for these two electrodes the % of Sb fluctuates from 1.26 to 1.55 (Table III). When the Zr content is decreased in the catalyst to 9.49 (Ti/SbRuZr 3), the potential to start OER is displaced to more positive potentials, which could contradict the fact that this input is inhibited increasing the Zr composition in the anode. However, this experiment indicates that Sb element also mitigates the occurrence of OER when combined with Zr, since its composition rose to 1.60 at % for this electrode (refer to Table III). This behavior is confirmed when the Sb content is lowered down to 1.07 at % for Ti/SbRuZr 4, and the least positive potential is obtained for OER, although it is worthwhile mentioning that the Zr at % drops drastically to 2.76. In addition, the Ti/SbRu anode in Fig. 3a which presents a similar Sb composition (without containing Zr) to Ti/SbRuZr 4 corroborates this trend. These evaluations suggest that when Sb2O5 and ZrO2 are combined in the catalyst, the increase of both compounds is effective to suppress the OER, revealing an additional role for Sb. While in the absence of ZrO2, Sb2O5 catalyzes the OER (Ti/SbRu). It has been reported that in order to enhance the activity of RuO2 catalyst, its first coordination shell can be altered by substituting attached surface O atoms by Cl or S; or either the replacement of Ru atoms in the second coordination shell by another metal atom, as adopted from homogeneous catalysis (i.e. active field of mixed-oxide catalysis).32 In line with the second approach, multiple theoretical studies have been conducted with ab initio DFT methods to evaluate the ligand effects of metallic species (i.e. Cr, Ir) on the OER electro-catalysis for RuO2(110).32,33 To this concern, Exner et al. have concluded on a thermodynamic basis that the free adsorption energy for the oxygen bond to coordinate unsaturated Ru atom (i.e. active center of RuO2 catalyst) can be diminished by 0.8 and 1.0 eV when Ru 2f atoms in the second coordination shell are selectively replaced by Ir or Cr, respectively. This drop in the bond strength represent substantial abatements in the thermodynamically part of OER reaction barrier (i.e. Gibbs free-energy loss). In connection with the above findings concerning the suppression of OER by combination of Sb2O5 and ZrO2 within RuO2 structure, it is plausible to suggest that when Sb is only included in the catalyst, the same catalytic effect for OER is observed as that reported for Ir or Cr.32 Note that Sb is contained in compositions lower than 1.5 at. %, whence there is a high probability that it targets the substitution of Ru 2f atoms in the second coordination shell; while the sole addition of Zr (Ti/RuZr) in contents around 12.48% could not only replace Ru 2f atoms, but also the coordinatively unsaturated Ru atoms, thus, inhibiting the electro-catalysis of RuO2 active centers. When both Zr and Sb are included in the catalyst (i.e. ternary materials), there is a generalized inhibitory effect toward OER due to the high composition of these elements which most likely substitute unsaturated Ru atoms, except for Ti/SbRuZr 4 catalyst where this substitution is diminished, as observed in their lower compositions (Table III). However, these ideas need to be strongly corroborated on a theoretical basis, since they contradict the experimental findings reported above, concerning the formation of co-deposited phases for RuO2 and ZrO2 (e.g. non solid solution).
