
Abstract
The role of neuronal inflammation developing during the formation of amyloid plaques and Lewy bodies is investigated. The influence of various exogenous and endogenous factors on the development of neuroinflammation is established, but the role of various infectious agents in the development of this process is much less studied. Today, the existence of a universal trigger mechanism of the neurodegenerative process is obvious: a specific pathogen of a bacterial or viral nature (including long-term persistent in nervous tissue in a latent state), reactivating, penetrates into certain cerebral structures, where it is influenced by either Aβ or resident macrophages of the central nervous system, which, in turn, are activated and induce the release of proinflammatory cytokines, leading to the development of neuronal inflammation, autophagy and neurodegeneration. The reactivation of latent infection, such as herpes, in APOE4 carriers significantly increases the risk of development of Alzheimer’s disease. Class-II genes of the HLA locus (HLA II) may be related to the progression of neurodegenerative diseases. An increase in iron levels in the glia is induced by inflammation, which leads to neurodegeneration. Disruption of the homeostasis of redox-active metals, iron and copper, is an integral part of the pathogenesis of Alzheimer’s disease and Parkinson’s disease. The developing neuroinflammation leads to intensification of the processes of peroxidation, oxidation of metals and the development of ferroptosis.
Similar content being viewed by others


INTRODUCTION
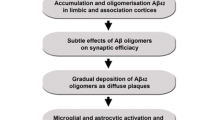
Neurodegenerative diseases are a large group of heterogenic states accompanied by the progressive death of particular groups of neurons and the development of atrophic changes in respective parts of the nervous system, impairing mainly cognitive and motor functions. The key to solving the mystery of neurodegeneration is not so much understanding the genetic patterns of its formation, but rather understanding different stages and triggers of this process. The more than 115-year history of the study of Alzheimer’s disease (AD) has resulted in the appearance of at least 14 hypotheses and theories of disease development, the amyloid hypothesis being the most popular of them. In recent years, medicinal substances targeting the amyloid element in the pathogenesis of Alzheimer’s disease have been actively studied, including those based on human monoclonal antibodies: antibodies against the β-amyloid (Aβ) protein. As a result, the first drug capable of reducing the level of Aβ in the brain of AD patients by destroying amyloid plaques and decreasing the rate of progression of cognitive and functional disorders at the early stages of AD was recorded in the United States in June, 2021 [5]. In spite of the fact that the efficiency of such therapy is yet insufficient for the recovery of patients, it opens up wide prospects for registration and introduction of other drugs with a similar mechanism of action but probably with a greater effectiveness [2]. At the same time, the following question still remains: why were the drugs that seemed to affect the “cause of the disease,” i.e., aggregated amyloid forms, inefficient at the stage of dementia? The answer to this question lies in other, less studied hypotheses of AD, which are probably not independent but represent the stages of a multistage pathological process. One such stage is neuroinflammation that develops during the formation of Aβ deposits, i.e., amyloid plaques [8].
All physicians remember from their student days the signs of inflammation described by Cornelius Celsus: rubor et tumor, cum calore et dolore, which Galenus supplemented with one more important sign: functio laesa. However, rubor, local hyperthermia and microcellular edema in the brain cannot be visualized, and the brain parenchyma has no nociceptors at all. At the same time, neuroinflammation is a peculiar form of inflammatory response: the process of activation of the elements of glial tissue, mainly microgliocytes and astrocytes, in response to various external factors (infectious, traumatic, dysmetabolic). Being central nervous system (CNS) resident macrophages, microgliocytes thereby perform the “phagocyte response of a body to irritant agents,” as I.I. Mechnikov described the process of immune response. It is accompanied by the formation from outgrowth (inactive) microglia of an amoeboid-like (active) microglial phenotype characterized by the predominance of phagocytic activity and the secretion of cytokines.
Neuroinflammation definitely plays a role in the development of neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, and is one of the proven elements of the pathogenesis of multiple sclerosis, schizophrenia and depression [3]. In turn, neuroinflammation can be caused by quite different factors. The role of chronic stress and the induction of inflammation by released cortisol has been shown. A high fat, so-called “western diet” and overnutrition proved to be important for the development of neuroinflammation. Western diet is a broad term defining the modern model of nutrition characteristic of western communities, which is based on ultra-processed, ready-to-eat foods made of refined substances rich in simple carbohydrates (mainly simple sugars), salt, fats [mostly saturated fatty acids (SFA)] and cholesterol. This nutrition model is also depleted of grain and cellular tissue and has low levels of monounsaturated and polyunsaturated fatty acids, including anti-inflammatory omega-3, -6 and -9 acids. Moreover, it has a substantial effect on the function and commensal microbiota of the gut, thereby indirectly resulting in the malabsorption of nutrients and vitamins from food [42].
Numerous studies have shown that such nutrition has a destructive effect on general health, causing hypercholesterolemia, obesity, nonalcoholic fatty liver disease, hyperglycemia, insulin resistance and elevated arterial blood pressure (AP), generally maintaining the metabolic syndrome. These conditions cause and intensify systemic inflammation characterized by the enhanced activity of peripheral proinflammatory cytokines. Using experimental models it was shown how 14 weeks on a diet with a high content of fats enriched in saturated fatty acids resulted in enhanced phosphorylation of serine residues of the τ protein, which are associated with neurofibrillary pathology in the hippocampus. Diets with a high content of both saturated fatty acids and sucrose accelerated these processes already after 7 days of feeding. Three to four weeks of the “western diet” considerably increased the amyloid and τ levels in the hippocampus of rats. A four-month change in nutrition in the mouse AD model caused a twofold increase in the Aβ-40 and Aβ-42 levels in the hippocampus [66]. Proinflammatory factors cause polarization of the microglia and MDM (proinflammatory monocyte-derived macrophage), while free fatty acids and Aβ activate TLR4 (toll-like receptor 4) on the microglia and MDM, which leads to the loss of their capacity for Aβ phagocytosis. The depolarization of AQP4 (aquaporin-4) on astrocytes impairs Аβ clearance by the glymphatic system. In parallel with the above, free fatty acids and 27-hydroxycholesterol alter the composition of lipid rafts in the neuronal membranes, where the amyloidogenic process occurs, increasing the cleavage of amyloid precursor protein to Aβ. The accumulated Аβ is not removed from the brain to a sufficient extent because of the reduced level of LRP1. Activated glial and immune cells express proinflammatory molecules such as TNF-α, IL-1β, IL-6 and iNOS, increasing neuroinflammation and, together with Аβ accumulation, cause neurodegeneration and AD development [10, 65].
The cause of neuroinflammation, which is much less studied and most promising for research, is the variable effects of different infectious agents on nervous tissue.
Astrocytes, being structural components of the neurovascular unit, facilitate the full functioning of the blood–brain barrier protecting the brain, inter alia, against infectious agents. One of the pathways for the penetration of causative agents into the central nervous system through the blood–brain barrier is largely the same as in HIV (the so-called Trojan-horse mechanism) or via direct damage to epithelial cells. The second pathway is perineural, typical, e.g., of the herpes group viruses with respect to the mechanism of retrograde axonal transport. In this case, the herpes simplex virus-1 (HSV-1) persistent for a long time in the trigeminal ganglion can reactivate and penetrate into the CNS under certain conditions. The periodic activation of HSV-1 with transaxonal movement of newly replicated viruses is usually accompanied by manifestation of the infection as rashes on the skin and on the oral and nasal mucosa. However, transaxonal migration is also possible in the opposite direction: to the CNS structures [38], resulting in invasion of the pathogen into the pons of the brain, the olfactory bulb, the amygdale, the hippocampus, and the optic thalamus. In this case, the pathogen can manifest itself in no way, in spite of latent intrathecal limited inflammation. In another case, the manifested forms of encephalitis develop. In particular, it is well-known that the main area affected in case of herpetic encephalitis is precisely the limbic system, which is probably due to HSV-1 invasion through the olfactory bulb from the olfactory epithelium [49].
Pathogen penetration causes the development of microglial activation and formation of reactive astrogliosis: proliferation and hypertrophy of astrocytes, while the intensity of glial fibrillary acidic protein (GFAP) expression is determined by the duration and intensity of the effect of the damaging factor (Fig. 1). The activation of glial cells results in the release of inflammation mediators: interleukins (IL-1β, IL-6, IL-8), TNF-α, and gamma interferon (IFN-γ). Precisely the same response is determined immunohistochemically in AD, to a greater extent along the periphery of amyloid plaques [56]. At the same time, reactive astrocytes surrounding the amyloid plaques lose their neurotrophic functions and acquire neurotoxic properties due to the production of proinflammatory cytokines (IL-1, IL-6, TNF-α), which has an effect on neurodegeneration in Alzheimer’s disease [53]. Microgliocytes produce reactive nitrogen and oxygen species, while astrocytes become a source of toxic glutamate. The developing glutamate–calcium excitotoxicity causes progressive neurodegeneration.

Heterogeneity of the astroglial response (with respect to GFAP expression) in the area of amyloid plaques in brain tissue (arrows) in a person who died of Alzheimer’s disease. Immunohistochemical method, DAB; magnification 400 [9].
At present, it has been convincingly shown that the level of GFAP in blood plasma, which reflects the intensity of reactive astrogliosis, significantly increases even before the first symptoms of AD appear. The blood level of GFAP is much higher in people without cognitive disorders but with preclinical Alzheimer-type pathological changes in the form of Аβ accumulation, in contrast to people with no symptoms or Аβ accumulation (p < 0.01). In addition, the plasma concentrations of GFAP correspond to the severity of the disease, progressively increasing at the preclinical stage (the stage of moderate cognitive disorders) and reaching the maximum values in patients with dementia [17].
In view of the similarity of responses to a latent viral infection and accumulating amyloid protein in AD, one more question arises: is the formation of Аβ aggregates a response to pathogen invasion into the CNS? In reality, Аβ can be considered as a protein with physiological immune properties, which has an antiviral and antimicrobial activity at certain concentrations [19]. Аβ is the most ancient protein of living organisms: its age is more than 400 million years, and it is found in 70% of vertebrates [41]. There are a lot of data in favor of the fact that Аβ exhibits protective properties against infectious agents [29], contributes to the recovery of neural plasticity after brain injury, is involved in the regulation of synaptic transmission [50] and facilitates functioning of the blood–brain barrier [14].
A pathomorphological study performed using the autopsy material of more than 1 thousand deceased patients with AD and progressive supranuclear palsy and persons without apparent neurodegenerative pathology showed the high levels of viral DNA and RNA of herpes viruses (HHV-6, HHV-7 and, to a lesser extent, HSV-1) only in the group of deceased AD patients. It is no less important that the viral load calculated for HHV-6 correlated with the severity of dementia, the number of neurons and the density of amyloid plaques in the cerebral cortex. Experimental studies demonstrated a probable relationship between HSV-1 and the formation of amyloid plaques. The point is that the amyloid protein has a sequence similar to a certain extent to that of HSV-1 glycoprotein B. There is a hypothesis that the viral protein is a matrix for Аβ transcription. The studies by M.A. