Abstract
A low-cost, thin, graphitized carbon electrode with electrodeposited platinum and polyaniline has been characterized for electrochemical oxidation of aliphatic alcohols in alkaline medium and for possible further incorporation of atomic metals and alloys. Our previously developed atomic metal polyaniline composites used solid platinum as the electrode material. The replacement of this solid platinum with a small amount of electrochemically annealed Pt on a carbon support was optimized for alcohol oxidation. An HPLC procedure for analysis of oxidation products of 1-propanol has also been utilized for the characterization of the composite electrode.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
The preparation of composite materials that function as electrocatalysts is an active field of research. The in-situ incorporation of metal clusters on an atomic scale, such as Aun and AunPdm (1 ≤ n,m ≤ 8), in a polyaniline matrix has been developed1–5 and their use for oxidation of alcohols in alkaline media has been demonstrated.6–8 It has been shown that the number of atoms, and the order in which they are added, tracks their electrocatalytic activity in excellent agreement with calculated electron affinity.9,10 In order to generate maximum interaction between the active atomic metal centers embedded in polyaniline (PANI), the close coupling and synergistic effects of the composite components in the support cannot be overlooked. Carbon supported platinum is widely used as a composite electrode due to its known activity toward alcohols,11 and composites of carbon-platinum-polyaniline have been studied in acidic media.12 Among all the different preparation procedures of Pt clusters - e.g. dispersement in paste and powders,11 ion exchange,13 impregnation,13 colloidal precipitation,13 vapor deposition, potential-step,11,14 potential cycling,13 and chronoamperometric15 - the potentiostatic electrodeposition method16,17 remains one of the best due to its high reproducibility and controllability. The forms of carbon that are ideal for potentiostatic deposition include glassy carbon,18 nanotubes,16 pyrolytic,14,17 graphite cloth, fiber, and paper.15 Platinum is known to have the highest activity for the oxygen reduction reaction (ORR) in alkaline media.19
Size and structural effects have remained the main issue for loading Pt nanoparticles into the different types of carbon support but an indirect influence of the support may also affect the rate of metal dispersion, the accessibility of active sites or even its own catalytic activity. One of the attractive features of composites prepared on atomic scale is dramatic reduction of the amount of precious metals in the composite electrode. In the case of atomic gold, such reduction is estimated to be on the order of 10,000. However, all of our previous preparations have been done on a solid Pt substrate coated with PANI matrix. Thus, the primary goal of this study has been replacement of expensive Pt with a less expensive electroactive material, e.g. carbon. However, a small amount of Pt is necessary for oxidation of alcohols in alkaline medium.7 The secondary question to be answered was therefore, what is the minimum coverage of Pt on the carbon electrode?
In this study, thin, electrically conductive synthetic graphite plates processed from petroleum coke were used as electrodes for platinum deposition. By varying the platinum loading onto the carbon support and keeping the amount of PANI constant, performance of the composite electrode was studied. The probe used for testing the catalytic activity of these electrodes was 1-propanol. HPLC was used for product analysis during electrooxidation of 1-propanol. The activity of the supported Pt catalyst toward oxidation of 1-propanol was compared before and after polymerization of thin PANI film. We outline a method for preparation and annealing of a carbon-platinum-polyaniline (C/Pt-PANI) composite. The planned use of such an electrode is for insertion of catalytically active metal clusters on the atomic scale. Clusters of gold containing 1–7 atoms of metal deposited onto platinum-supported PANI showed odd-even changes in catalytic activity toward oxidation of aliphatic alcohols and enhanced activity compared to polycrystalline gold.4,6,7 The loss of catalytic activity of gold supported only on carbon in alkaline media makes the presence of platinum on the carbon support a requirement.6,20–22
Experimental
Chemicals
All chemicals were used as received. Potassium tetrabromoplatinate, K2PtBr4 (99%) and tetrafluroboric acid, HBF4 (48%) from Strem Chemicals, perchloric acid (70%) from Macron Fine Chemicals, aniline (99.5+%), 1-propanol (99.5+%) and propionic acid (99%) from Aldrich, sulfuric acid (95-98%) from BDH Chemicals, potassium hydroxide from J. T. Baker, propionaldehyde (99+%) from Acros Organics and 2-propanol (70%) from EMD.
Electrodeposition
Electrochemical experiments were done with an electrochemical workstation (CH Instruments 406, Austin, TX) in a three-electrode cell. The double-junction reference electrode was Ag/AgCl in 1 M KCl//1 M KNO3. All potentials mentioned in this work are referred to this electrode (E = 0.235 V vs. SHE). Thin synthetic graphite plates from Advanced Carbon Engineered Solutions (North Bay, Ontario) with bulk density of 1.88 g/cm3 and specific resistivity of 10 μΩm were used as counter and working electrodes. Before use, each 3.5 cm × 1.0 cm × 0.05 cm electrode was polished with alumina lapping pads (30, 12 and 0.05 μm), obtained from Allied High Tech Products Inc. and was thoroughly rinsed with 2-propanol. The working electrode was then encased in Coleflex heat shrinkable tubing, and a geometric area of 0.5 cm × 0.7 cm was cut out. In order to ensure proper wetting of the exposed porous carbon material, each working electrode was held under vacuum in plating solutions for approximately 20 minutes, or until air bubbles were no longer seen forming on the surface of the electrode. Immediately after this step, either platinum was electropolymerized from 0.001 M or 0.0085 M K2PtBr4 in 1 M HClO4 or polyaniline was deposited from solutions of either 0.25 M aniline/1 M HClO4 or 0.4 M aniline/2 M HBF4. Platinum depositions were carried out at 60°C to decrease deposition time. Unlike the chloroplatinate salts, potassium tetrabromoplatinate solutions are more stable in acid and do not hydrolyze.23 The K2PtBr4 stability in 1 M HClO4 was confirmed by UV absorbance measurements before use. Platinum was deposited on carbon, C/Pt, or on PANI, C/PANI-Pt, at a constant potential of −0.1 V, as specified in the Results. The polyaniline was either electropolymerized directly on carbon, C/PANI, or on platinum, C/Pt-PANI, at constant potential of 0.8 V until a charge density of 1.43 C/cm2 was reached and was kept constant throughout this work. After each deposition, the electrode was rinsed with deionized water and cyclic voltammograms (CVs) were recorded in 1 M HClO4 at 20 mV/s between +0.9 V and −0.2 V.