To date, it is not clear whether the content of Zr or Sb is more crucial to perform the CER, whence the electrochemical behavior of these materials needs to be assessed in chloride media. In order to corroborate the effect of Zr content in the active chlorine evolution on ternary oxides, CV experiments were carried out in chloride media. Ti/SbRu catalyst was not considered since it was demonstrated that it has no effect on CER (not shown). Fig. 4 shows CVs conducted on Ti/Ru, SbRuZr 1, Ti/SbRuZr 2, Ti/SbRuZr 3 and Ti/SbRuZr 4 electrodes in 0.1 M NaCl. A comparison with the experiments shown in Fig. 3 reveals clear differences in the OER performed in sulfate media, particularly the behavior observed in the presence of chlorides indicates that CER occurs more facile than oxygen evolution on each electrode (i.e. lower overpotential), whereby it can be assumed that the main reaction pathway has changed with the change of electrolyte, thus, the charge of these experiments (Fig. 5) could be mainly related to CER inputs. Although it is not possible to determine how much CER contributions exceed to OER with the current experiments. The behavior obtained in Fig. 4 shows that at a fixed current, the lower the potential is, the higher the electro-catalytic activity will be. Therefore, it is observed that the CER catalysis increases for the ternary electrodes as the Zr content is increased regardless of the Sb composition in the anode, whereby the best catalysis for this reaction is obtained for Ti/SbRuZr 1 which contains a Zr at % of 14.95 (Table III), and the second lowest content for Sb (1.26 at %). These results indicate that the ZrO2 composition is way more critical than the Sb2O5 content to catalyze the CER, although both combined compounds assist to suppress the OER. Additionally, it is necessary to consider the