
Abstract
Tris-pyridineoximate iron, cobalt, and nickel(II) pseudoclathrochelates with apical ferrocenyl substituent were obtained in the reasonable yields (50–70%) in a boiling ethanol by the template condensation of 2-acetylpyridineoxime with ferrocenylboronic acid on the corresponding M2+ ion as a matrix. The composition and structure of new ditopic compounds, isolated in the forms of their ionic associates with perchlorate anion, were determined using elemental analysis, UV-vis spectroscopy, MALDI-TOF mass spectrometry, and NMR spectroscopy. According to the magnetometry data, the iron(II) pseudoclathrochelate is a diamagnetic compound, while the temperature dependences of magnetic susceptibility of the nickel and cobalt(II) complexes are characteristic of the high-spin systems with S = 1 and 3/2, respectively. As follows from the X-ray diffraction data for the iron and nickel(II) pseudoclathrochelates, the Ni–N distances (2.15–2.17 Å) are characteristic of the high-spin Ni2+ complexes, while they in its iron(II)-containing analog, slightly exceed of 2 Å, thus suggesting the low-spin state of this ion.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The encapsulation of a metal ion within a cavity of the rigid three-dimensional ligand determines a number of interesting physical and chemical properties of the resulting cage complexes of this cation [1], namely the unusually high chemical stability of the complex in various media and in various degrees of oxidation of its metal center, including the stabilization of unusual low [1, 2] or rare high [3–6] oxidation states, nonclassical manifestations of the Jahn–Teller effect in these (pseudo)macrobicyclic compounds, temperature spin transitions, etc. For cobalt(II) clathrochelates [1] and pseudoclathrochelates [7], slow magnetic relaxation was previously found in the absence of an external magnetic field; they exhibit [8–16] the properties of monomolecular magnets, which in some cases characterized by exceptionally high (for cobalt(II) complexes) magnetization reversal barriers. The magnetic properties of metal complexes of this type largely depend on a number of factors, including their electronic and spatial (chemical) structure, the nature of donor atoms, and the geometry of the coordination polyhedron, the presence of an intrinsic nuclear magnetic moment of the encapsulated metal ion and its surrounding nuclei, and even on much less obvious influence of the supramolecular organization [1] and polymorphism [9] of their crystals. All of the above factors determine the possibility of using compounds of this type to create molecular magnetic materials and devices. In this regard, of significant fundamental and practical interest is the directed design of new polytopic (multicenter) molecular (pseudo)cage complexes of d-metals with slow magnetic relaxation and the ability to externally switch the degree of interaction between their magnetically and redox-active metal centers. Thus, binuclear pseudoclathrochelate complexes can be obtained by cross-linking with 3d-metal-containing Lewis acids, in particular, ferrocenylboronic acid. It has been assumed that the magnetic interaction between the metal centers of the resulting hybrid molecule can be controlled by selective oxidation or reduction of the redox-active fragment, which is included in the apical substituent at the clathrochelate cage. Therefore, we have obtained new ferrocenylborate tris-pyridineoximate iron, cobalt, and nickel(II) pseudoclathrochelates; the crystal and molecular structures of iron and nickel(II) complexes were obtained by the single X-ray diffraction experiments, and their magnetic properties were studied using the magnetometry method.
EXPERIMENTAL
Materials and methods. We used commercial reagents purchased from Sigma-Aldrich: FeCl2⋅4H2O, Ni(ClO4)2 ⋅ 6H2O, NaHCO3, NaClO4·H2O, hydroxylamine hydrochloride, 2-acetylpyridine, ferrocenylboronic acid {FсB(OH)2}, sorbents, and organic solvents without additional purification. Co(ClO4)2·6H2O was obtained according to the procedure reported [17]. 2‑Acetylpyridineoxime (AcPyOxH) was obtained according to the known procedure [18].
Elemental analysis (C, H, N) was performed on a Carlo Erba microanalyzer (model 1106). The iron content was determined spectrophotometrically. The content of cobalt and nickel was determined by the X‑ray fluorescence method. All these analyses were carried out at the Laboratory of Microanalysis of the A.N. Nesmeyanov Institute of Organoelement Chemistry, Russian Academy of Sciences.