Testing of C/Pt and C/Pt-PANI electrodes
Cyclic voltammograms were recorded in solutions of 0.5 M 1-propanol in 1 M KOH at 50 mV/s using Pt foil as counter electrode. All CVs were recorded between +0.4 V and −0.7 V until a stable voltammogram was obtained.
HPLC analysis
HPLC measurements were performed on Agilent 1100 Series HPLC system (Agilent Technologies, USA) with HC-75 (H+) column, 7.8 × 305 mm, particle size 9 μm (Hamilton, USA). The mobile phase for isocratic elution was 5 mM H2SO4 in water at a flow rate of 0.7 ml/min. The separation column was thermostated to 80°C and a variable wavelength detector was used to measure the absorbance at 190 nm. For system control and data evaluation, Agilent ChemStation software, rev. B.04.02 SP1, (Agilent Technologies, USA) was employed. Volume of sample injected on the column was 100 μl. Peaks were identified using standard addition method. Sensitivity of the method to 1-propanol (0.27 AU·L/mol) was significantly lower than sensitivity to propionic acid (7.97 AU·L/mol). Sensitivity to unknown product of aldol condensation as a marker of propionaldehyde was approximately 85 AU·L/mol, i.e. ten times higher than sensitivity to propionic acid.
Electrooxidation of 1 M 1-propanol in 1 M KOH was performed in a U-type glass cell with a frit separating the working and auxiliary electrode compartments of 3 and 15 ml, respectively. Platinum mesh (6.25 cm2) was used as auxiliary electrode in the larger compartment. Both compartments were filled with the same solution. Oxidation of 1-propanol for HPLC analysis was conducted by applying potential step sequence described in the Results. This sequence of pulses was repeated 360 times making the cumulative time at the oxidation potential, −0.26 V, a total of one hour. Samples were analyzed after 12 hours of storage at room temperature.
SEM characterization
Further characterization of the electrodes was carried out with scanning electronic microscope (SEM) Hitachi SU8230 cold field-emission SEM (Tokyo, Japan).
Results and Discussion
Our previous experience with preparation of Pd-PANI composites on Pt has shown that the deposition order of the components determines the location of the metal inside the composite.24 Therefore, starting with the synthetic graphite as the support, the sequence of platinum and PANI depositions was investigated. The result is shown in Figure 1. The depositions were carried out at constant potential and controlled by applied charge. Platinum was deposited from 0.001 M K2PtBr4 in 1 M HClO4 at potential E = −0.1 V and PANI was deposited from 0.2 M aniline in 1 M HClO4 at potential E = 0.8 V. The charge density used for the deposition of polyaniline was 1.43 C/cm2, and the charge density for Pt was 5.38 × 10−2 C/cm2.