influence of the average crystallite size (Microstructural characterization section) upon the increase of the electroactive area, which could remarkably contribute to the CER catalysis along with ZrO2 composition. To this concern, Table II displays electroactive areas estimated using Randles-Sevcik equation (Eq. 4), and the parameters involved during Fe(CN)64−/Fe(CN)63− oxidation on the different DSAs. As observed, the incorporation of Zr to the Ti/Ru material significantly increases the active area, while Sb addition slightly diminishes this area. When both metals (Zr, Sb) are combined in the ternary catalyst, the electroactive surface is considerable higher than for the Ti/Ru electrode. Note that the highest (Ti/SbRuZr 4) and lowest (Ti/SbRuZr 3) electroactive areas for the ternary anodes are obtained for the lowest and highest content of Sb (refer to Table III), respectively; indicating that the increase of Sb composition is a detriment for the electroactive area of ternary catalysts (Ti/SbRuZr).
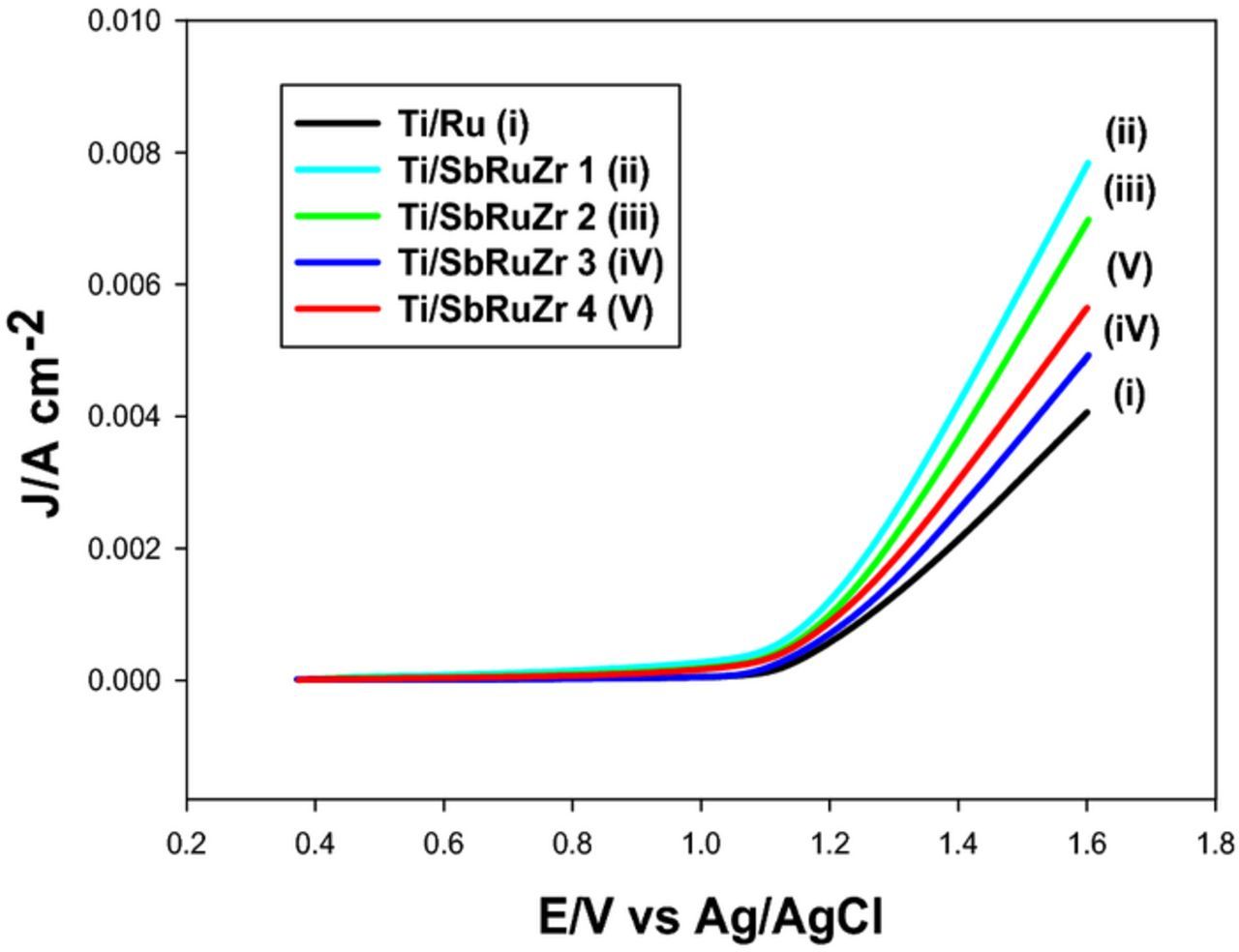
Figure 4. Cyclic voltammetry experiments conducted in electrolytes containing 0.1 mol L−1 NaCl using a scan rate of 20 mV s−1 for multiple DSAs indicated on the plot.
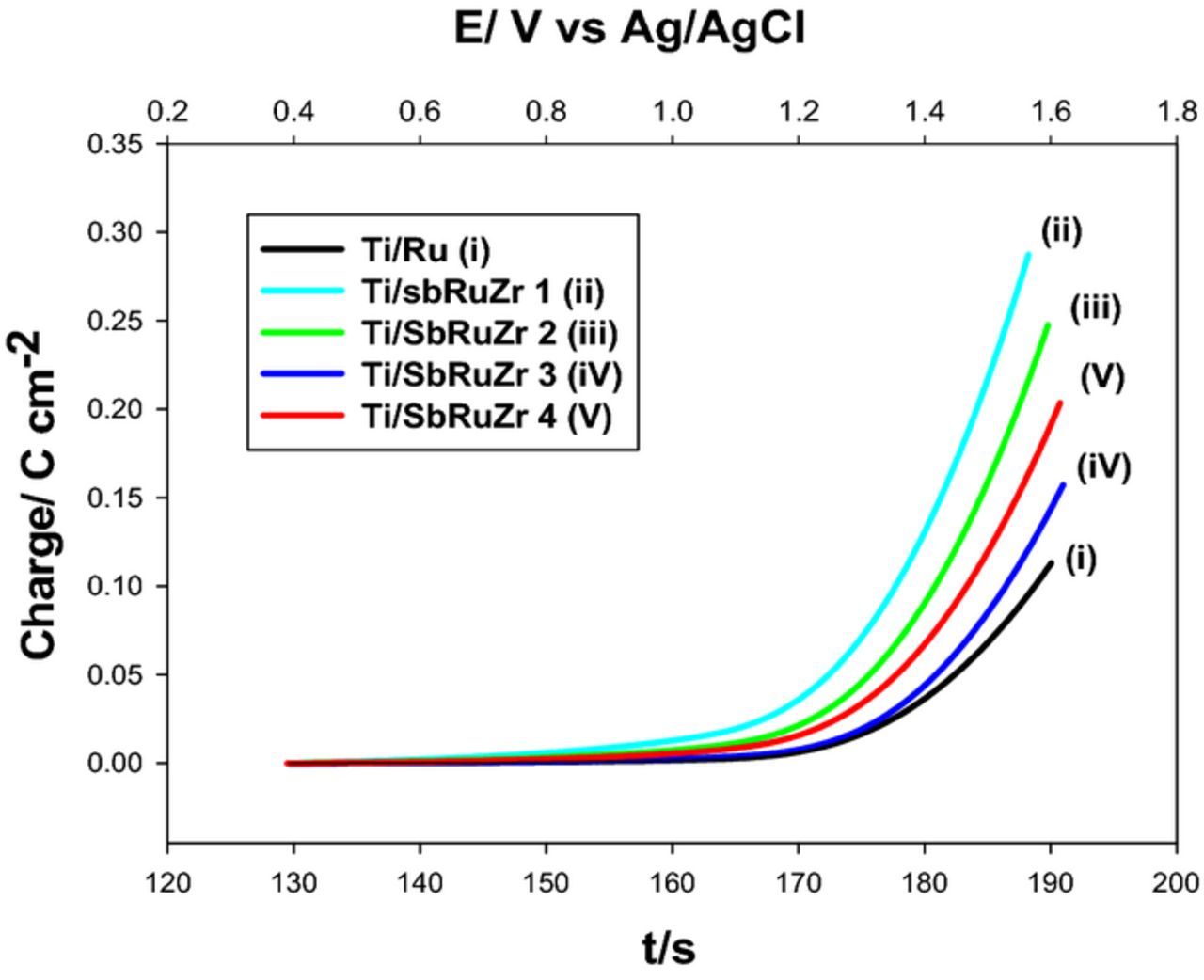
Figure 5. Charge as a function of time and potential calculated from cyclic voltammetry performed in electrolytes containing 0.1 mol L−1 NaCl for multiple DSAs indicated on the plot.
Again, explanations to this behavior are supported on the basis of DFT studies conducted to analyze the chlorine evolution reaction on RuO2(110) by Exner et al.,34 which is a slightly different mechanism to the active chlorine formation evaluated in the present study. According to this work, the chlorine evolution reaction preferentially follows a Volmer-Heyrovsky mechanism, which is enhanced by