Article Text

Abstract
Objective To study the detailed nature of genomic microevolution during mixed infection with multiple Helicobacter pylori strains in an individual.
Design We sampled 18 isolates from a single biopsy from a patient with chronic gastritis and nephritis. Whole-genome sequencing was applied to these isolates, and statistical genetic tools were used to investigate their evolutionary history.
Results The genomes fall into two clades, reflecting colonisation of the stomach by two distinct strains, and these lineages have accumulated diversity during an estimated 2.8 and 4.2 years of evolution. We detected about 150 clear recombination events between the two clades. Recombination between the lineages is a continuous ongoing process and was detected on both clades, but the effect of recombination in one clade was nearly an order of magnitude higher than in the other. Imputed ancestral sequences also showed evidence of recombination between the two strains prior to their diversification, and we estimate that they have both been infecting the same host for at least 12 years. Recombination tracts between the lineages were, on average, 895 bp in length, and showed evidence for the interspersion of recipient sequences that has been observed in in vitro experiments. The complex evolutionary history of a phage-related protein provided evidence for frequent reinfection of both clades by a single phage lineage during the past 4 years.
Conclusions Whole genome sequencing can be used to make detailed conclusions about the mechanisms of genetic change of H. pylori based on sampling bacteria from a single gastric biopsy.
- Helicobacter Pylori
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
-
Helicobacter pylori causes serious diseases following chronic infection of the human stomach.
-
Infection can persist over many years, during which genomic evolution is driven by high rates of mutation and recombination.
What are the new findings?
-
Coinfection of a single host with two strains of H. pylori can be revealed by sequencing the genomes of several isolates from a single biopsy.
-
When coinfection occurs, it can last for many years without one or the other strain being lost.
-
During that time, recombination between strains happens which has a much more profound effect than mutation or recombination with the same infecting strain.
How might it impact on clinical practice in the foreseeable future?
-
Performing similar studies in larger numbers of individuals will provide detailed insights on how H. pylori uses the diversity it generates to chronically infect the human stomach. A better understanding of within-host genomic evolution is necessary to design effective therapies against H. pylori.
Introduction
Helicobacter pylori is a host-specific bacterial pathogen that establishes a chronic infection in the human gastric mucosa, resulting in a variety of gastroduodenal diseases ranging from superficial gastritis and peptic ulcer to gastric cancer and mucosa-associated lymphoid tissue lymphoma.1 ,2 Several studies on within-host evolution have shown that recombination can be a potent force of genomic diversification, especially in the presence of mixed infection with multiple strains.3–6 H. pylori isolates sampled sequentially from the same patients have been compared, so as to estimate mutation and recombination rates as well as the size of recombined fragments.3–5 In one of these studies, the evolutionary distance between pairs of sequential isolates was significantly correlated with the period of time between isolation, but not with the age of hosts.4 This suggested that the pairs had shared common ancestors a relatively short time before the first isolation. This hypothesis was confirmed by a comparison of simultaneously isolated pairs of genomes from 40 South Africans, where the majority of time to the most recent common ancestor (TMRCA) was greater than 3.5 years.6
The mode of evolution of H. pylori within individual hosts has not been investigated in detail, and a number of questions remain unanswered. For example, it is unknown whether recombination rates are similar for different strains, and whether the amount of DNA that strains import varies over time and depends on specific challenges due to changes in the gastric environment or immune selection. It is also unknown whether recombination is primarily facilitated by chronic mixed infection, or whether exchange takes place with lineages that transiently colonise the stomach before being outcompeted by the dominant strains. Experiments performed in vitro suggested that recombination rates can vary significantly between strains even when all other conditions are identical.7–9 They also identified that recombined regions occasionally contained short unaffected gaps (ie, identical to the recipient rather the donor strain), and these have been termed ‘interspersed sequences of the recipient’ (ISR); but the relevance of these laboratory experiments to within-host evolution remains undetermined.