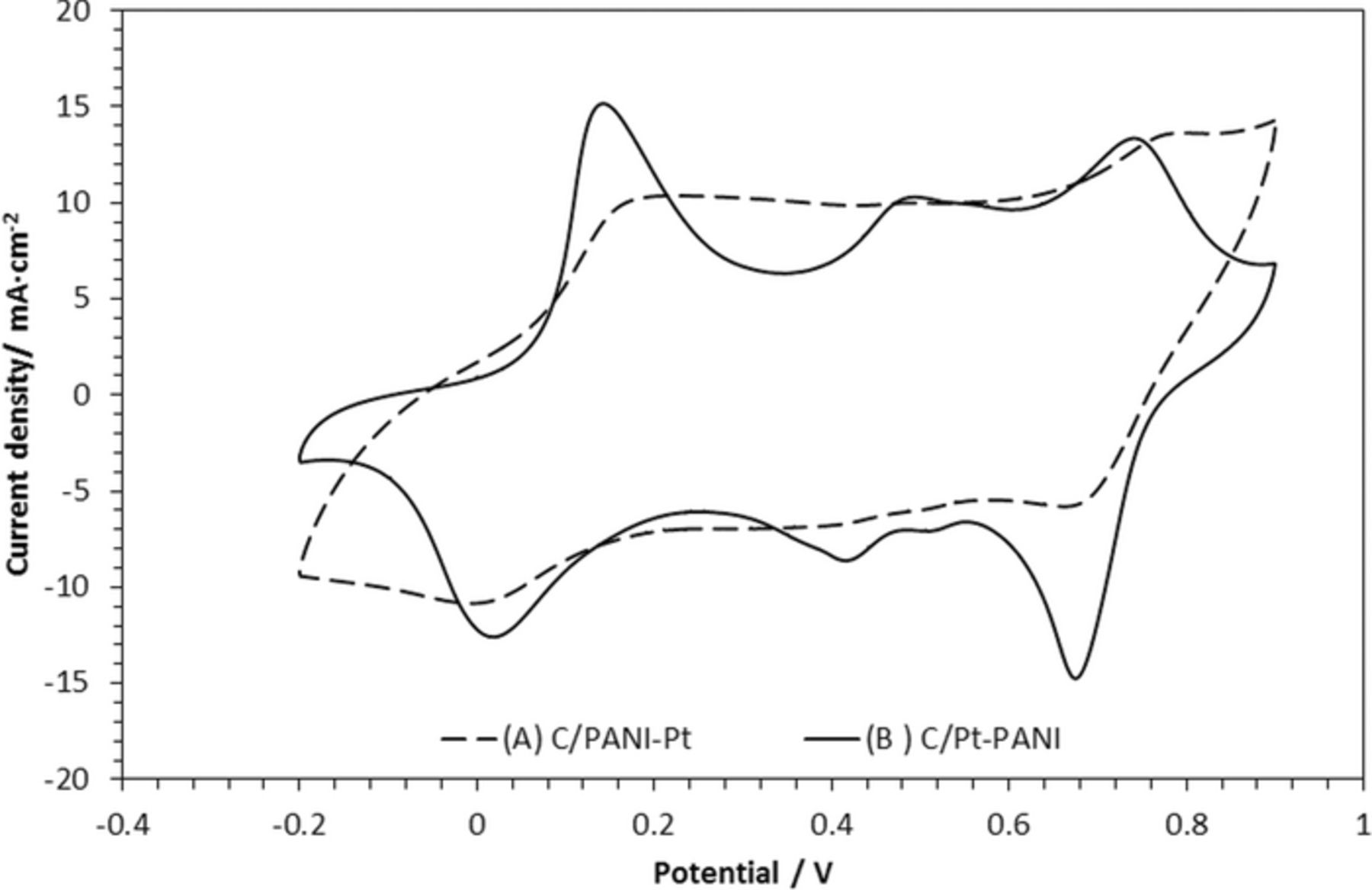
Figure 1. Effect of the deposition sequence of Pt and PANI on the voltammetry of the carbon electrodes: (A) PANI followed by Pt deposition on carbon support, C/PANI-Pt (B) Platinum deposition followed by aniline polymerization on carbon support, C/Pt-PANI. Cyclic voltammograms were recorded in 0.1 M HClO4 with 20 mV/s.
The cyclic voltammetry of C/Pt-PANI electrode resembles that of PANI film deposited directly onto a polycrystalline platinum electrode.25 Distinct oxidation/reduction peaks around 0.15 V/0.05 V can be seen for the expulsion/insertion of H+ ion attributed to the transition between leucoemeraldine and emeraldine form of polyaniline, and around 0.75 V/ 0.70 V for the ClO4− anion insertion/expulsion attributed to the redox transition between emeraldine and pernigraline forms. The middle redox peaks arise from the response of the hydroquinone and benzoquinone species due to overoxidation of the PANI.26,27 In contrast, the PANI redox transitions in the C/PANI-Pt are suppressed by the presence of the Pt-particles. Furthermore, peaks in the potential range of hydrogen adsorption/desorption, are broad but symmetrical, indicating polycrystalline structure of the deposited Pt. The overlap of hydrogen sorption-desorption processes with the redox transition of PANI results in a pseudocapacitive behavior of the electrode; this observation agrees with that already reported.28 Based on this distinguished difference in behavior, all further studies were completed solely with the C/Pt-PANI composite electrode on which the behavior is dominated by PANI electrochemistry in an acidic medium.
Characterization of C/Pt and C/Pt-PANI
It has been generally accepted that the physico-chemical properties of the supporting carbon material and size and distribution of metal particles are important characteristics. Prior to the Pt-deposition the carbon electrodes were characterized by SEM and cyclic voltammetry. Figure 2A shows the SEM image of the polished synthetic graphite plate (see Experimental section). It shows open surface structure that governs the low resistance and low double layer capacity observed in the CV in Figure 2C curve 1. The SEM image obtained after Pt deposition demonstrates that the growth of platinum agglomerates is uneven with asymmetric, irregular edges (Figure 2B). In agreement with the accepted nucleation and growth theory, the irreversible growth of the nanocrystals is achieved through atomic addition until the reaction is terminated.29 Figure 2C curve 2, shows a sharp peak corresponding to the underpotential deposition of protons on the Pt surface at potentials ranging from −0.25 to 0 V. Before the hydrogen evolution, on the reverse scan of the same potential range, the oxidative desorption of protons from the Pt surface generates a broad peak. The anodic region bound by E > 0.8 V and the single peak at ∼0.5 V correspond to oxidation and reduction of Pt respectively. The CV shown as curve 3 changed profoundly after an electrochemical annealing treatment was applied in steps (of 10 cycles) between −1.0 and +1.0 V with a scan rate of 1 V/s. From the SEM micrograph shown in Figure 2D, it appears that during this treatment the nanoclusters coalesce, resulting in larger hemispherical (1.0 μm) platinum agglomerates. The corresponding CV is shown in Figure 2C, curve 3. It agrees with a voltammogram of the polycrystalline platinum electrode.25 The distinct peaks due to strong hydrogen adsorption at 0.1 V and weaker hydrogen adsorption at −0.1 V indicate a high surface quality of deposited Pt.