substituting the top most Ru layer of the fully O-covered RuO2 (110) surface (i.e. debilitating the metal-O bonding) with a different metal atom (i.e. platinum oxide). This diminishes the Gibbs energy loss by 50 meV, and modifies the elementary reaction step with highest energy, considered the adsorption of chlorine for the O-covered RuO2 catalysts to the recombination step to form gaseous chlorine for the Pt-modified RuO2 surface. As described above for the CV experiments, the addition of Zr is in similar molar ratios compared to Ru for most ternary catalysts (Table I), except for Ti/SbRuZr 3 and Ti/SbRuZr 4, whence it is suggested that Zr replaces Ru atoms from the second coordination shell and surface Ru atoms during synthesis. In line with the theoretical results presented by Exner et al.,34 the substitution of topmost Ru layer by ZrO2 would generate the CER electro-catalysis, unlike the occurrence for OER. However, this insight needs a comprehensively evaluation since a solid solution is not formed between RuO2 and ZrO2 phases, as previously described in Microstructural characterization section.
Other voltammetry results of DSA electrodes containing SnO2 instead ZrO2 (Ti/RuO2-Sb2O5-SnO2) in NaCl and Na2SO4 electrolytes have shown that chlorine evolution reaction (CER) occurs more facile than oxygen evolution,35 and depending on Cl− concentration this reaction could initiate at lower overpotentials. Under this premise, a comparison of the onset potential for CER and charge (Q, number of mol electrolyzed to carry out the CER) was determined for each electrode in 0.1 mol L−1 NaCl. As observed in Fig. 5, a similar trend to that shown in Fig. 4 is obtained for the charge as a function of potential and time, confirming that the increase of ZrO2 content enhances the catalytic behavior toward Cl2-active evolution.
In order to further clarify the effects of ZrO2 and Sb2O5 on the RuO2 catalytic activity toward Cl2-active, their rate formations (k Cl2-active) were individually determined for each anode in 0.1 mol L−1 NaCl solution at 17 mA cm−2 (Chemical analysis section). The efficiency of these catalysts was followed (i.e. evolution of Cl2-active concentration) as a function of electrolysis time. These experiments were conducted at low current densities in order to mitigate the occurrence of OER, which can arise as a side competitive reaction at higher currents. The results obtained from this analysis reveal that the Cl2-active formation on each anode follows zero order kinetics (Eqs. 6 and 7):
![Equation ([6])](https://content.cld.iop.org/journals/1945-7111/163/9/H818/revision1/d0006.gif)
![Equation ([7])](https://content.cld.iop.org/journals/1945-7111/163/9/H818/revision1/d0007.gif)
where  denotes the Cl2-active concentration during electrolysis (μmol L−1),
denotes the Cl2-active concentration during electrolysis (μmol L−1),  is the initial concentration of former species (μmol L−1),
is the initial concentration of former species (μmol L−1), represents the formation rate of Cl2-active (μmol L−1 min−1) and t is a given time during electrolysis (min). Figure 6 shows the
represents the formation rate of Cl2-active (μmol L−1 min−1) and t is a given time during electrolysis (min). Figure 6 shows the  values calculated for each electrode. As observed, there are remarkable differences in the electro-catalytic activity to perform the Cl2-active evolution on ternary catalysts. This result confirms that the Ti/SbRu anode does not catalyze the CER if compared with the pure phase of RuO2 (Ti/Ru), which agrees with the CV analysis shown in Fig. 3 where the Ti/SbRu catalyst enhances the OER. Note also that the Ti/Ru anode presents the largest overpotential to perform the CER (Fig. 4). In contrast, the ZrO2 presence in Ti/Ru increases twice the
values calculated for each electrode. As observed, there are remarkable differences in the electro-catalytic activity to perform the Cl2-active evolution on ternary catalysts. This result confirms that the Ti/SbRu anode does not catalyze the CER if compared with the pure phase of RuO2 (Ti/Ru), which agrees with the CV analysis shown in Fig. 3 where the Ti/SbRu catalyst enhances the OER. Note also that the Ti/Ru anode presents the largest overpotential to perform the CER (Fig. 4). In contrast, the ZrO2 presence in Ti/Ru increases twice the  values, significantly affecting the CER in the system, e.g. Ti/RuZr and ternary electrodes. In summary, the ZrO2 and Sb2O5 presence (Ti/SbRuZr 1, Ti/SbRuZr 2, Ti/SbRuZr 3 and Ti/SbRuZr 4) on Ti/RuO2 suggests that the former reaction depends on ZrO2 concentration, and it is enhanced when its content is increased. A low composition of ZrO2 (Ti/SbRuZr 4) generates a similar formation rate to generate Cl2-active compared to Ti/RuZr anode, revealing the important effect of this element to improve the electro-catalytic properties of DSAs. All these results adequately agree with the electrochemical tests conducted in sulfate and chloride media.
values, significantly affecting the CER in the system, e.g. Ti/RuZr and ternary electrodes. In summary, the ZrO2 and Sb2O5 presence (Ti/SbRuZr 1, Ti/SbRuZr 2, Ti/SbRuZr 3 and Ti/SbRuZr 4) on Ti/RuO2 suggests that the former reaction depends on ZrO2 concentration, and it is enhanced when its content is increased. A low composition of ZrO2 (Ti/SbRuZr 4) generates a similar formation rate to generate Cl2-active compared to Ti/RuZr anode, revealing the important effect of this element to improve the electro-catalytic properties of DSAs. All these results adequately agree with the electrochemical tests conducted in sulfate and chloride media.