MALDI-TOF mass spectra in the positive and negative regions were recorded on a BrukerAutoflex II (BrukerDaltonics) MALDI-TOF-MS mass spectrometer in the reflecto-mol mode. Ionization was caused by a UV laser with a wavelength of 337 nm. The samples were placed on a steel plate, 2,5-dihydroxybenzoic acid was used as a matrix. The measurement accuracy was 0.1%.
1H and 13C{1H} NMR spectra of solutions in CD2Cl2 and CD3CN were recorded on a Bruker Avance 600 and a Varian Inova 400 spectrometers. The measurements were performed against the residual signals of these solvents: CD2Cl2 (1H 5.32 ppm, 13C 54.00 ppm), CD3CN (1H 1.94 ppm, 13C 1.32 ppm). Data processing was carried out using the Mestrenova 12.0.0 program.
UV-vis spectra of solutions of the complexes in acetonitrile and methylene dichloride were recorded in the ranges of 190–1000 and 230–1000 nm, respectively, on an Agilent Cary 60 spectrophotometer. The individual Gaussian components of these spectra were calculated using the Fityk program [19].
Magnetic measurements were carried out on a Quantum Design PPMS-9 magnetometer. A finely dispersed powder sample of the respective complex was suspended in organic oil and hermetically sealed in a plastic bag. The static magnetic susceptibility was measured in the temperature range 3–300 K in an external magnetic field of 5 kOe. The diamagnetic correction of the bag and oil was taken into account; the diamagnetic contribution of this complex was estimated using the Pascal constants [20]. The magnetization of the sample was measured at an external magnetic field strength in the range of 0–5 T at temperatures of 2, 4, and 6 K. The dynamic magnetic susceptibility was measured in the frequency ranges of 10–100, 100–1000, and 1000–10 000 Hz in alternating magnetic fields with an amplitude of 5, 3, and 1 Oe, respectively. The measurements were performed both in zero and in a magnetic field of 1 kOe in the temperature range of 2–16 K.
[Fe(AcPyOx)3(BFc)](ClO4) (1). 2-Acetylpyridineoxime (0.27 g, 1.98 mmol), NaClO4·H2O (0.46 g, 3.3 mmol), NaHCO3 (0.28 g, 3.3 mmol), and ferrocenylboronic acid (0.17 g, 0.73 mmol) were dissolved/suspended in ethanol (3 mL) in argon atmosphere. The reaction mixture was stirred for 5 min, and FeCl2·4H2O (0.13 g, 0.66 mmol) and ethanol (4 mL) were added. The resulting mixture was boiled for 30 min, the dark red precipitate formed was filtered off, washed with ethanol (12 mL, in 3 portions), diethyl ether (10 mL, in 2 portions), and hexane (5 mL). The product was extracted with methylene chloride (20 mL, in 4 portions). The filtered extract was evaporated to dryness, and the solid residue was dried in vacuum. Yield, 0.36 g (72%).
For C31H30N6O7BClFe2 anal. calcd. (%): C, 49.17; H, 3.97; N, 11.10; Fe, 14.81.
Found (%): C, 48.95;H, 3.87; N, 11.18; Fe, 14.75.
Mass-spectrum (MALDI-TOF), m/z: 657 [M–\({\text{ClO}}_{4}^{ - }\)]+. 1H NMR (CD2Cl2, δ, ppm): 2.66 (s, 9Н, CH3), 4.25–4.48 (m, 9Н, Fc), 7.00 (d, 3Н, 6-Py), 7.48 (t, 3Н, 5-Py), 7.93 (d, 3Н, 3-Py), 8.05 (t, 3Н, 4-Py). 13C{1H} NMR (CD2Cl2, δ, ppm): 13.69 (s, CH3), 69.65, 70.54, 73.12 (all br. s, Fc), 124.96 (s, 3-Py), 126.13 (s, 5-Py), 138.39 (s, 4-Py), 153.36 (s, 6-Py), 158.59 (s, 2-Py), 160.32 (s, C=N). Electronic absorption spectrum (CH3CN, λmax, nm) (ε × 10–3, mol–1 L cm–1): 200 (80), 226 (12), 249 (23), 278 (7.3), 290 (14), 299 (7.4), 358 (4.7), 431 (2.3), 463 (5.2), 484 (3.8), 519 (15).
[Co(AcPyOx)3(BFc)](ClO4) (2). 2-Acetylpyridineoxime (0.27 g, 1.98 mmol), NaClO4·H2O (0.18 g, 1.3 mmol), NaHCO3 (0.28 g, 3.3 mmol), and ferrocenylboronic acid (0.17 g, 0.73 mmol) were dissolved/suspended in ethanol (3 mL) under argon atmosphere. The reaction mixture was stirred for 5 min, and Co(ClO4)2·6H2O (0.24 g, 0.66 mmol) and ethanol (4 mL) were added. The resulting mixture was refluxed for 40 min; the resulting light brown precipitate was filtered off, washed with ethanol (30 mL, in 6 portions), diethyl ether (10 mL, in 2 portions), and hexane (5 mL). The product was extracted with methylene chloride (20 mL, in 4 portions). The filtered extract was evaporated to dryness, and the solid residue was dried in vacuum. Yield, 0.26 g (52%).
For C31H30N6O7BClCoFe anal. calcd. (%): C, 48.98; H, 3.95; N, 11.06; Fe, 7.37; Co, 7.77.
Found (%): C, 48.79;H, 3.88; N, 10.97; Fe, 7.25; Co, 7.60.
Mass-spectrum (MALDI-TOF), m/z: 660 [M–\({\text{ClO}}_{4}^{ - }\)]+. 1H NMR (CD2Cl2, δ, ppm): –2.78 (br. s, 3Н, 3-Py), 2.53 (br s, 9Н, CH3), 14.62 (br s, 3Н, 4-Py), 24.87 (br s, 5Н, Cp (unsubst.)), 27.20 (br s, 2Н, β-Cp), 56.67 (br s, 2Н, α-Cp), 80.35 (br s, 3Н, 5-Py), 397.23 (br s, 3Н, 6-Py). Electronic absorption spectrum (CH3CN), λmax, nm) (ε × 10–3, mol–1 L cm–1): 201 (48), 221 (25), 249 (8.6), 262 (2.7), 277 (15), 341 (5.3), 415 (0.6), 426 (0.8).
[Ni(AcPyOx)3(BFc)](ClO4) (3). 2-Acetylpyridineoxime (0.28 g, 2.0 mmol), ferrocenylboronic acid (0.15 g, 0.64 mmol), NaClO4·H2O (0.08 g, 0.58 mmol), and NaHCO3 (0.1 g, 1.17 mmol) were dissolved/suspended in ethanol (5 mL) under argon atmosphere. The reaction mixture was stirred for 5 min, and Ni(ClO4)2⋅ 6H2O (0.21 g, 0.58 mmol) and ethanol (5 mL) were added. The resulting mixture was refluxed with stirring for 1 h. The bright orange precipitate formed was filtered off, washed with ethanol (10 mL, in 2 portions), diethyl ether (14 mL, in 2 portions), and hexane (7 mL). The product was extracted with methylene chloride (9 mL, in 3 portions). The filtered extract was evaporated to dryness, and the solid was dried in vacuum. Yield, 0.28 g (64%).
For C31H30N6O7BClFeNi anal. calcd. (%): C, 48.98; H, 3.95; N, 11.06; Fe, 7.37; Ni, 7.77.
Found (%): C, 48.94; H, 3.84; N, 10.95; Fe, 7.50; Ni, 7.60.
Mass-spectrum (MALDI-TOF), m/z: 659 [M–\({\text{ClO}}_{4}^{ - }\)]+. 1H NMR (CD2Cl2, δ, ppm): –24.00 (br s, 9Н, CH3), 3.72 (br m, 9Н, Fc), 15.46 (br s, 3Н, 4-Py), 44.13 (br s, 3Н, 5-Py), 59.23 (br s, 3Н, 3-Py), 137.32 (br s, 3Н, 6-Py). Electronic absorption spectrum (CH2Cl2, λmax, nm) (ε × 10–3, mol–1 L cm–1): 252 (21), 263 (2.6), 288 (13), 312 (11), 353 (0.8), 381 (0.7), 776 (0.011), 828 (0.008), 898 (0.016).
X-ray diffraction. X-ray diffraction studies of single crystals [Fe(AcPyOx)3(BFc)](ClO4)·2CHCl3 (1·2CHCl3) and [Ni(AcPyOx)3(BFc)](ClO4)·2CH2Cl2 (2·2CH2Cl2) obtained by slow evaporation of solutions of the corresponding pseudoclathrochelates in chloroform–heptane and methylene chloride–hexane mixtures, respectively, were carried out on a Bruker APEX2 DUO CCD diffractometer (MoKα radiation, graphite monochromator, ω-scanning). The structures were solved using the ShelXT program [21] and refined in full-matrix least squares using the Olex2 program [22] in the anisotropic approximation by F2hkl. The positions of hydrogen atoms were calculated geometrically and refined in the isotropic approximation using the “rider” model. The main crystallographic data and refinement parameters are presented in Table 1. Structural data for these complexes were deposited at the Cambridge Crystallographic Data Center (CCDC nos. 2124333 and 2124334; http://www.ccdc.cam.ac.uk/).
RESULTS AND DISCUSSION
Tris-pyridineoximate iron, nickel, and cobalt(II) pseudoclathrochelates 1–3 with apical ferrocenyl substituent at their cross-linking boron atom were obtained in the reasonable yields (50–70%) by Scheme 1 in a boiling ethanol as a solvent by the template condensation of 2-acetylpyridineoxime with ferrocenylboronic acid on the corresponding M2+ ion as a matrix. NaHCO3 was added to neutralize the realizing H+ ions, and perchlorate anion was chosen as a counterion because it forms the stable ionic associates with these bulky semiclathrochelate cations.
The composition and structure of the obtained new ditopic compounds were obtained using elemental analysis, UV-vis spectroscopy, MALDI-TOF mass spectrometry, and NMR spectroscopy; their magnetic characteristics are determined by the method described in the Experimental part.
The positive range of the MALDI-TOF mass spectra of the obtained ferrocenylboron-capped 3d-metal tris-pyridineoximates contain the intense peaks of the corresponding pseudoclathrochelate cations [M–ClO4–]+, which are characteristic of the ionic associates of this type [23]. These peaks have the characteristic isotopic distributions, which were in good agreement with those theoretically calculated.
The number, position, and multiplicity of signals in the solutions 1H and 13C{1H} NMR spectra of solutions of the obtained compounds, as well as the ratio of the integrated signal intensities in the 1H NMR spectra, confirmed the composition and symmetry of their molecules. The spectra of the diamagnetic iron(II) complex show characteristic signals of the methyl group and the heterocyclic fragment of the chelating ligand synthon, as well as characteristic signals of the apical ferrocenyl substituent at the boron atom. Cobalt and nickel(II) complexes with the same pseudoencapsulating ligand are high-spin paramagnetic compounds, which follows from the high chemical shifts of the ligand proton signals in their 1H NMR spectra (up to almost 400 ppm in the case of cobalt(II) compounds). In the latter case, according to [11], the pseudocontact contribution to the chemical shifts prevails, which also affects the position of the proton signals of the apical ferrocenyl substituent (it causes a shift of these signals to the weak-field region and a significant increase in the difference between the positions of the proton signals in its Ср-cycles). The corresponding chemical shifts δ1Н increase for the protons of those groups that are closer to the encapsulated paramagnetic Co2+ metal center. In the 1H NMR spectrum of nickel(II) complex 3, on the contrary, the contact contribution is predominant [11], which mainly affects the signals of the protons of the chelating ligand synthon, while practically not affecting the signals of the protons of the apical group. Indeed, the proton signals of the ferrocenyl substituent in this spectrum have the form of an unresolved multiplet, which is slightly (by ~0.6 ppm) shifted to high fields as compared to those in the 1H NMR spectrum of its diamagnetic Fe(II)-containing pseudoclathrochelate analog.
