Article Text

Abstract
Objectives We aimed to assess the expression of the CCL24 chemokine in systemic sclerosis (SSc) and to evaluate the possible pathogenic implications of the CCL24/CCR3 axis using both in vitro and in vivo models. We further investigated the efficacy of an anti-CCL24 monoclonal antibody (mAb), CM-101, in inhibiting cell activation as well as dermal and pulmonary inflammation and fibrosis in experimental animal models.
Methods We used ELISA and fluorescence immunohistochemistry to determine CCL24 levels in serum and CCL24/CCR3 expression in skin biopsies of SSc patients. Skin fibroblasts and endothelial cells treated with CCL24 or SSc serum with or without CM-101 were used to follow cell activation and differentiation. Prevention and treatment in vivo bleomycin (BLM)-induced models were used to evaluate experimental dermal and pulmonary fibrosis progression following treatment with the CM-101 mAb.
Results CCL24 circulating levels were significantly elevated in SSc patients. CCL24/CCR3 expression was strongly increased in SSc skin. Blockade of CCL24 with CM-101 significantly reduced the activation of dermal fibroblasts and their transition to myofibroblasts induced by SSc serum. CM-101 was also able to significantly inhibit endothelial cell activation induced by CCL24. In BLM-induced experimental animal models, CM-101 profoundly inhibited both dermal and pulmonary fibrosis and inflammation.
Conclusions CCL24 plays an important role in pathological processes of skin and lung inflammation and fibrosis. Inhibition of CCL24 by CM-101 mAb can be potentially beneficial for therapeutic use in SSc patients.
- systemic sclerosis
- pulmonary fibrosis
- inflammation
- chemokines
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Systemic sclerosis (SSc) is a multifactorial inflammatory and fibrotic disease involving chemokines in its pathophysiology.
What does this study add?
We revealed the elevation of CCL24 in SSc circulation and skin and established its involvement in fibroblast activation and differentiation into myofibroblasts.
How might this impact on clinical practice or future developments?
Using CM-101, a monoclonal antibody that selectively targets CCL24, we significantly decreased the inflammatory and fibrotic features of bleomycin-induced experimental dermal and pulmonary fibrosis mouse models. Our data support a potential therapeutic effect of CM-101 in SSc patients.
Introduction
Systemic sclerosis (SSc or scleroderma) is a connective tissue disease characterised by excessive fibrosis and extracellular matrix deposition in the skin, lung, and other visceral organs.1 Despite recent advances in understanding the disease pathogenesis, the pathogenic processes underlying the various phenotypic manifestations, intensification of symptoms and clinical expression of the disease are still not well understood. Disease progression is characterised by an early inflammatory onset followed by tissue fibrosis and proliferative vasculopathy. In the clinic, these events are leading to skin fibrosis, interstitial lung disease, myocardial insufficiency, vascular obliteration, distal ulcerations and gangrene. The early inflammatory phase involves the immune cell network including lymphocytes, eosinophils and monocytes, as well as endothelial and endothelial progenitor cells. In the advanced phase, fibroblasts and myofibroblasts take the lead to generate tissue fibrosis.2–4
Chemokines are a group of small signalling proteins that induce migration and activation of cells.5 During inflammation, immune cells release chemokines that promote cellular migration, induce immune cell and fibroblast activation, trigger differentiation of T-cells to the T helper phenotype and induce transition of fibroblasts to profibrotic myofibroblasts.6 7 In SSc pathogenesis, chemokines have been proposed to foster migration and activation of inflammatory and fibrotic cells, inducing the secretion of cytokines that promote collagen and matrix deposition in the affected organs.8 Indeed, patients with SSc exhibit increased systemic levels of proinflammatory chemokines including CCL2, CCL5, CCL3, CXCL8, CXCL9, CXCL10 and CXCL16 (Chemokine c-c and c-x-c motifs ligand).9–12 Some were also shown to be correlated to limited or diffuse cutaneous disease phenotype and/or to organ-specific pathology as lung disease or skin vascular inflammation.13–15
CCL24 (Chemokine c-c motif ligand 24, eotaxin-2) is a chemokine that promotes immune cell trafficking and activation as well as profibrotic activities through the CCR3 receptor (C-C chemokine receptor type 3).16 Both CCL24 and CCR3 were shown to be involved in lung and skin inflammation and fibrosis in previous studies.17–19
CCR3 is robustly expressed on eosinophils and recent data suggested that eosinophilic inflammation might be involved in the pathogenesis and progression of SSc. In SSc patients, eosinophil counts, but not total leukocytes, were significantly higher than in patients with other connective autoimmune diseases. Eosinophil counts correlated positively with both interstitial lung disease severity and modified Rodnan skin thickness score.20 Notably, CCR3 was demonstrated to be expressed on oral and dermal fibroblasts where it modulates wound healing and tissue remodelling processes.17 21 A recent study by Lee et al also showed overexpression of CCR3 on monocyte populations isolated from SSc patients.22
CCL24 was shown to be involved in proinflammatory reactions, specifically contributing to the type 2 immune reaction involving Th2 lymphocytes and M2 macrophages that were shown to be present in skin lesions of SSc patients.23 24 Accordingly, CCL24 was found to play a dominant role in inducing profibrotic effects and to be overexpressed in fibrotic lungs and bronchoalveolar lavage fluid from patients with idiopathic pulmonary fibrosis (IPF), a disease sharing similar lung dysfunction features with SSc.18 25 26 Furthermore, CCL24 was shown to promote collagen production in human lung fibroblasts27 and to be constitutively expressed by dermal fibroblasts.28
Altogether, these studies may suggest a potential role of CCL24/CCR3 signalling in the pathogenesis of SSc.