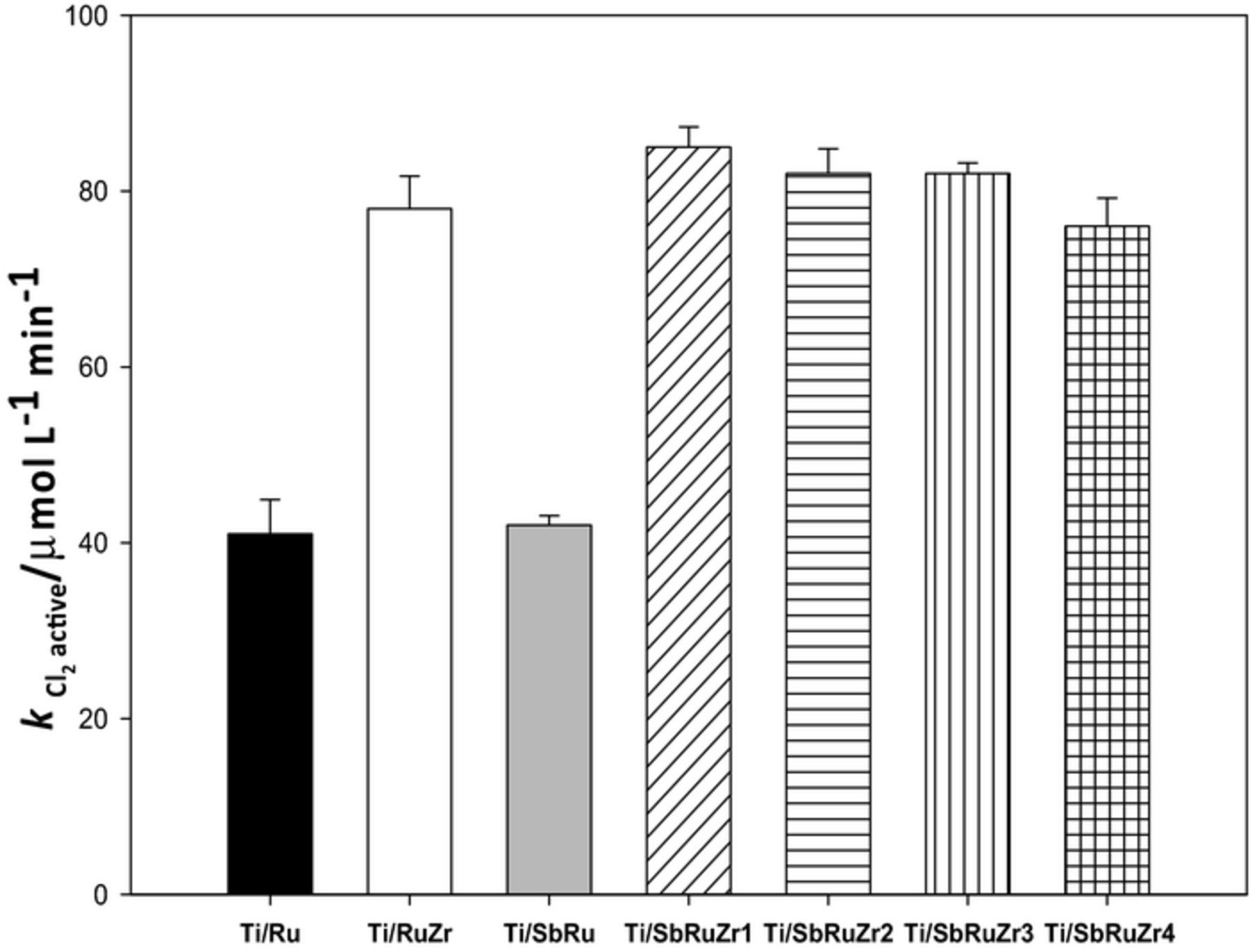
Figure 6. Rate formations of Cl2-active species (k Cl2-active) determined for different DSAs using an iodometric method with UV-Vis spectroscopy.