Here we sequenced the genomes of 18 H. pylori isolates obtained at the same timepoint from a Chinese patient with chronic gastritis and nephritis. This is the most comprehensive sequencing effort of H. pylori from a single patient ever reported, and one of the most comprehensive for any bacterial pathogen, similar, for example, to recent studies in Staphylococcus aureus.10 ,11 Comparisons of these genomes enabled us to study their genetic relationships and within-host evolution.
Materials and methods
Isolation of H. pylori
H. pylori colonies were isolated from a gastroscopic antral biopsy specimen of a 53-year-old male patient with chronic gastritis and nephritis (Mesangial proliferative nephritis), a resident of Zhejiang province, China. The patient underwent gastroscopy in 2012 and 2013. In 2012, the first endoscopy showed chronic superficial gastritis. The second follow-up endoscopy in 2013, showed chronic superficial gastritis, with a histologic diagnosis of mucosa mild chronic inflammation with mild intestinal metaplasia. The patient had not received antibiotics during the 3 months before the first gastroscopy, and did not take any antisecretory medication. Biopsy specimens taken in 2012 were homogenised and inoculated on Campylobacter Agar base (CM0935, Oxoid) plates containing 10% defibrinated sheep blood and H. pylori supplement SR0147E (Oxoid). Plates were incubated in microaerobic (5% O2, 10% CO2 and 85% N2) conditions at 37°C for 48 h or 72 h. Eighteen single colonies with characteristic morphology were obtained and confirmed by urease, catalase and oxidase tests. Each colony was further purified with another round of single-colony isolation.
Genome sequencing, assembling and annotation
Genomic DNA was extracted using the QIAamp DNA Mini Kit (QIAGEN, Germany) according to the manufacturer's instructions. Libraries were prepared using the IlluminaTruSeq DNA Sample Preparation Kit. Eighteen isolates were sequenced using the IlluminaMiSeq sequencing system by running 2*150 cycles according to the MiSeq System User Guide. De novo assembly was applied to the paired-end short-insert library sequencing data of 18 H. pylori isolates, while optimal assembly parameter k-mer were selected from 13 to 127 in odd numbers. Two isolates (3 and 12) were assembled using SOAPdenovo V.1.05,12 and the remaining 16 isolates were assembled using Velvet V.1.2.07,13 while the optimal hash length of 18 assemblies was selected. Annotation of the resulting contigs was performed using RAST (Rapid Annotations using Subsystems Technology),14 while tRNAs and rRNAs were identified using tRNAscan-SE15 and RNAmmer.16 The 18 genomes were aligned using progressiveMauve,17 and the stripSubsetLCBs script was used to extract 357 genomic regions shared among all of them, and of length >500 bp. The concatenated length of these shared regions was 1344 kbp. According to NCBI submission requirement, draft genome assemblies were split into smaller contigs if any sequence contained more than 10 continuous Ns, and the contigs shorter than 200 bp were discarded before the submission to NCBI.
Genome sequence analysis
The shared aligned regions were first used to construct the phylogeny (shown in figure 1) using the neighbour-joining method.18 ClonalFrame V.1.219 was applied separately to genomes from the two clades, using a total of 20 000 iterations with the first half discarded as burn-in and the second half sampled every 10 iterations. Using a previous estimate of the H. pylori within-host molecular clock of 1.38×10−5 mutation per site per year,6 the clonal genealogies of the two clades were shown on a yearly time scale in figure 2. For each recombination event inferred by ClonalFrame, the minimum distance to potential donors from either clade was computed (figure 3), thus providing an estimate for the number of inter-clade and intra-clade events.20–22 For each recombination event, a neighbour-joining tree was computed in the affected region (four examples are shown in figure 4). Mapping of recombination events to the previously completed and annotated genome F5723 was performed using MuMMER24 in order to assess which genes had been exchanged (see online supplementary figures S2 and S3). Finally, the sequences of the common ancestors of the two clades was imputed in ClonalFrame,19 and a pairwise comparison was performed as previously described,22–25 to assess the level of homology in different parts of the genomes (figure 5).

Neighbour-joining tree of the 18 isolates.

ClonalFrame time-scaled tree of the 18 isolates, with numbers of inferred recombination events indicates on branches.

Analysis of recombination events’ likely origins. The top panel shows events found in clade A, and the bottom panel shows the events found in clade B. The X-axis shows the distance of the best match of the import to a sequence in clade A, and the Y-axis shows the distance of the best match of the import to a sequence in clade B. The labels a, b, c and d correspond to the events highlighted in figure 4.

Neighbour-joining trees of the four exemplar recombined regions indicated with arrows in figure 3.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
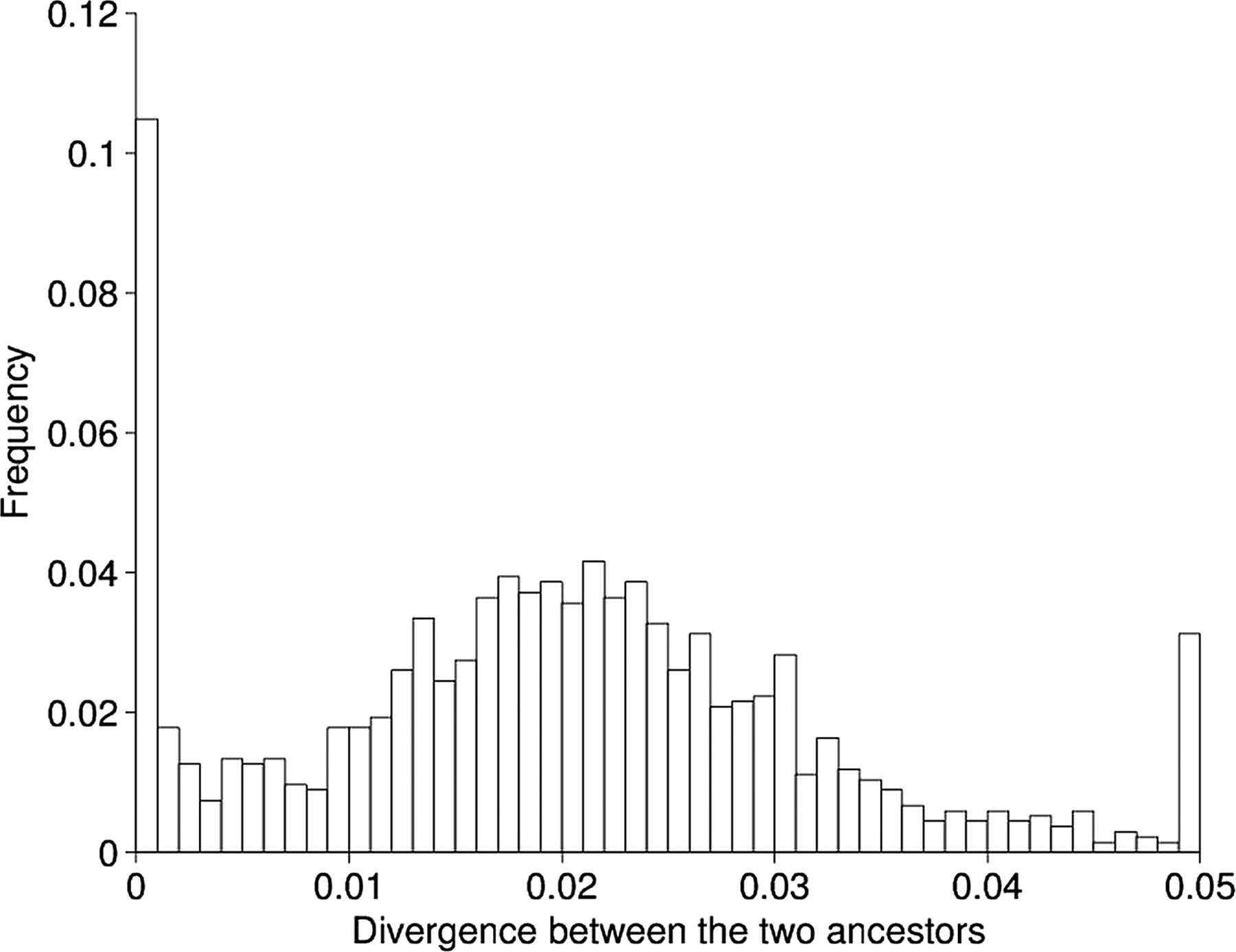
Site-by-site divergence levels between the two imputed ancestral sequences for clades A and B.
Restriction modification systems analysis
Blastn26 was used to search in the 18-genome sequences for the restriction modification (R-M) genes identified by previous studies and listed in the REBASE database (http://rebase.neb.com/rebase/rebase.seqs.html).27 Hits with coverage above 90% and E-value below 1e-10 were selected for the analysis of the differential gene content in the two clades. SNP patterns in the genes shared by all 18 genomes were analysed based on their alignment in MAUVE.17
Antibiotic susceptibility testing
Susceptibility of the H. pylori isolates to the six antibiotics: amoxicillin (Amo), clarithromycin (Cla), furazolidone (Fur), levofloxacin (Lev), tetracycline (Tet) and metronidazole (Met) was tested via agar dilution method using reference standards obtained from the National Institutes for Food and Drug Control. Ten microlitres of bacterial suspension (108 CFU/mL) from each isolate was inoculated onto Mueller-Hinton agar plates (Oxoid) containing 5% sheep blood and various concentrations of the above antibiotics, and incubated at 37°C for 3 days under microaerophilic conditions. Reference strain ATCC43504 NCTC1163728 was used as a quality control. The resistance break points to Amo, Cla, Fur, Lev, Tet and Met were set at ≥2, ≥1, ≥2, ≥2, ≥2 and ≥8 μg/mL, respectively, following previous studies.29 ,30
Data availability
The draft genomic sequences of H. pylori isolates have been deposited in the NCBI GenBank database (accession numbers have been shown in online supplementary table S1).
Results
Isolation of H. pylori and whole genome sequencing
We selected 18 single-colony isolates (designated 1–18) from the cultural plates of a gastroscopic antral biopsy sample taken from a 53-year-old Chinese patient with chronic gastritis and nephritis. Sequencing was performed using the IlluminaMiSeq sequencing system by running 2*150 cycles according to the MiSeq System User Guide. The overall properties of these 18 H. pylori genomes are summarised in online supplementary table S1. Most of the draft genomes were assembled in less than 100 contigs, and the GC content (about 39%) and total assembly genome lengths (about 1.6 Mbp) were as expected for H. pylori.31
A neighbour-joining tree based on the shared genome shows that the isolates are divided into two distinct clades (figure 1). Clade A contains 7 isolates, while clade B is made of the remaining 11. Since H. pylori has a pan-mictic population structure, we conclude that these two clades represent distinct infections of the same host by two distinct strains from the local gene pool. Analysis of the multilocus sequence typing loci revealed that both clades belong to the hspEAsia population (see online supplementary table S2).32
Although all the strains were isolated at the same time, the branches above genomes 4 and 16 are short compared to the root-to-tip distances for other strains. This suggests that they may be hybrids between the two strains that originally colonised the stomach. The shorter branch length is exactly what would be expected from a method like neighbour-joining, which does not account for recombination.
Clonal genealogy of the two clades
We used ClonalFrame to infer the clonal genealogy for the two clades in a way that accounts for recombination.19 ClonalFrame was applied separately to the two clades to avoid making the assumption that they shared the same evolutionary parameters. The ratio, r:m, represents the ratio of rates at which substitutions are being introduced by recombination and mutation, and is a convenient summary of their relative effects;33 r:m was estimated to be 2.3 and 19.8 in clades A and B, respectively. Recombination, rather than mutation, therefore, is the main driver of evolution in both clades, but it was almost an order of magnitude more important than mutation in clade B. This difference was due to a higher rate of recombination relative to mutation in clade B (ρ/θ=1.05) compared to that in clade A (ρ/θ=0.09).The average tract length of recombination was similar in both clades (δ=895 bp (572; 1250)).
Once the recombination events are accounted for, clades A and B have heights of 5.9×10−5 and 3.9×10−5 mutations per site, respectively. The within-host mutation rate has recently been estimated for H. pylori at 1.38×10−5 per site per year,6 which is significantly higher than in many other bacterial pathogens,34 but in good agreement with previous studies in H. pylori.3–5 This implies that the TMRCA for clades A and B are 4.2 and 2.8 years, respectively (figure 2).These estimates are consistent with a previous study which found an average TMRCA of 3.61 years for pairs of genomes from a single infection in 40 different hosts.6 This result does not mean that the two infections only happened a few years ago, but rather, that they happened at least that long ago, with genetic diversity having since been restricted by genetic drift and possibly immune selection. The host was 53 years old which suggests that the infections may indeed have happened before the TMRCAs, and evidence for this is provided by the comparison of ancestral sequences in the section after next.
Recombination events and their origins
Individual recombination events were inferred by ClonalFrame on each of the branches of the two clonal genealogies corresponding to the two infections (figure 2). The parameter ν reflects the average amount of polymorphism brought in via recombination, which was estimated to be 0.026 (0.025; 0.027) in clade A and 0.024 (0.023; 0.026) in clade B (values in square brackets are the 95% credibility intervals). The two estimates are similar to each other and slightly higher than the average nucleotide divergence between strains from the two lineages (π=0.0212). This is consistent with many of the combination events identified by ClonalFrame being exchanges between the two clades.
We applied previously described methodology to assess the likely origin of each recombination event.20–22 Briefly, the recombined fragments were extracted from the ClonalFrame output files and compared with the homologous sequence from the genomes from both clades (minus the strains affected by the recombination event). A match was considered to be found if the sequence was identical or contained a single nucleotide difference with the recombined segment. In clade A, a total of 51events were extracted. Two events matched only in clade A, 32 events matched only in clade B, two events matched both, and 15 matched neither. In clade B, a total of 297 events were extracted; 114 events matched only in clade A, 50 events matched only in clade B, 62 events matched both, and 71 matched neither. More events were detected between clades than within clades, although the latter type of events does not create as many differences as the former. Within-clade recombination is, therefore, harder to detect, so our results do not necessarily mean that recombination happens more frequently between, rather than within, clades. The events that do not match either clade could still be coming from one or the other clade, since the diversity of both clades was only partially sampled, and comparison was made only with present strains (whereas recombination may be ancient).
For each recombination event inferred, the distance of the best match in each of the two clades was represented as a scatter plot (figure 3). The area at the bottom-left corresponds to events that have an ambiguous origin (ie, could be one or the other clade); the events on the left, but not the bottom, have a clear origin in clade A, and the events on the bottom, but not the left, have a clear origin in clade B. In clade A, many events have a clear origin in clade B as previously reported. In clade B, many events are from clade A, but also quite a few from clade B and also quite a few ambiguous, again as described immediately above. In both clades, several events were also found that seemed to come neither from clade A or B (middle to top-right in figure 3).