CM-101 is a humanised monoclonal antibody (mAb) that targets the human chemokine CCL24. Previous studies have shown that administration of CM-101, by binding CCL24 with high affinity, reduced proinflammatory responses in animal models of several diseases including atherosclerosis, rheumatoid arthritis and multiple sclerosis.29–31 This experimental evidence supports the ability of CM-101 to significantly interfere with proinflammatory processes in vivo.
In light of these results, we aimed to test the expression levels of CCL24 both in the circulation and in the skin of SSc patients, compared with healthy individuals. In addition, we aimed to assess the ability of the CCL24 blocking antibody CM-101 to interfere with profibrotic activities induced by CCL24 both in vitro and in experimental models of dermal and pulmonary fibrosis in vivo.
Results
Elevated levels of CCL24 and increased expression of CCR3 in SSc patients compared with healthy subjects
Initially, we aimed to test whether the circulating levels of CCL24 were altered in SSc patients compared with healthy individuals. Serum levels of CCL24 were significantly increased in all 37 SSc patients compared with healthy controls (n=23) (1003±110 and 262±32 pg/mL, respectively, p<0.0001, non-parametric Mann-Whitney U test). Notably, in diffuse SSc patients (n=27), CCL24 levels were fourfold higher than in healthy controls (1072±146 and 262±32 pg/mL, respectively, p<0.0001, non-parametric Mann-Whitney U test), while in limited SSc patients (n=10), a threefold elevation (816±94 pg/mL) was found compared with healthy volunteers (p<0.0001, non-parametric Mann-Whitney U test) (figure 1).
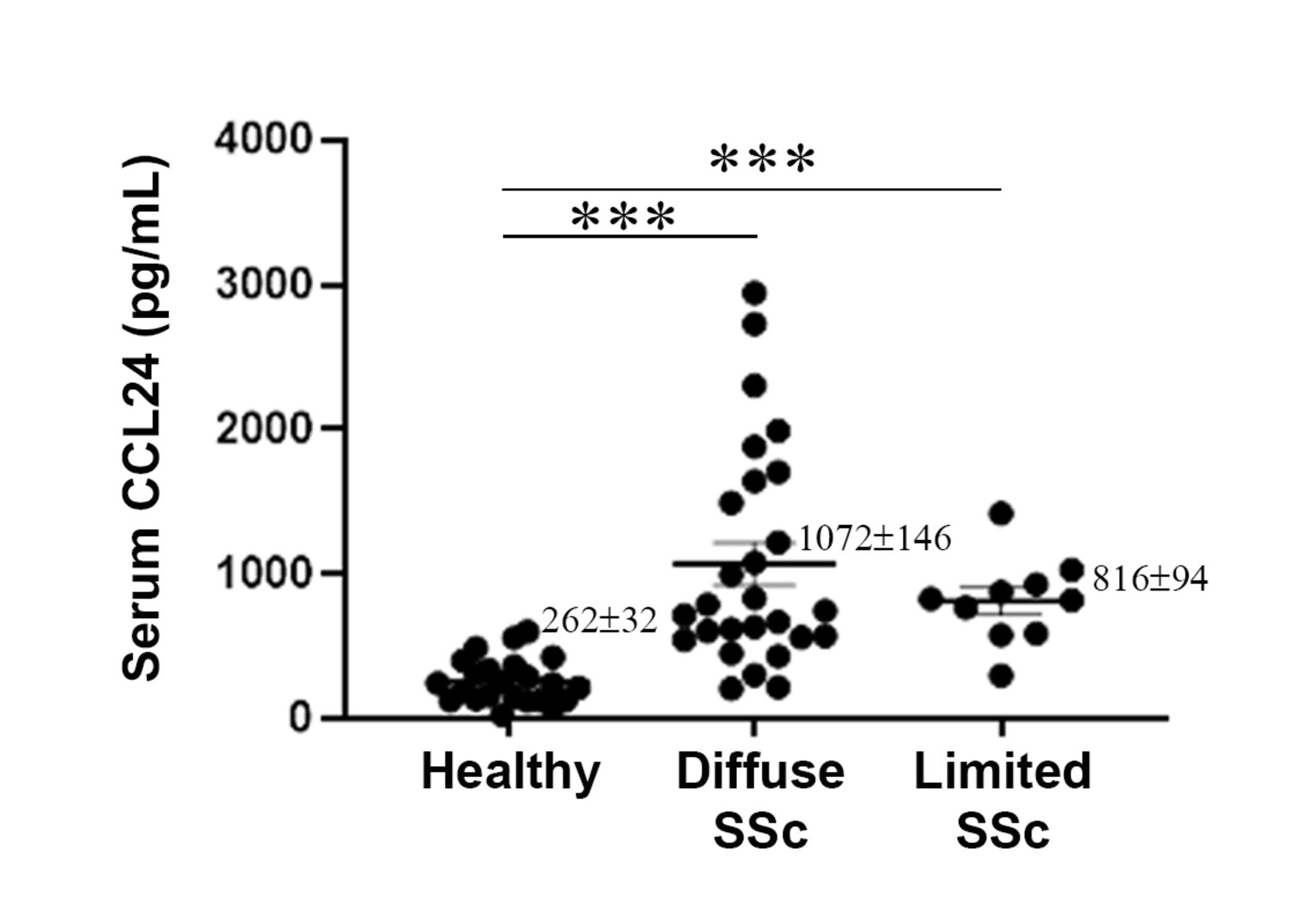
Elevation of CCL24 in systemic sclerosis (SSc) serum. CCL24 serum levels in healthy volunteers (n=23), patients with limited SSc (n=10) and patients with diffuse SSc (n=27) were measured using a commercial Enzyme-Linked Immunosorbent Assay (ELISA) kit. Mean concentration of CCL24 were 262±32, 816±94 and 1072±146 for healthy controls, limited SSc and diffuse SSc patients, respectively. Each dot represents a subject. Bars represent the mean±SEM. ***P<0.0001 (non-parametric Mann-Whitney U test).
Next, we wanted to assess the expression of CCL24 and its receptor CCR3 in skin biopsies of early diffuse SSc patients. Using immunofluorescence staining of skin sections, we demonstrated that the expression of both CCL24 and CCR3 is considerably upregulated in SSc compared with healthy control skin (figure 2A–D). Indeed, CCL24 and CCR3 expression were negligible in healthy skin (figure 