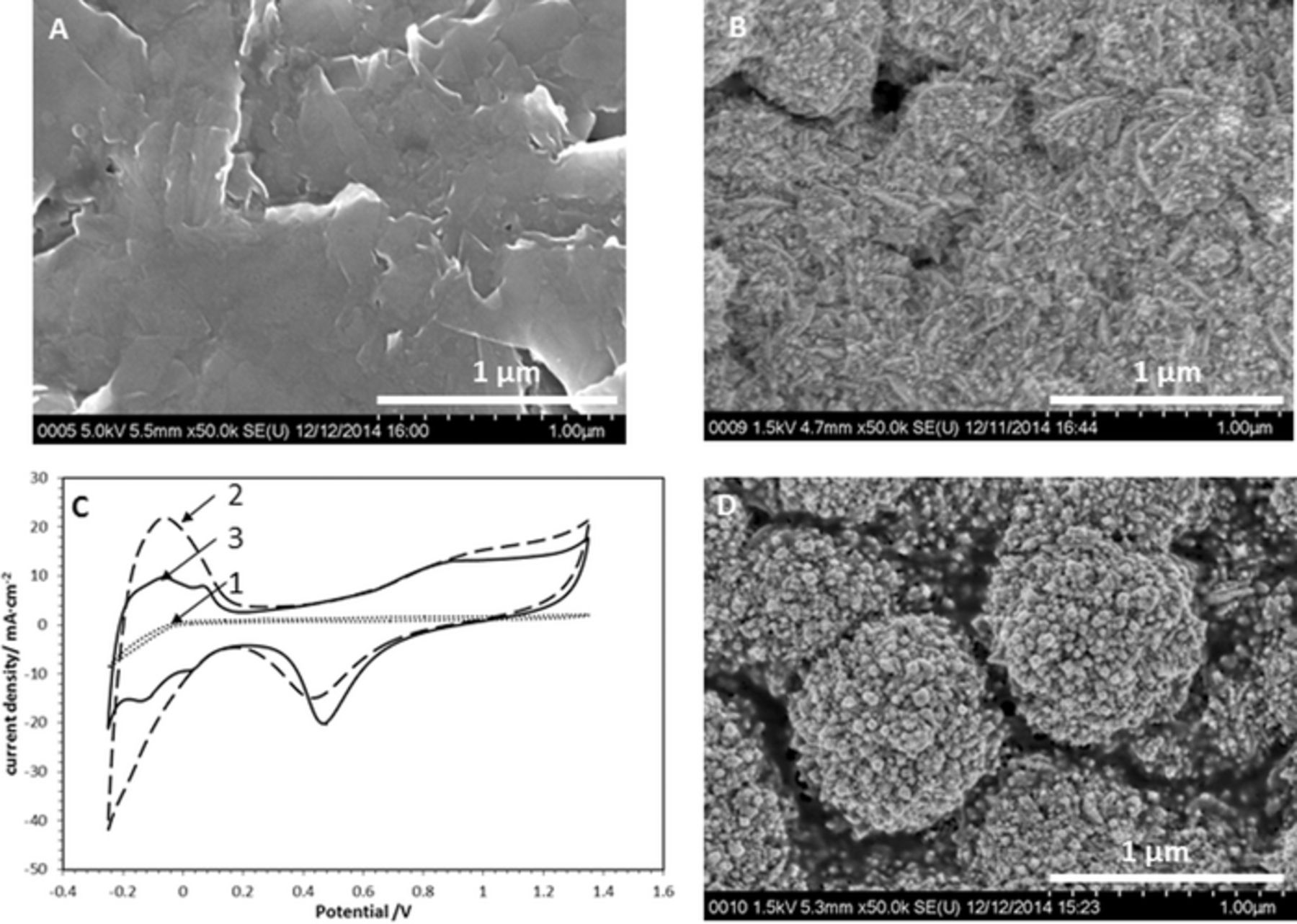
Figure 2. Platinum electrodeposited on carbon support: SEM images of polished carbon substrate (A), of C-support after Pt deposition from 0.0085 M K2PtBr4 in 1 M HClO4 at constant potential of E = −0.1 V using 1.71 C/cm2 (B), and after applied electrochemical annealing treatment in 1 M H2SO4 (D). CVs of electrodes shown as SEM images in (A), (B) and (D) were recorded sequentially as curve (1), (2) and (3), in 1 M H2SO4 with 20 mV/s, shown in (C).
In order to examine the effect of the deposited platinum on the electrochemical surface area (ESA), voltammograms were recorded after the electrochemical annealing step, Fig. 3. The inset in this figure shows a CV recorded after annealing, with the shaded region indicating the area of H-atom adsorption peaks. The integration of the charge within this region followed the method applied by Stevens and Dahn.30 Figure 3 shows the dependency of the ESA upon the applied deposition charge densities, QPt, from 0.07 to 7.14 C/cm2.
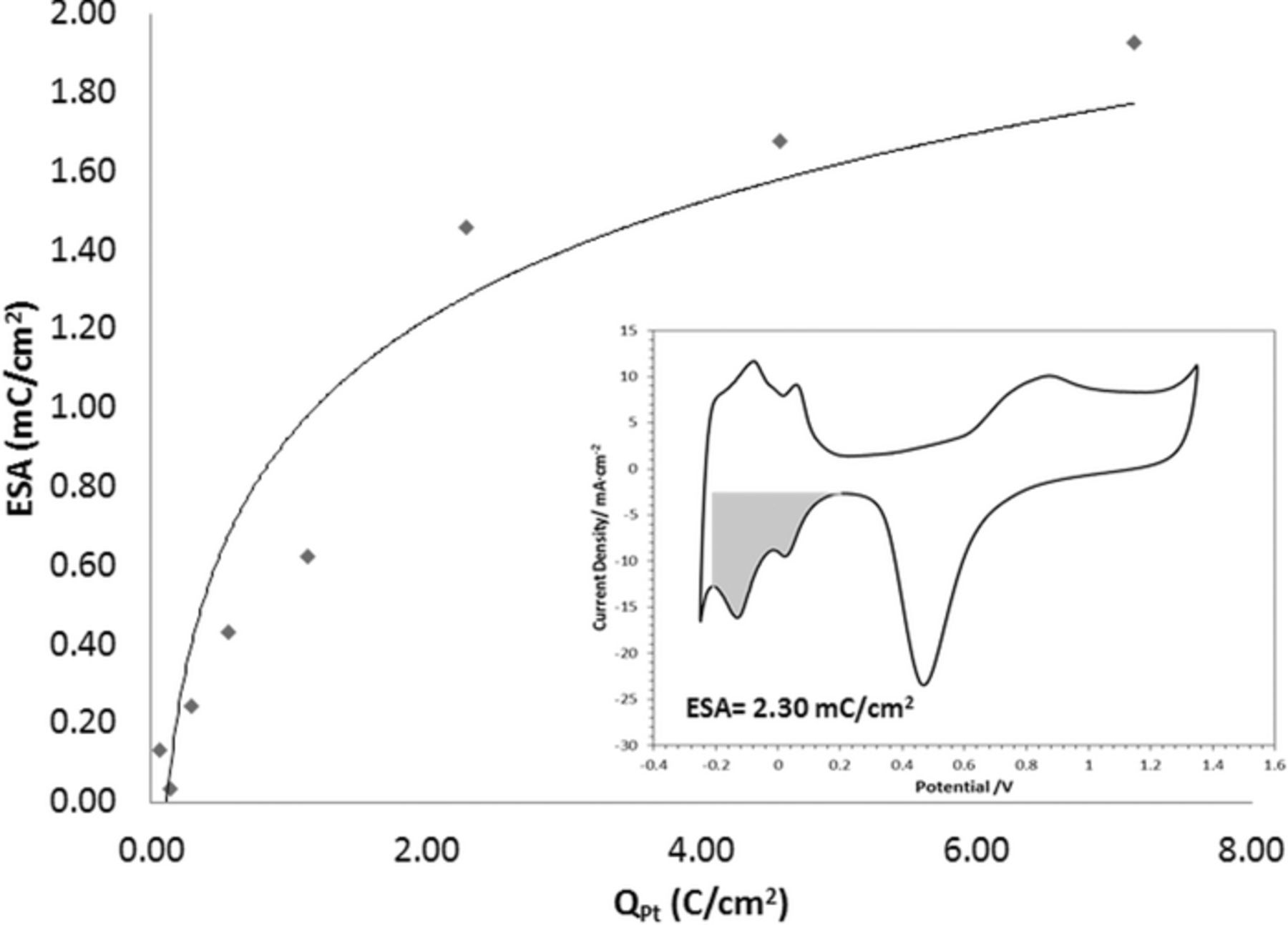
Figure 3. Dependence of ESA on the charge used for Pt deposition. Inset: Stable CV obtained for Pt-loading with QPt = 2.30 C/cm2. All platinum depositions were carried out at 60°C and followed the plating conditions given in Fig. 2.
With the increased Pt loading, the ESA begins slowly to level off. The diminishing increase of the ESA may result from an interplay between primary and the secondary nucleation centers (on the surface of the electrodeposited Pt nanoparticles) leading to complex nanograined deposits. The latter limits the number of specific crystal faces that can be formed on the platinum during sulfate ion adsorption.31
In order to examine the effect of Pt-loading on PANI activity, the polymerization of aniline was always carried out from the same electrolyte at constant potential E = 0.8 V and controlling the thickness of the deposition using a charge density of 1.42 C/cm2. It was observed that the shape of the CV shown in Figure 1 as curve B does not undergo fundamental changes by varying the Pt-loading. The average charge density under the oxidation peak at 0.15 V for all the different Pt-loadings was determined to be 90.0 ± 1 mC/cm2. This reveals the expected consistency for PANI deposition.