The solution UV-vis spectrum of the diamagnetic complex 1 in acetonitrile (Fig. 1a) in its visible range contains three less intense (ε ~ 2–5 × 103 mol–1 L cm–1) bands with maxima from 430 to 485 nm, as well as a more intense (ε ~ 1.5 × 104 mol–1 L cm–1) band with maximum at approximately 520 nm. All these bands were assigned to the metal-to-ligand charge transfer Fed → Lπ* bands [24]. Set of the bands, in the UV range of this spectrum is assigned to the π–π* transitions both in the chelating pyridineoximate ligand fragments and in the ferrocenyl substituent at its cross-linking boron atom. The low-intensive (ε ~ 30–150 mol–1 L cm–1) bands of d–d transitions in the visible range in the range 350–450 nm, characteristic of the ferrocene derivatives [24], are masked by the aforementioned intense metal-to-ligand charge transfer bands. The spectrum of a parent ferrocenylboronic acid (Fig. 1d) contains the intense (ε ~ 3–30 × 103 mol–1 L cm–1) charge transfer bands and the low-intensive (ε ~ 30–120 mol–1 L cm–1) bands of d–d transitions in the range 200–240 nm and at approximately 410 and 450 nm, respectively.

Decomposition of electronic absorption spectra of acetonitrile solutions of (a) iron 1, (b) cobalt 2, (c) nickel(II) 3 complexes and (d) initial ferrocenylboronic acid into their Gaussian components. The insets show fragments of these spectra in the near UV, visible, and near IR ranges ((d) and (c), respectively).
UV-vis spectrum of its Co(II)-encapsulating analog 2 (Fig. 1b), which is formed by the same ferrocenyl-containing pseudocaging ligand, contains similar ligand-centered bands in the UV range, while two much less intensive (ε ~ 6–8 × 102 mol–1 L cm–1) bands at 415 and 426 nm, were assigned to the d–d transitions in this apical ferrocenyl substituent. Spectrum of their Ni(II)-containing analog 3, besides the sentence intensive ligand-centered bands, also contains the low-intensive (ε ~ 8–16 mol–1 L cm–1) broad bands in the range 770–900 nm assigned to the d–d transitions of its hexacoordinate Ni2+ ion [24, 25].
The aforementioned spin state of the obtained 3d‑metal pseudoclathrochelates was confirmed by the experimental magnetometry data. It was found that the iron(II) complex 1 is a diamagnetic compound within the studied temperature range 3–300 K. The temperature plot of the magnetic susceptibility for the high-spin nickel(II) complex 3 with the electronic d8 configuration (Fig. 2, curve 1) suggests the constancy of χT = 1.0 cm3 K mol–1 to 10 K, which corresponds to a pure spin value expected for a system with electron spin S = 1. The decrease in χT at temperatures below 10 K apparently results from the saturation effect under the influence of an external magnetic field (the Zeeman effect) on the population of the quantum magnetic levels of the encapsulated Ni2+ ion. On the contrary, in the case of high-spin cobalt(II) pseudoclathrochelate 2 with the d7 electron configuration, the χT values at temperatures above 100 K are in the range of 2.5–2.6 cm3 K mol–1 (Fig. 2, curve 2), which is higher than the purely spin value of 1.87 cm3 K mol–1 for a system with the electron spin S = 3/2. This indicates the presence of a spin-orbit coupling in the case of the complex 2: the decrease in χT below 100 K is usually explained by the effect of zero-field splitting, which is a consequence of the aforementioned spin-orbit coupling of the crystal field of the ligands. These magnetic dependences are characteristic of the high-spin cobalt(II) pseudoclathrochelates of this type possessing a high magnetic anisotropy [11]. On the other hand, the decisive influence of the electrostatic field due to ferrocene on the increase in a magnetic anisotropy value and, as a consequence, on the effective energy barrier of a magnetization reversal has been earlier observed for the dysprosium ferrocenoylacetonate [26].

Temperature plots of χТ for the complexes 2 (2) and 3 (1) in a magnetic field of 5 kOe.
This is also consistent with the data of X-ray diffraction analysis of iron and nickel(II) tris-pyridine oximate pseudoclathrochelates (Fig. 3). Their single crystals obtained by slow evaporation of solutions in chloroform–heptane and methylene chloride–hexane mixtures contain two solvate molecules of the corresponding chlorine-containing organic solvent per one formula unit of the complex, i.e. are crystal solvates of composition 1·2CHCl3 and 3·2CH2Cl2.