2A,C). Conversely, in SSc skin, we observed significantly higher numbers of CCL24-positive mononuclear cells, captured in specific dermal locations (p<0.001 vs healthy skin, figure 2A,B). In particular, these cells, highly expressing CCL24, were identified during the process of extravasation and migration into dermal tissue, within the vessel lumen, passing between CD31-positive microvascular endothelial cells, as well as in the perivascular areas, where they were found to express CCL24 in a granular pattern (figure 2A). A strong CCR3 expression was detected in SSc dermal microvascular endothelial cells and spindle-shaped fibroblasts and epidermal keratinocytes (figure 2C). Densitometric analysis of immunofluorescence staining intensity confirmed that CCR3 expression was significantly increased in SSc skin biopsies with respect to controls (p<0.001, figure 2D). Furthermore, double immunofluorescence with the myofibroblast marker α-smooth muscle actin (α-SMA) revealed the presence of numerous profibrotic myofibroblasts strongly expressing CCR3 in SSc dermis (figure 2E). Taken together, these observations suggest that the CCL24/CCR3 axis may play a role in SSc pathophysiology.

Expression of CCL24 and CCR3 in systemic sclerosis (SSc) skin tissue. (A–D) Immunofluorescence analyses of CCL24 and CCR3 on skin sections from patients with early diffuse SSc (n=5) and healthy controls (HC; n=5). Representative images of skin sections double-immunostained for CCL24 (A) or CCR3 (C) (both green) and the pan-endothelial cell marker CD31/platelet-endothelial cell adhesion molecule (PECAM-1) (red) are shown. Nuclei were counterstained using 4′,6-diamidino-2-phenylindole (DAPI) (blue). Arrowheads indicate spindle-shaped dermal fibroblasts. Scale bar: 50 µm. (B) Quantitative analysis of CCL24-positive cells counted in 10 randomly chosen microscopic high-power fields (hpf; ×40 original magnification) of the dermis per sample (n=5 for both HC and SSc). (D) Densitometric analysis of CCR3 immunofluorescent staining intensity on digitised images of skin sections (n=5 for both HC and SSc) expressed as optical density in arbitrary units (au). Data are mean±SEM. Student’s t-test was used for statistical analyses. (E) Representative images of SSc skin sections double-immunostained for CCR3 (green) and α-SMA (red) and counterstained with DAPI (blue). Both single and overlay images are shown. Note the strong expression of CCR3 in numerous α-SMA-positive dermal myofibroblasts (arrowheads; shown at higher magnification in the insets). Scale bar: 50 µm. α-SMA, α-smooth muscle actin.
CM-101 attenuates CCL24-induced migration of dermal fibroblasts
We, next, aimed to test the involvement of CCL24 in SSc pathogenesis and whether blocking CCL24 activity can halt the inflammatory and fibrotic processes leading to reduced tissue damage. For this purpose, we used CM-101, a mAb that targets CCL24, thus blocking its activity. We first evaluated the capacity of CM-101 to interfere with the migration and activation of dermal fibroblasts by using commercially available normal human dermal fibroblasts (NHDF cells). CCL24 induced 3.2 fold increase of NHDF chemotaxis (p<0.05). Preincubation of CM-101 with CCL24 led to a significant, dose-dependent, reduction of migration index to 1.2, 1.4 and 2.4 when 100, 50 and 10 nM CM-101 was added (p<0.01, p<0.01 and p<0.05, respectively), respectively (figure 3A).

CM-101 attenuates fibroblast and endothelial cell activation and inhibits fibroblast migration and transition to myofibroblasts. (A) CM-101 attenuates normal human dermal fibroblast (NHDF) migration towards CCL24. Migration experiments were performed using a traswell assay. In some experimental conditions, CCL24 was pretreated with CM-101 as indicated. Data are expressed as % migrating cells per high power field. Bars represent the mean±SD level of migration using migration index from three independent experiments. *P<0.05 and **p<0.01. n.s., not significant. (B) Representative FACS (Flow cytometry) plots demonstrating α-SMA expression in NHDF cells incubated with systemic sclerosis (SSc or scleroderma) serum with or without CM-101 (10 or 5 µg/mL). Similar results were obtained with five different serum samples. Graph represents mean of average α-SMA expression in NHDF cells treated with five SSc serum samples. The results were calculated for each individual serum sample, comparing α-SMA expression prior to and following CM-101 treatment (*p<0.05, **p<0.01 for 5 µg/mL and 10 µg/mL CM-101, respectively). Basal α-SMA expression in serum-free treated cells yielded low detection levels (4.6%±1.2%, data not shown). (C) NHDF cell activation following treatment with SSc serum, with or without preincubation with CM-101, was assessed by fluorescent calcium release (Fluo-4). (D) Activation of HUVEC endothelial cells following treatment with SSc sera from two donors (D1 and D2) was assessed by vascular cell adhesion molecule 1 (VCAM-1) expression. Activation levels were tested with or without preincubation of patient sera with CM-101 (50 or 10 µg/mL). Results are shown as mean±SEM. *P<0.05 and **p<0.01 (Student’s t-test). α-SMA, α-smooth muscle actin; IgG, control commercial human immunoglobulin G.
CM-101 reduces SSc serum-induced dermal fibroblast activation and transition to myofibroblasts and interferes with endothelial cell activation
In SSc, the conversion of fibroblasts into myofibroblasts is evident by the expression of α-SMA and is a detrimental event fostering the progression to advanced fibrosis. In order to evaluate the influence of CM-101 on fibroblast to myofibroblast transition, NHDF cells were incubated with sera from SSc patients that were either untreated or preincubated with CM-101. The expression of α-SMA was measured to evaluate myofibroblast transition and cell activation. As shown in figure 3B, treating cells with SSc serum induced cell activation and upregulation of α-SMA expression. This effect was significantly attenuated by 50% when cells were treated with SSc serum preincubated with CM-101 (10 µg/mL). These results suggest that CM-101 may attenuate the transition of fibroblasts to myofibroblasts induced by SSc sera (figure 3B). To further assess the effect of CM-101 on the activation of dermal fibroblasts, we examined cell activation using calcium influx release following incubation with SSc sera. Preincubation of the sera with 50 or 10 µg/mL CM-101 resulted in significantly reduced calcium influx release by 50% and 25%, respectively, compared with cells activated with sera without antibody (both p<0.01) (figure 3C).
To assess, the effect of CM-101 on endothelial cell activation, we measured the expression of VCAM-1, an adhesion molecule known to be highly expressed on activated endothelial cells. VCAM-1 was quantified using flow cytometry following human umbilical vein endothelial cells (HUVEC) activation with SSc sera. Following preincubation of the sera with CM-101 at a concentration of 10 or 50 µg/mL, VCAM-1 expression was reduced by an average of 36% and 61%, respectively, in comparison with cells treated with patient serum alone (both p<0.01) (figure 3D).