Behavior of the electrodes in 1-propanol
The platinum loaded carbon support in the absence and presence of PANI in 1 M KOH was compared in Figure 4A and 4C. The hydrogen adsorption/ desorption peak potentials were shifted in the alkaline medium toward the more negative potentials as compared to Fig. 1. The pH change provides more insights into the Pt-loading effect on the carbon support; at the higher Pt-content the shape of the CV curves is better resolved. The electrode surface becomes more sensitive to other electrochemical reactions such as the oxygen reduction around −0.3 V that may coincide with the hydrogen peroxide oxidation/reduction. The C/Pt-PANI electrode also mirrors that behavior. Peaks in the same potential range that can likely be assigned to the formation of Pt(OH)x and PtOx,32 are also seen on the reverse scan. Oxidation of 0.5 M n-propanol in 1 M KOH on C/Pt and C/Pt-PANI electrodes is compared in Fig. 4B and 4D. It is observed that with increased platinum loading, there is an increase in activity toward 1-propanol oxidation with the potential remaining around −0.2 V. Furthermore, at a higher charge density of 4.5 C/cm2, the peaks corresponding to hydrogen adsorption on platinum can also be seen between −0.4 and −0.7 V (curve 4 of Fig. 4B and 4D). Comparing the absence and presence of PANI (Fig. 4B and 4D), having a PANI film present tends to slightly suppress the peak currents, but does not disturb the general trend due to platinum loadings.
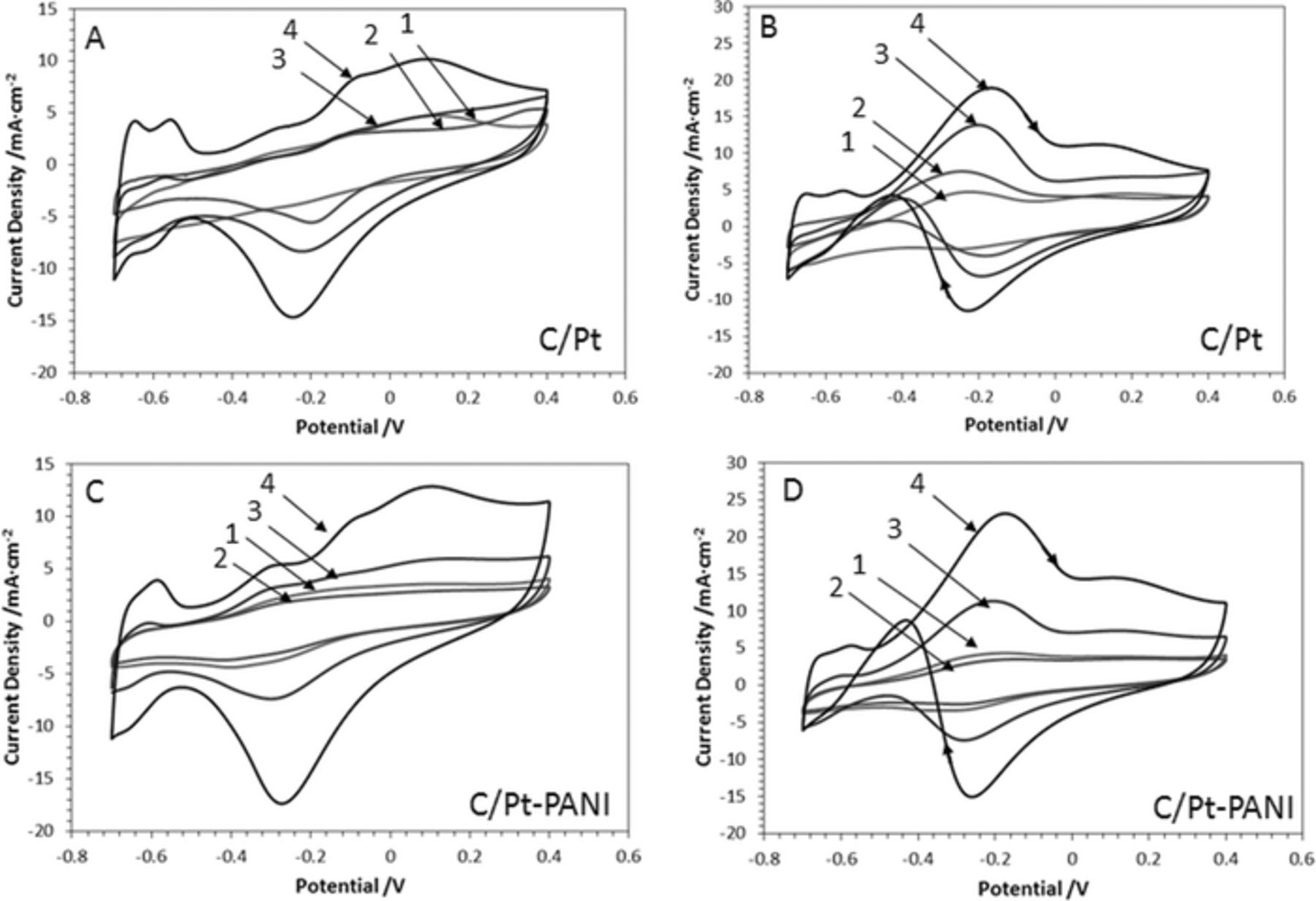
Figure 4. Voltammograms of C/Pt and C/Pt-PANI in 1 M KOH are shown in (A) and (C). The response of the same electrodes to 0.5 M 1-propanol/1 M KOH are shown in (B) and (D). Platinum deposition charge densities were: (1) 0.29 C/cm2, (2) 1.14 C/cm2, (3) 2.29 C/cm2, (4) 4.57 C/cm2. All CVs were recorded at 50 mV/s.