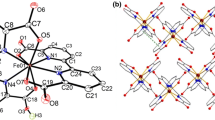
General view of the pseudoclathrochelate cations [Fe(AcPyOx)3(BFc)]+ (M = Fe2+) and [Ni(AcPyOx)3(BFc)]+ (M = Ni2+). Hydrogen atoms are not shown for clarity, and the remaining atoms are shown as thermal vibration ellipsoids (p = 50%).
According to the X-ray diffraction data, the Fe2+ and Ni2+ ions, which are encapsulated by the same pseudomacrobicyclic ligand, are in the different spin states at 120 K, which unambiguously follows from an analysis of the geometric parameters of the corresponding MIIN6 coordination polyhedra (Table 2). The Ni–N distances (2.15–2.17 Å) for the nickel(II) ion which is surrounded by six donor nitrogen atoms of three chelating pyridineoximate synthons in the pseudoclathrochelate cation [Ni(AcPyOx)3(BFc)]+ are characteristic of the high-spin complexes of this ion with nitrogen-containing heterocyclic ligands [27], while in the cation [Fe(AcPyOx)3(BFc)]+ they slightly exceed 2 Å, which indicates the low-spin state of the Fe2+ ion [28–30]. In addition, their MIIN6 coordination polyhedra have a geometry close to intermediate between a trigonal prism (TP, distortion angle φ = 0°) and a trigonal antiprism (TAP, φ = 60°); however, the degree of this distortion depends on the nature of the encapsulated metal ion. Its geometry in the [Fe(AcPyOx)3(BFc)]+ cation is closer to TP, as expected for the low-spin iron(II) ion, while in the [Ni(AcPyOx)3(BFc)]+ cation it is closer to TAP; the corresponding distortion angles φ are 35.3(2)° and 27.3(2)°, respectively (Table 1). For a more reliable description of such TP–TAP distortion, it is convenient to use the so-called “symmetry measures” [31]. The lower the value of the “symmetry measure” corresponding to the chosen ideal polyhedron, such as TP or TAP, the more accurately the coordination polyhedron can be described by this polyhedron. The values of such “symmetry measures” estimated from X-ray diffraction data for complexes 1·2CHCl3 and 3·2CH2Cl2 using the Shape 2.1 program [29] (Table 2) make it possible to describe the geometry of the FeN6 coordination polyhedron in iron(II) pseudoclathrochelate as a distorted TP, and the NiN6 polyhedron in its nickel(II)-containing analog as a distorted TAP.
CONCLUSIONS
Thus, we have obtained the first ferrocenylborate tris-pyridineoximate iron, cobalt, and nickel(II) pseudoclathrochelates and the crystal and molecular structures of iron and nickel(II) complexes of this type were obtained by the single crystal X-ray diffraction experiments. Their spin states and temperature plots of magnetic properties were studied using the magnetometry method.
REFERENCES
Y. Z. Voloshin, I. Belaya, and R. Kramer, Cage Metal Complexes clathrochelates revisited, Springer, 2017.
Y. Z. Voloshin, V. V. Novikov, Y. V. Nelyubina, et al., Chem. Commun. 54, 3436 (2018).
S. Tomyn, S. I. Shylin, D. Bykov, et al., Nat. Commun. 8, 14099 (2017).
S. I. Shylin, M. V. Pavliuk, L. D’Amario, et al., Chem. Commun. 55, 3335 (2019).
S. I. Shylin, M. V. Pavliuk, L. D’Amario, et al., Faraday Discuss. 215, 162 (2019).
S. I. Shylin, J. L. Pogrebetsky, A. O. Husak, et al., Chem. Commun. 57, 11060 (2021).
O. A. Varzatskii and L. V. Penkova, S. V. Kats (Menkach), et al., Inorg. Chem. 53, 3062 (2014).
V. V. Novikov, A. A. Pavlov, Y. V. Nelyubina, et al., J. Am. Chem. Soc. 137, 9792 (2015).
A. A. Pavlov, Y. V. Nelyubina, S. V. Kats, et al., J. Phys. Chem. Lett. 7, 4111 (2016).
S. V. Dudkin, A. S. Belov, Y. V. Nelyubina, et al., New J. Chem. 41, 3251 (2017).
A. A. Pavlov, S. A. Savkina, A. S. Belov, et al., Inorg. Chem. 56, 6943 (2017).
O. A. Varzatskii, S. V. Kats, A. A. Pavlov, et al., Inorg. Chim. Acta 471, 413 (2018).
A. A. Pavlov, S. A. Savkina, A. S. Belov, et al., ACS Omega 3, 4941 (2018).
A. A. Pavlov, D. Y. Aleshin, S. A. Savkina, et al., ChemPhysChem 20, 1001 (2019).
D. Yu. Aleshin, A. A. Pavlov, S. A. Belova, et a., Russ. J. Inorg. Chem. 64, 1532 (2019). https://doi.org/10.1134/S0036023619120027
A. S. Belov, Y. Z. Voloshin, A. A. Pavlov, et al., Inorg. Chem. 59, 5845 (2020).
M. J. Hynes and M. T. O’Shea, Dalton Trans. 2, 331 (1983).
C. B. Aakeröy, A. S. Sinha, K. N. Epa, et al., Chem. Commun. 48, 11289 (2012).
M. Wojdyr, J. Appl. Crystallogr. 43, 1126 (2010).
G. A. Bain and J. F. Berry, J. Chem. Educ. 85, 532 (2008).
G. M. Sheldrick, Acta Crystallogr. Sect. A 64, 112 (2008).
O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, et al., J. Appl. Crystallogr. 42, 339 (2009).
S. V. Kats, O. V. Severinovskaya, O. A. Varzatskii, et al., Macroheterocycles 8, 314 (2015).
A. B. P. Lever, Inorganic Electronic Spectroscopy (Elsevier, Amsterdam, 1984).
S. V. Dudkin, S. A. Savkina, A. S. Belov, et al., Macroheterocycles 11, 418 (2018).
P. S. Koroteev, Z. V. Dobrokhotova, A. B. Ilyukhin, et al., Dalton Trans. 45, 6405 (2016).
H. Ohtsu and K. Tanaka, Inorg. Chem. 43, 3024 (2004).
M. A. Halcrow, Spin-Crossover Materials: Properties and Applications (John Wiley & Sons Ltd., Chichester, United Kingdom, 2013).
A. F. Asachenko, M. A. Topchiy, G. E. Zelinskii, et al., Russ. J. Inorg. Chem. 65, 1494 (2020). https://doi.org/10.1134/S0036023620100022
A. L. Pomadchik, A. S. Belov, E. G. Lebed, et al., Russ. J. Inorg. Chem. 65, 1503 (2020). https://doi.org/10.1134/S0036023620100162
S. Alvarez, Chem. Rev. 115, 13447 (2015).
ACKNOWLEDGMENTS
MALDI-TOF mass spectra were obtained using the equipment of the Center for Collective Use of the Frumkin Institute of Physical Chemistry and Electrochemistry RAS. X-ray diffraction studies were carried out using the scientific equipment of the Center for Research on the Structure of Molecules of the A.N. Nesmeyanov Institute of Organoelement Compounds RAS with the support of the Ministry of Science and Higher Education of the Russian Federation.
Funding
Magnetic measurements for the obtained cage complexes of 3d metals were carried out on the equipment of the Center for Collective Use of the Kurnakov Institute of General and Inorganic Chemistry RAS and were supported by the Russian Science Foundation, grant no. 21-73-20145. Spectral measurements were carried out within the framework of the State Assignment of the Kurnakov Institute of General and Inorganic Chemistry RAS in the field of fundamental scientific research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest.
ADDITIONAL INFORMATION
The article was prepared based on the materials of the XXVIII International Chugaev Conference on Coordination Chemistry, Olginka, Tuapse oblast, Russia, October 3–8, 2021.
Additional information
Translated by V. Avdeeva
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Belova, S.A., Belov, A.S., Efimov, N.N. et al. Synthesis, Structure, and Magnetic Properties of Ditopic Ferrocenylboron-Capped Tris-Pyridineoximate Iron, Cobalt, and Nickel(II) Pseudoclathrochelates. Russ. J. Inorg. Chem. 67, 1151–1157 (2022). https://doi.org/10.1134/S0036023622080034
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0036023622080034