CCL24 is a significant driver of inflammation and fibrosis in bleomycin-induced dermal fibrosis mouse models
The evidence that the significant NHDF activation induced by CCL24 could be dampened using a CCL24 blocking antibody in vitro, led us to explore the importance of CCL24 in dermal fibrosis. For this purpose, we employed the commonly used bleomycin (BLM)-induced dermal fibrosis experimental murine model. We first employed the BLM model on CCL24 knockout mice compared with wild-type (WT) mice. Mice lacking the CCL24 gene showed a reduced response in all tested disease parameters. Histological assessment revealed reduced dermal thickness as well as attenuated infiltration of immune cells into the bronchoalveolar lavage (BAL) fluid (p<0.05) (figure 4A–C). α-SMA expression in the skin lesions taken from knockout mice compared with WT was significantly reduced as evident by quantitative real-time PCR analysis and similar to the levels detected in healthy mice (p<0.05) (figure 4D). The reduction in α-SMA further supports the attenuated process of fibroblast activation in the skin and is complementary to the in vitro data shown in figure 3. Next, we performed a prevention model, in which CM-101 was administered together with the first BLM injection before the onset of the pathological skin lesions and throughout disease progression. We observed a significant effect on histopathological skin features in mice treated with BLM and CM-101 when compared with those treated with BLM and phosphate-buffered saline (PBS). CM-101 prevented the increase in both dermal thickness (figure 4E) and skin collagen content (figure 4F). Dermal thickness showed statistically significant differences both for 0.5 and 2.5 mg/kg doses of CM-101, as compared with BLM and PBS treated groups (31% and 57% reduction of dermal thickness, respectively, both p<0.001) (figure 4E). The prophylactic effect of CM-101 was also evident by a significant dose-dependent reduction of skin collagen concentration (57% and 64% reduction at 0.5 mg/kg and 2.5 mg/kg of CM-101, respectively, both p<0.001) (figure 4F).
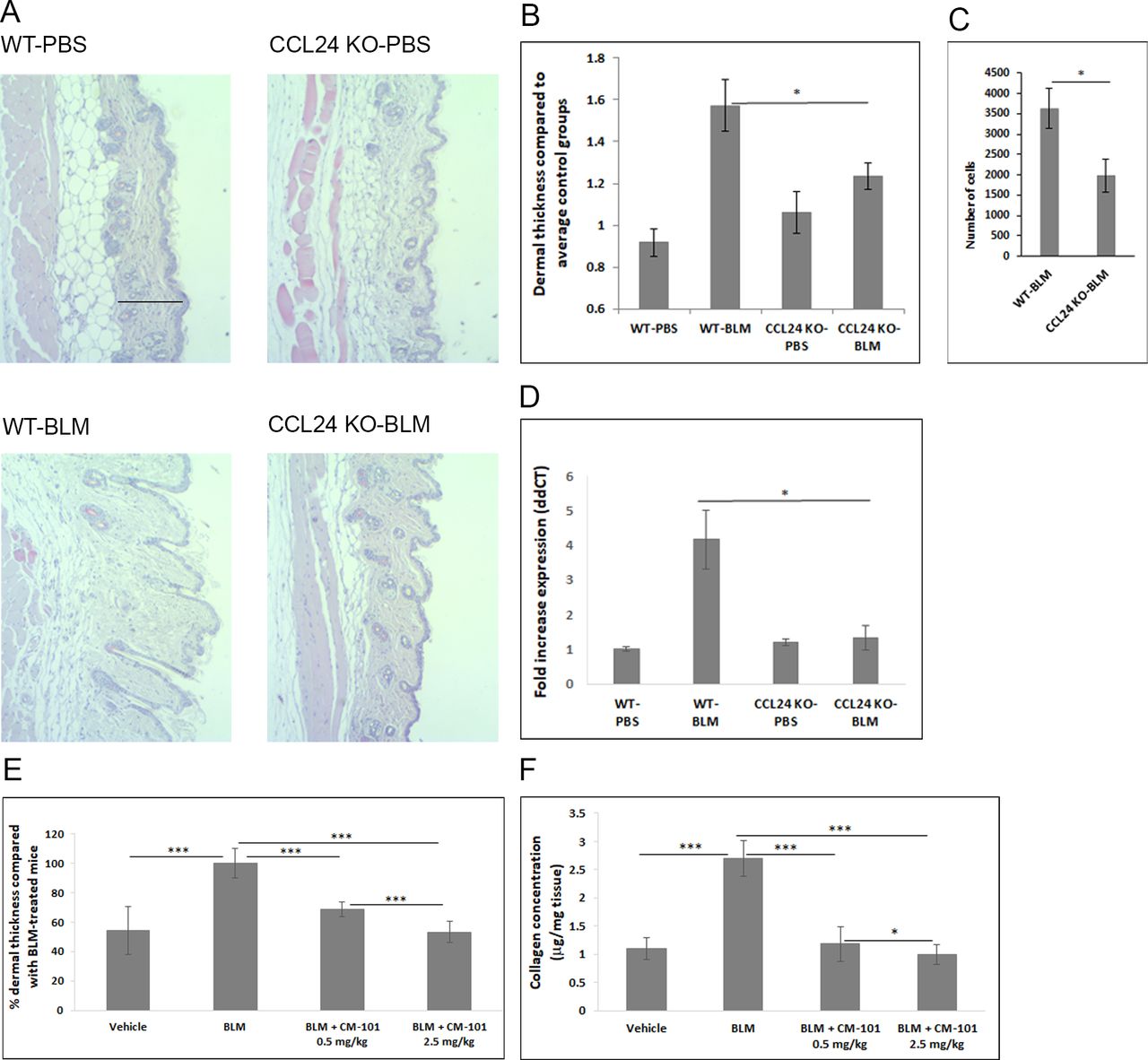
CCL24 gene inactivation and anti-CCL24 monoclonal antibody both prevent fibrosis and inflammation in the bleomycin (BLM)-induced dermal fibrosis mouse model. CCL24 knockout (KO) and wild-type (WT) mice were injected subcutaneously with BLM for 21 days daily. (A) Representative images of H&E-stained skin sections. (B) Dermal skin thickness in the CCL24 KO mice following BLM injections compared with WT mice treated with BLM. (C) Inflammatory infiltrate into the BAL fluid in the CCL24 KO mice compared with WT mice. (D) Expression of α-SMA assessed by real-time PCR in skin samples from BLM model compared with healthy WT mice. CCL24 KO mice retained low expression of α-SMA following daily BLM injections for 21 days. (E and F) BLM prevention model included four arms (n=8 mice each). Vehicle-injected control group, BLM group treated with BLM + PBS and two groups injected with BLM together with CM-101 in two different doses: BLM + CM-101 0.5 mg/kg and BLM + CM-101 2.5 mg/kg. (E) Dermal thickness was assessed in mice treated with CM-101 0.5 or 2.5 mg/kg two times per week for 21 days in parallel to daily subcutaneous BLM administration. (F) Skin collagen content (µg per mg skin tissue) was measured by Sircol assay in skin lysates from BLM-induced dermal sclerosis mice treated with CM-101 or PBS. Bars represent the mean±SEM. *P<0.05 and ***p<0.001 (Student’s t-test). α-SMA, α-smooth muscle actin; PBS, phosphate-buffered saline.