Product analysis
A most direct test of the electrocatalytic efficiency is the electrolysis and analysis of the products of 1-propanol oxidation on C/Pt and C/Pt-PANI electrodes. For that purpose an HPLC analysis of the electrolyte containing oxidation products from 1 M 1-propanol in 1 M KOH was developed. The characteristic feature of oxidation of propanol in strongly alkaline medium is the blockage of the working electrode with oxidation product resulting in greatly reduced current efficiency and characteristic cyclic voltammograms (insets of Figure 5A and 5B). The peak around −0.2 V observed during the forward scan indicates the catalytic oxidation of 1-propanol. The products of the oxidation are adsorbed on the electrode surface in the region of 0.0 to 0.4 V and block further oxidation. A periodic application of "cleaning" pulse removes the blocking products and restores the electrolysis. Thus, electrolysis was carried out using pulsing regime shown in Figure 5C. The repetitive pulses were initiated at −0.26 V (for 10 s) then stepped to 0.4 V (for 0.1 s) and terminated at −0.6 V (for 0.2 s). The resulting chronoamperometric response for the last applied oxidation pulse at −0.26 V in the series is shown in Figure 5A and 5B. Electrocatalytic activity for C/Pt-PANI electrodes is generally somewhat lower than that for the bare C/Pt. That can be explained by the limited mass transport of propanol due to the layer of PANI. At the same time, electrodes with higher Pt loading tend to show higher catalytic activity as can be seen in Fig. 5, curves 1–4.
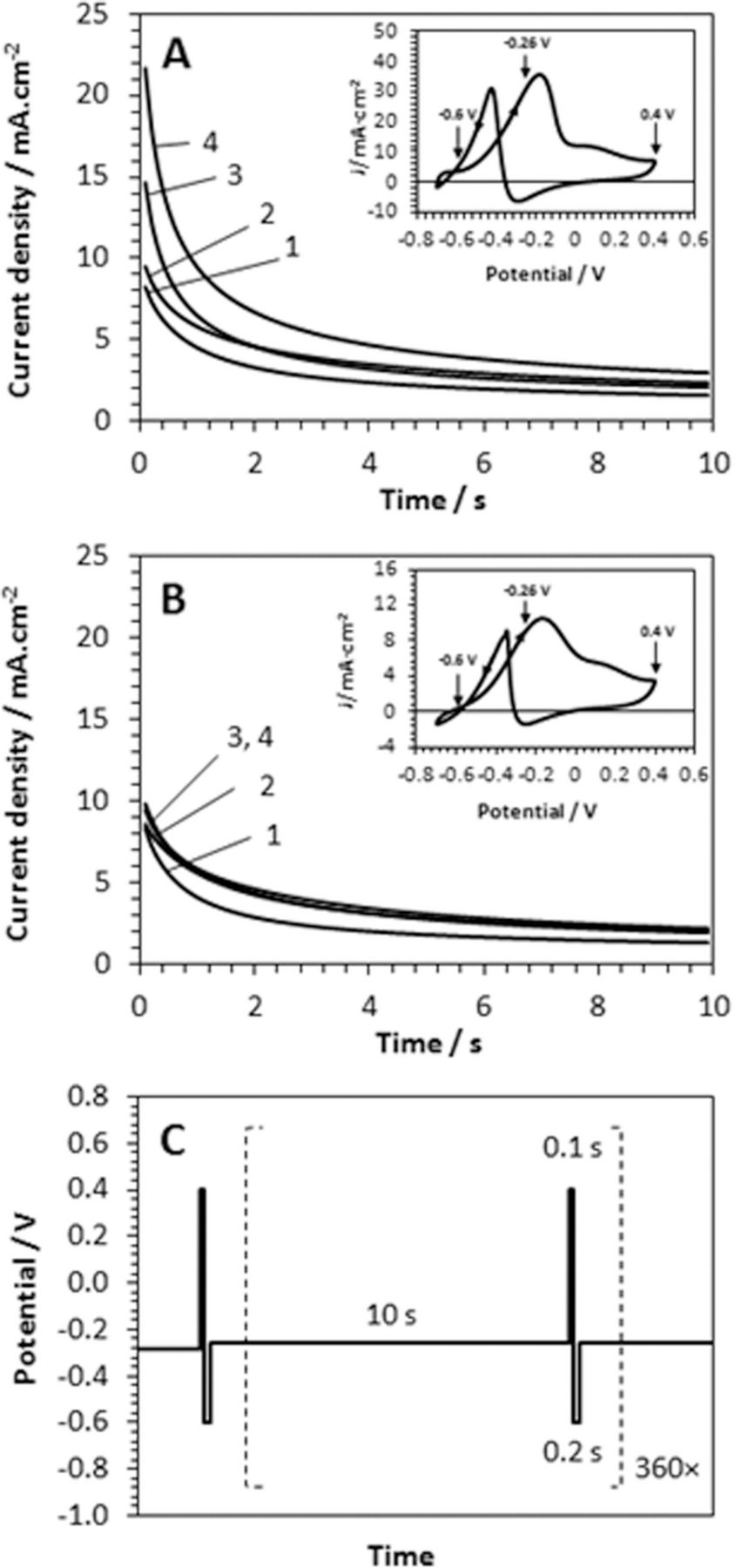
Figure 5. Pulse oxidation of 1 M 1-propanol on (A) C/Pt and (B) C/Pt-PANI in 1 M KOH. Charge density used for the deposition of Pt was Q = (1) 0.29, (2) 1.14, (3) 2.29 and (4) 4.57 C/cm2. Insets show corresponding CVs recorded for Q = 4.57 C/cm2 with scan rate 50 mV/s. (C) Diagram of applied potential pulses.