Accelerated life test for DSA
Electrochemical stability of DSA in a corrosive environment or electrolyte of interest is an overriding factor to consider in order to evaluate its long-term performance, along with its efficiency to catalyze the CER or OER. Thus, accelerated life tests (ALT) were conducted for the Sb2O5 doped-Ti/RuO2-ZrO2 anodes (Ti/SbRuZr 1, Ti/SbRuZr 2, Ti/SbRuZr 3 and Ti/SbRuZr 4), and comparison catalysts (Ti/Ru, Ti/RuZr and Ti/SbRu) using the procedure described in Electrochemical analysis section. It is worthwhile mentioning that in ALT, the anode lifetime is defined as the time at which its potential suddenly becomes more positive during oxygen evolution reaction, occurring under galvanostatic mode. As observed in Fig. 7, the voltage of the Ti/Ru material slowly increases during the early stages of testing (∼5 000 s), attaining a pseudo-stationary potential around 3 V. After this time, the anode voltage rapidly augments to higher values than 6 V (∼9 000 s). Similar behaviors are observed for Ti/SbRu and Ti/RuZr anodes, their voltages are relatively constant around 3 V during the first 5 000 s, and they increased up to 6 V after 7 200 and 9 360 s, respectively. This indicates that Zr addition improves electrode stability during the occurrence of oxygen evolution reaction in acid solution, however, Sb incorporation generates an antagonistic effect on this stability. On the other hand, all ternary anodes (Ti/SbRuZr 1-Ti/SbRuZr 4) present failure times (∼12 000 s) considerably longer than Ti/Ru, Ti/SbRu and Ti/RuZr, suggesting a stability enhancement due to the combined presence of Sb and Zr species into the catalysts. Among ternary electrodes, Ti/SbRuZr 3 (Zr/Ru 0.5) displays the highest stability up to 22 860 s.
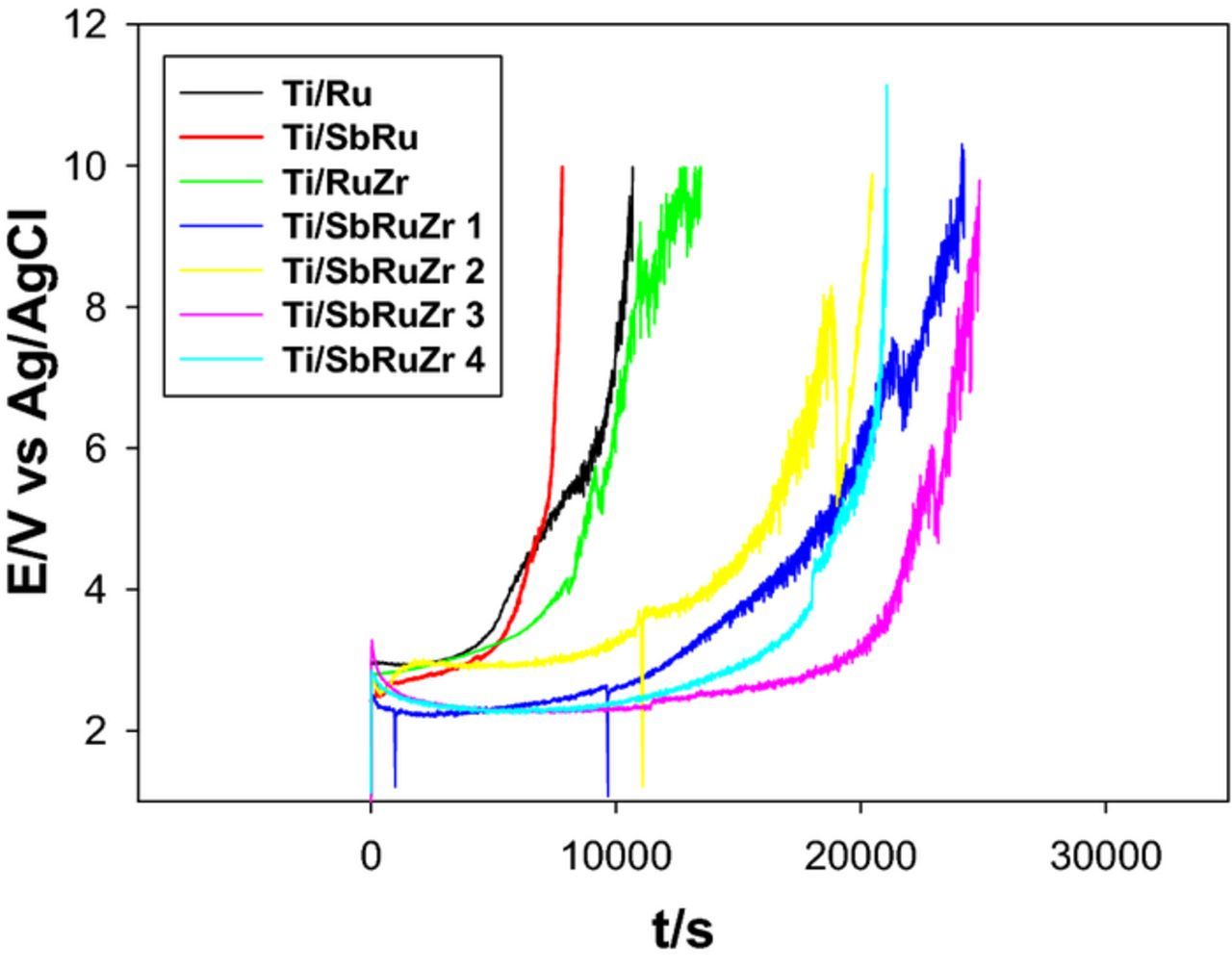
Figure 7. Variation of anode potential as a function of time during accelerated life test conducted for different DSAs and Ti/Ru catalyst at 1 A cm−2 in 1 mol L−1 H2SO4.
In literature, electrode failure has been reported as the consequence of several complex mechanisms: (1) erosion due to gas bubbles physically formed on its surface and external pores, (2) dissolution of electrocatalyst by electrochemical oxidation of RuO2, generating soluble products (e.g. RuO4) into the electrolyte, and (3) passivation of substrate as a result of Ti oxidation, which grows due to oxidizing agents to form an insulating TiO2 layer.36,37 In the present work, the color of electrolyte solution (volume of 150 mL) turned light blue at the end of ALT for all electrodes, whence it is suggested that ruthenium dissolution leading to the formation of soluble Ru species is most likely the main mechanism causing electrode failure.
Conclusions
DSAs (Sb2O5-doped Ti/RuO2-ZrO2) with different ZrO2 contents were prepared by Pechini method, and microstructurally and electrochemically characterized to evaluate their catalysis to perform the CER in chloride media. XRD data revealed co-deposited phases for RuO2 and ZrO2 structures within ternary catalysts, while SEM micrographs showed substantial changes in film morphology and a decrease of average crystallite size as ZrO2 content was increased. CV analysis demonstrated that one of the roles of Sb2O5 is increasing the coating conductivity, and its sole presence within the RuO2 structure catalyzed the OER compared to the Ti/Ru and ternary electrodes. Additionally, the electrochemical evaluations suggested that when Sb2O5 and ZrO2 were combined in the catalysts, the increase of both compounds was effective to suppress the OER. CV experiments along with an iodometric method indicated that although the chlorine evolution reaction preferentially occurred by the suppression of OER, the ZrO2 composition was more critical than the Sb2O5 content to catalyze the CER, even at low compositions. Accelerated life tests performed with all DSAs exposed that all ternary anodes (Ti/SbRuZr 1-Ti/SbRuZr 4) presented failure times (∼12 000 s) considerably longer than Ti/Ru, Ti/SbRu and Ti/RuZr electrodes (<9 400 s), suggesting a stability enhancement due to the combined presence of Sb and Zr species into the catalysts.
First-principle calculations will be the motivation of forthcoming analyses intended to comprehensively investigate the underlying chemistry of the experimental findings reported in the present work, particularly the catalytic effects of RuO2 and ZrO2 mixtures, and whether they are able to display this behavior as co-deposited phases.
Acknowledgments
Financial support is greatly appreciated from SECITI-CDMX-MEXICO (Grant No. 2016-272), and Universidad de Antioquia (Sostenibilidad Program 2014-2015, Colombia). J. Vazquez-Arenas also acknowledges the Cátedras-CONACYT (Mexico) program via project No. 1456. R. Palma-Goyes would like to thank COLCIENCIAS, Convocatoria No. 567-2012 for the financial support to pursue his Ph.D., as well as project No. 111565842980 from COLCIENCIAS, convocatoria 658, 2014.