In a second set of experiments, the efficacy of CM-101 was tested using a treatment model where CM-101 was administered only after the onset of fibrotic signs, 8 days following the first BLM injection.32 Histological assessment of skin lesions stained with H&E and Masson’s trichrome revealed significant elevation of dermal thickness and collagen deposition following 21 days of BLM administration. This elevation was significantly reduced when mice were treated with CM-101 (figure 5A). Both skin thickness and collagen deposition were significantly reduced in the 2.5 mg/kg CM-101-treated group as compared with the mouse group treated with BLM alone (p<0.001 and p<0.01, respectively) (figure 5B,C). Another feature that characterises the BLM model and is similar to human SSc is the development of bronchoalveolar inflammation.33 To evaluate the effect of CM-101 on lung inflammation, we collected BAL fluid and assessed the number of white blood cells (WBC) and mononuclear cells, which represent the cell population that is directly involved in the inflammatory process. Treatment with BLM for 21 days significantly increased WBC and mononuclear cells in BAL fluid (p<0.05). The number of WBC and mononuclear cells was decreased significantly following CM-101 treatment compared with the BLM-administered group (p<0.05 and p<0.01, respectively) (figure 5D,E). To further characterise the effect that CM-101 had on skin fibrosis, myofibroblast activation was evaluated by quantification of α-SMA levels in the skin of mice treated with BLM, in presence or absence of CM-101 (figure 5F). Western blot analysis of skin homogenates from CM-101-treated mice revealed a significant 66% reduction in α-SMA compared with vehicle-treated group (p<0.05) (figure 5F).

CM-101 treatment attenuates skin fibrotic remodelling in the bleomycin (BLM)-induced dermal fibrosis mouse model. Model includes four arms (n=8 mice each). Vehicle-injected control group, two BLM groups injected with BLM + PBS and one BLM + CM-101 2.5 mg/kg. BLM baseline (8 days) assessed disease stage when CM-101 treatment initiated at day 8 and BLM (21 days) representing the end stage of this model. (A) Representative images of H&E and Masson’s trichrome (MT) staining of skin sections from BLM-induced dermal sclerosis mice. (B) Dermal thickness shows increase thickness following BLM treatment and reduction of thickness when mice were further treated with CM-101. (C) Skin collagen concentration was measured in skin lysates using Sircol assay. (D and E) BAL fluid was assessed by flow cytometry for total white blood cell (WBC) and mononuclear cell counts. (F) Skin tissue homogenates from BLM (21 days) and BLM + CM-101 2.5 mg/kg (50 µg total protein per lane) were analysed by western blot using anti-α-SMA primary antibodies. β-tubulin was measured as a loading control. Graph reports densitometry quantification values of α-SMA levels in skin extracts (n=5 mice in each group). Bars represent the mean±SEM. *P<0.05, **p<0.01 and ***p<0.001(Student’s t-test). α-SMA, α-smooth muscle actin; PBS, phosphate-buffered saline.
CM-101 inhibits lung fibrosis and inflammation in the BLM-induced pulmonary fibrosis mouse model
There are major similarities in the features of the lung disease in patients with SSc and IPF. In both diseases, there is evidence of inflammatory and fibrotic processes dominating the pathological picture with the involvement of similar cellular components.34 CCL24 was reported in the past as being significantly and differentially expressed in lungs of IPF patients.25 26
Although there are no animal models that precisely exemplify pulmonary fibrosis, the intratracheal injected BLM model is considered a well-established experimental model, especially for testing pharmaceuticals compounds.35 We, thus, explored the experimental IPF model in C57BL/6 mice given a single intratracheal administration of BLM with or without CM-101.
Overall, we found that CM-101 had a significant antifibrotic and anti-inflammatory effect in the experimental BLM-induced lung fibrosis model as compared with vehicle-treated animals that included either PBS or irrelevant immunoglobulin G (IgG).
BLM animals treated with PBS revealed massive immune cell infiltration, extensive fibrosis and severe tissue injury as compared with significantly reduced histopathological changes in H&E-stained and Masson’s trichrome-stained lung sections from the CM-101-treated group (figure 6A). Quantifying fibrosis using Ashcroft score36 revealed a 40% reduction of fibrosis following CM-101 treatment compared with the BLM + PBS-treated group (figure 6B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CM-101 attenuates lung fibrosis and inflammation in the bleomycin (BLM)-induced pulmonary fibrosis mouse model. Model includes four arms (n=11 mice each). Vehicle-injected control group and BLM baseline to assess disease stage when CM-101 treatment initiated at day 10. BLM group injected with BLM + PBS and BLM + CM-101 2.5 mg/kg (from day 10, two times per week). (A) Representative images of H&E and Masson’s trichrome (MT) staining of lung sections from vehicle control mice, BLM baseline, BLM and BLM + CM-101 2.5 mg/kg treatment groups. (B) BLM + CM-101 2.5 mg/kg treated mice show significant reduction of fibrosis measured by Ashcroft score compared with BLM group. (C) Similar model was conducted and compared lung collagen concentration in lung lysates from vehicle control group, BLM injected with BLM + PBS, BLM + CM-101 2.5 mg/kg group and BLM + IgG 2.5 mg/kg group. Collagen amount was measured using Sircol assay. (D) The analysis of bronchoalveolar lavage fluid to assess infiltration of immune cells into the lung was done by counting of white blood cells (WBC) using COBAS 6000. Bars represent the mean±SEM. *P<0.05, **p<0.01 and ***p<0.005 (Student’s t-test). IgG, immunoglobulin G; PBS, phosphate-buffered saline.
In addition, CM-101-treated mice exhibited significantly reduced levels of lung collagen. Indeed, collagen content was decreased to similar levels as in healthy animals (16.9±2 and 14.8±4.77 for no BLM and BLM + CM-101 groups, respectively), whereas a twofold increase in lung collagen content was observed in the BLM + PBS-treated and BLM + IgG-treated groups (31.1±3.22 and 30.2±3.21, respectively) (both p<0.001, figure 6C). To further evaluate the inflammatory status, we performed BAL fluid immune cell count. We found robust attenuation of immune cell infiltration to the BAL fluid obtained from the BLM animals treated with CM-101 compared with BLM animals treated with either PBS or IgG, as testified by significantly reduced WBC counts (p<0.01, figure 6D).
Discussion
Chemokines are fundamental effectors involved in diverse inflammatory processes and act as a potent chemoattractant that regulate cell trafficking, angiogenesis and even collagen production.8 Dysregulation of chemokine pathways has a significant impact on the pathogenesis of several autoimmune diseases37 38 participating in both disease development and progression. Specifically, it was shown over the years that chemokines play a significant role in the pathogenesis of SSc,8 promoting the transformation of fibroblasts into myofibroblasts, fostering migration and activation of inflammatory cells and impairing angiogenesis and vasculogenesis in these patients.10 39–41
CCL24 is a chemokine that was previously shown to be involved in proinflammatory and profibrotic processes. It was demonstrated to induce proliferation and collagen synthesis of human lung fibroblasts27 and could serve as a differentiating marker in pulmonary fibrosis compared with pneumonitis.42 Here, we show, for the first time, that CCL24 levels are significantly increased in serum samples and skin biopsies from SSc patients. In diffuse SSc skin, CCL24 expression was evident in perivascular mononuclear cell infiltrates as well as in cells in transition through the vessel wall from the bloodstream to the dermal tissue. The receptor for CCL24, CCR3, was also strongly expressed on SSc dermal microvascular endothelial cells and α-SMA-positive myofibroblasts. Conversely, skin sections from healthy donors showed negligible staining for either CCL24 or CCR3. These collective observations led us to assume that the CCL24/CCR3 axis may play an important role in driving migration of inflammatory cells from the periphery into inflamed fibrotic skin and that it may act locally to foster fibroblast activation. Neutralising CCL24 using the novel CM-101 antibody had a robust effect on both fibrosis development in vivo and on fibroblast activation in vitro. We observed a dose-dependent decrease in fibroblast migration towards CCL24 following treatment with CM-101. Moreover, adding CM-101 to sera from SSc patients was able to inhibit serum-induced dermal fibroblast activation, significantly reducing α-SMA expression and calcium influx in these cells. Of note, the finding that CM-101 was able to significantly attenuate such activation in the presence of sera that contains a wide panel of chemokines and cytokines, suggests that CCL24 may be a major contributor to fibroblast activation. In addition, the ability of CM-101 to attenuate CCL24-induced endothelial cell activation, measured by their VCAM-1 expression, further supports the broad effect of CCL24 to induce the activation of different cell types that are known to be involved in SSc pathogenesis as well as the broad applicability of CM-101 in inhibiting these activities.43 Collectively, our in vitro observations suggest that CCL24 is a prominent trigger of fibroblast and endothelial cell activation and that the blocking antibody CM-101 is potently capable of mitigating these processes.