Figure 6 shows HPLC chromatograms of samples taken after one hour of pulsed electrooxidation using C/Pt and C/Pt-PANI electrodes. As a result of 1-propanol (peak 2) oxidation, propionic acid (peak 1) was detected. Another expected product, propionaldehyde, could not be solely identified due to the competing aldol condensation reaction.33 However, the presence of peak 3 and several minor peaks prove propionaldehyde being one of the products. These peaks were also found in chromatograms of samples prepared by adding propionaldehyde to 1 M KOH and 1 M 1-propanol (data not shown). The peak 1 (propionic acid) is approximately two times higher for C/Pt than for C/Pt-PANI electrode. That indicates that PANI layer on the Pt electrode does not contribute to catalytic activity of the oxidation. The ratio of peak 1 to peak 3 is roughly the same for both electrodes indicating that oxidation on either electrode is governed by the same mechanism.
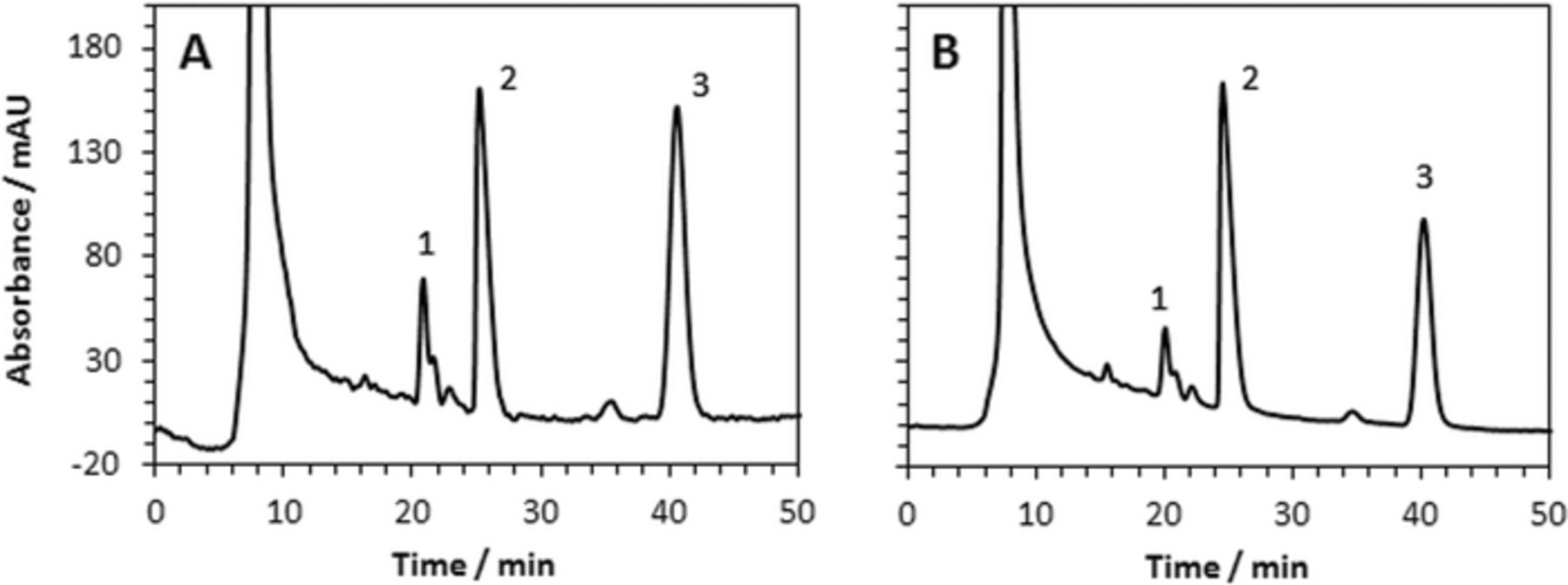
Figure 6. HPLC chromatograms of sample collected after pulse electrolysis discussed above (Figure 5). (A) C/Pt, (B) C/Pt-PANI. On both electrodes, Pt was deposited using 4.57 C/cm2. Peak identification: (1) propionic acid, (2) 1-propanol, (3) unknown product of aldol condensation of propionaldehyde.33
Conclusions
It was observed that the oxidation of 1-propanol on the C/Pt-PANI electrode depends on Pt-loading. As the amount of Pt increases, the electrochemical surface area of Pt changes and is stabilized by electrochemical annealing. The highest catalytic activity was determined to occur when a charge density of 4.5 C/cm2 was used. On the other hand, presence of PANI seems to have a slightly negative effect on the oxidation of 1-propanol due to hindrance of mass transport. This finding was confirmed using CV and by monitoring the products through HPLC. The main objective of this work, preparation of the composite electrode for deposition of atomic metals, has been achieved. It provides a low-cost alternative to solid Pt electrodes and represents the baseline experiment against which the performance of quantized atomic electrodes can be measured.
Acknowledgments
We thank the Advanced Carbon Engineered Solutions for providing the synthetic carbon plates (http://acesnorthbay.com/Contact.aspx). T. Křížek acknowledges the sponsorship of the Czech Fulbright Commission (Fulbright-Masaryk Scholarship).