To investigate these patterns further, local trees were computed for all recombination events. Figure 4 shows four exemplar regions, where recombination occurred in clade B, and with the labels a, b, c and d corresponding to those in figure 3B. Region a is a typical example of the many unambiguous imports from clade A to clade B. Strain 1 which is a member of clade B has clearly imported this region from clade A. In region b, two strains (1 and 15), both from clade B, have imported sequence which is otherwise characteristic of clade A. Since 1 and 15 are not closely related within clade B (figure 2), this region corresponds to two events, with the first strain importing from clade A and the second one importing also from clade A or from the first strain. For this reason, the two imports are at a low distance from members of both clades (figure 3B). In region c, it seems that strain 2 has imported something that is neither from clade A nor B, so that the distance is high to all potential donors (figure 3B). Strain 2 has several differences from all the others, and these are scattered in a region which is 895 bp long. The remaining genomes are virtually identical throughout this region, and so an alternative explanation would be that strain 2 is the only remaining representative of the clade B ancestral sequence. Finally, some genomic regions had a more complex evolutionary history, where it becomes difficult to deduce which recombination events happened. An example of this is region d which coded for a phage-related protein. Several recombination events must have happened to explain the difference between the clonal genealogy and the phylogeny observed in this region.
In vitro experiments found that about 10% of recombination events contained ISR, which are short stretches within imported sequence where the sequence is identical to the recipient rather than the donor.7–9 We searched for ISR in the clearest recombination events identified by recombination, that is, the ones that can be explained by a simple scenario of exchange between the two clades. Four examples of recombined regions which contained ISR are shown in online supplementary figure S1. In clade A, 3 out of 35 clear recombination events contained ISR, and in clade B, 16 out of 104 did. This is the first report of the detection of ISR in vivo, and the proportion of recombination events in which they occurred was similar to that reported in laboratory experiments.
Progressive genomic convergence of two H. pylori strains
Figure 2 suggests that recombination is progressive since events seem to be distributed on all the branches of the clonal genealogy. We hypothesised that the inter-clade recombination might result in genomic convergence between the two clades. To verify and quantify this, we compared the current average distance between genomes of the two clades with the distance between the two imputed common ancestors. Based on the ClonalFrame results, the ancestral sequence of the two clades was inferred, and the distance separating them was calculated and found to be equal to D=0.021308. This is slightly more than the current average pairwise distance between the genomes from the two clades (π=0.021213). Inter-clade recombination has therefore led to a slight convergence effect over the past ∼3 years, since the two clades shared common ancestors, which was more important than the divergence effect that occurred through mutation, and recombination with other strains.
The two imputed ancestral genome sequences were compared using previously described methodology22–25 which revealed a clear bimodality in the distribution of distance between the two genomes when looking along the genome (figure 5). This suggests that the two sources of infection were originally at a distance of ∼2.5% from each other, but that ∼15% of their genomes have recombined (prior to clade divergence) resulting in regions of high homology. This convergence would have happened as a result of recombination between the two strains in both directions, and we can estimate the rate at which this recombination occurred based on the amount of recombination we observed in the two clades; 4% of genomic positions have been recombined during the diversification of clade A, which has a sum of branch lengths equal to 12.01 years (figure 2). In clade B, 22% of positions recombined during 15.89 years of evolution. The expected convergence rate of the two ancestral strains is, therefore, equal to (4/12.01)+(22/15.89)=1.7% per year. This would suggest that the two strains have been coinfecting for ∼9 years before their common ancestors ∼3 years ago and, therefore, that infection with one of the two strains happened ∼12 years ago, whereas, the other strain was present before that for an indeterminable length of time.
Genes involved in recombination
The recombination events we found were mapped onto the complete annotated reference genome, F57, which is also a member of the hspEAsia population.23 All three types of recombination events were analysed in this way, namely events before the common ancestors, during the diversification of clade A, and during the diversification of clade B (see online supplementary figure S2). Outer membrane proteins were found to have recombined more than average (Fisher's exact test, p=0.0111), and especially the members of the hop family of genes (Fisher's exact test, p=0.0208) (see online supplementary figure S3). Previous studies revealed that recombination occurs more often in regions under positive selection, particularly the regions coding for proteins with a role in pathogenicity.35 The significantly increased recombination frequency was observed in gene encoding outer membrane proteins of H. pylori. Among outer membrane proteins, the hop subfamily encoding most known adhesins of H. pylori, is of particular interest.5 Our result is consistent with this previous report which found these genes to be more prone to within-host recombination than the remainder of the genome.