SSc patients present changes in inflammatory mediators, growth factors and profibrotic cytokines that all take part in the development of the disease. These factors influence several cell compartments within the target organs promoting tissue remodelling and damage. To evaluate the relevance of CCL24 and the effect of CM-101 in this complex array of factors, we employed the murine model of BLM-induced skin sclerosis that is considered a proper model to reflect SSc-related skin fibrosis.33 44 Mice lacking the CCL24 gene demonstrated robust attenuation of disease parameters compared with WT controls, which further supports the potential importance of the CCL24/CCR3 pathway in skin fibrosis. CM-101 treatment in the early phase of the inflammatory and fibrotic process, in a prevention mode, resulted in a dose-dependent reduction of skin thickness and collagen deposition. Moreover, in a treatment mode, when fibrosis and inflammation were already established, CM-101 was also shown to attenuate significantly the severity of skin fibrosis and thickness. Lung fibrosis and inflammation features of interstitial lung disease (ILD), accounting for major disease-related complications and mortality, were also significantly reduced in CM-101 treated animals. Collectively, the data show a broad effect of anti-CCL24 treatment, reducing tissue damage of both skin and lung.
Tissue fibrotic remodelling in SSc and IPF patients involves a relentless inflammatory component followed by replacement of the healthy tissue with fibrous, matrix-rich abnormal scarring.2 8 The robustness and complexity of the underlying pathogenic processes clearly suggest that multiple cellular pathways are operative in dictating the fate of the afflicted tissues in both conditions. In this context, CCL24 appears to promote several of those pathways as testified by the beneficial effects achieved in our in vivo models through its specific blockade using the CM-101 mAb.
To date, effective treatments for SSc patients that can alter established fibrosis and inhibit disease progression are limited. Alongside few medicinal treatments that are available, several new emerging therapies have shown benefit in SSc patients including both haematopoietic and also mesenchymal stem cell-based therapy protocols. However, in most of these options, liver toxicity, severe adverse effects and complications limit long-term effective treatment availability. Our findings provide the first evidence that an anti-inflammatory, antifibrotic approach selectively targeting the CCL24 pathway robustly attenuates experimental skin and lung inflammation and fibrosis. These preclinical data may support the translation to clinic of an anti-CCL24 antibody to attenuate the progression of fibrosis in SSc and IPF patients.
Acknowledgments
Partial data of this manuscript were introduced during the 4th Systemic Sclerosis World Congress, 18–20 February 2016, Lisbon, Portugal.
References
Footnotes
Handling editor Josef S Smolen
Contributors AM: Substantial contributions to the conception and design of the work. Drafting the work and final approval of the version to be published. MSS and NB: Analysis and interpretation of data for the work; drafting the work and revising it for publication. AK and VE: Data acquisition. MM: Immunostaining data acquisition, analysis and interpretation; drafting the work and revising it for publication. YL and JG: Revision for publication. MM-C: Revising the work critically for publication. All the authors were involved in the development, review and approval of the manuscript.
Funding This research has received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests AM, MSS, AK, NB and VE: ChemomAb employees. JG: ChemomAb consultant. MM-C: ChemomAb consultant, BMS, MSD, Actelion, Sanofi and Biogen.
Ethics approval Serum samples from systemic sclerosis patients were taken following Helsinki ethics committee approval (Meir Medical Center, Kfar-Saba, Israel). Archival skin specimens were taken under protocols approved by the local institutional review board at the Azienda Ospedaliero-Universitaria Careggi (AOUC), Florence, Italy. Studies involving animal models were approved by the National Board of Animal Studies in the Ministry of Health by the Kaplan Medical Center.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data of SSc patients and control participants belong to the Meir Medical Center, Kfar-Saba, Israel, and are not publicly available.