R-M systems
In order to understand the mechanisms involving the distinct rate of recombination between clades A and B, we analysed the R-M systems, one of the defences evolved in bacteria to recognise and cleave foreign DNA which plays an important role in transformation of H. pylori.36 One hundred and forty-nine R-M genes were found in the 18 genomes. Among them, 6 were shared by all 18 genomes, 11 were specific to clade A, and 6 were specific to clade B (see online supplementary table S3). Additionally, different SNP patterns were observed in the R-M genes shared by the two clades (see online supplementary table S4). Therefore, it is clear that the two clades have obvious differences in R-M systems, and these may explain their difference in recombination rates.
Results of antibiotic susceptibility testing
In order to know whether the different isolates were associated with different phenotypes, susceptibility to six antibiotics: Amo, Cla, Fur, Lev, Tet and Met was tested via an agar dilution method. All isolates were susceptible to Amo, Cla, Fur, Lev and Tet. Resistance to Met was, however, different between isolates (see online supplementary table S5). All isolates of clade A showed resistance to Met, and we found that they had a truncated RdxA, a known resistance mechanism.37 ,38 All isolates of clade B, on the other hand, were sensitive to Met, and had a complete RdxA (see online supplementary figures S4 and S5).
Discussion
Previous studies on within-host evolution of H. pylori have mainly been performed on two or three isolates cultured at one timepoint or, sequentially, from the same patient.3–6 ,39 ,40 Although this approach has provided estimates of average rates of recombination and mutation and recombination in the population, it has not provided information on how different strains use these processes to adapt within their host. The only study we are aware of, where several isolates were taken from the same host, was focused on genomic composition rather than homologous sequence comparison.41 A potential limitation of our study is that it was based on 18 genome sequences, so that the within-host diversity may only have been partially sampled. Assuming that the within-host population is uniform in the stomach, and that each genome is an independent random draw from this population, a strain representing only 10% of the population would have had a relatively small probability (p=0.15) of not being sampled in our study based on 18 genomes. To decrease this probability under the 0.05 significance threshold, 29 independent genomes would have been needed. If a strain was even less frequent, say, representing only 5% of the population, then there would have been almost equal probabilities of sampling or not in our study, whereas 59 genomes would have been needed to guarantee sampling.
Here we have shown that within a biopsy taken from the antral part of a single stomach, descendents of two separate infections were present, resulting in a phylogenetic tree with two widely distinct clades. This observation is consistent with reports of frequent infection with multiple strains.42–44 When coinfection happens, inter-strain recombination becomes possible, which leads to a much higher rate of within-host microevolution than would otherwise be possible by de novo mutation only, which causes the complex admixture patterns observed when comparing genomes from different human populations.32 ,45–49
All the isolates are distinct at the genomic level, and the two clades trace their common ancestors back to 2.8 and 4.2 years ago. Comparison of the ancestral sequences of the two clades revealed that recombination had been going on for an estimated 9 years prior to the common ancestors, meaning, that coinfection with the two strains started at least 12 years ago. This is an estimate of the time when coinfection with the two strains started, and it is likely that one or other strain was present for a long time before that, possibly since early childhood. It also assumes that the two strains have been recombining at the same rate since coinfection started, whereas, it seems probable that initially the second infecting strain would have been present at relatively low frequency, so that recombination may have been slower. The 12 years estimate may, therefore, be best seen as a lower bound for the amount of time during which coinfection happened. This result is compatible with the fact that the infected host was 53 years old, and suggests an absence of strong bottlenecks or clonal sweeps within the antral part of the stomach in the past 12 years.
Evolutionary changes have been predominantly caused by homologous recombination between the two clades, with one having a 10-fold higher rate of recombination than the other. In vitro experiments have shown that the frequency of transformation can vary significantly dependent on which donor and recipient strains were used.7–9 The two strains were in the same stomach, and should have had the same opportunities for recombination with one another, which suggests that the difference in recombination rate may be caused by differences in their intrinsic genetic properties.50 Our results suggested that the distinct rate of recombination may be due to the different R-M systems present in two clades. The overall effect of recombination was to cause the genomic convergence between the two infecting strains, both before and after the common ancestors of the two clades. The presence of recombination events on every branch of the genealogy implies that in stomachs with chronic mixed infections, recombination is a continuous process, involving the transfer at each event of a single short fragment.
The properties of imported tracts were similar to previous reports. The average import size was 895 bp, which was in good agreement with previous studies both in vitro7–9 and in vivo.3–5 Previous in vitro studies have reported frequent ISR resulting in sequences that are mosaics between donor and recipient.7–9 Investigating this property in vivo had not been previously possible because it requires knowledge of what donor and recipient sequences are likely to be, and here we were for the first time in a position to do so. We have shown that they, in fact, occur at a similar rate as in in vitro experiments, affecting about 10% of imports.
We found that a highly frequently exchanged gene coded a phage-associated protein (figure 4D). The full evolutionary history of this gene is impossible to determine, but it seems to have been transferred between the strains multiple times in the past 3 years. This result suggests that active phage infection and repeated transfer can occur within a single stomach without destabilising the bacterial population. Since H. pylori can acquire DNA by natural transformation, the importance of phage-mediated transfer to homologous recombination is unknown and deserves further study.
Met resistance is frequent in all H. pylori strains, and is often due to mutation or truncation of the RdxA. In our study, one of the clones has remained stably Met-resistant, despite competition with a Met-sensitive lineage, in the absence of antibiotic challenge. Thus, there appears to be weak selection for full-length versions of the RdxA.
In this study, we have shown that it is possible to use whole genome sequencing to make detailed conclusions about the mechanisms of genetic change of H. pylori based on sampling bacteria from a single gastric biopsy. Coming to conclusions about the role of natural selection is more difficult because we have not observed enough events to make reliable conclusions about which of the changes are likely to be adaptive. It is also difficult to speculate on the effect of the different recombination profiles of the two clones on their short or long-term evolutionary trajectories. Similar studies in large numbers of patients will provide unprecedented insight into the role of genetic exchange in allowing bacteria to survive in the uniquely hostile environment of the human stomach.
Acknowledgments
We thank Professor Ichizo Kobayashi for advice on the analysis of restriction modification genes.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figures
- Data supplement 2 - Online table S1
- Data supplement 3 - Online table S2
- Data supplement 4 - Online table S3
- Data supplement 5 - Online table S4
- Data supplement 6 - Online table S5
Footnotes
-
QC, XD, ZW and ZL are contributed equally.
-
Correction notice Shengqi Wang's affiliation and the Funding section have been updated since published Online First. The correct author name is Lihua He.
-
Contributors QC, XD, ZW, DF, SW and JZ conceived the study. QC, LH, YL, YY, XL, YG, MZ, JL and WW performed laboratory work. ZL, XD, MN, ZL, MZ and XB contributed to the bioinformatics assembly pipeline. XD, QC and ZL analysed the data. QC, XD, ZW, ZL, DF and JZ wrote the paper. All authors read and approved the final manuscript.
-
Funding This work was supported by the Science and Technology Program of Zhejiang Province Public Technology Social Development Project (No. 2010C33035), Key Projects in the National Science & Technology Pillar Program during the Twelfth Five-year Plan Period (No. 2012BAI06B02) and SKLID grant (No. 2014SKLID102). XD would like to acknowledge the NIHR for Health Protection Research Unit funding.
-
Competing interests None.
-
Patient consent Obtained.
-
Provenance and peer review Not commissioned; externally peer reviewed.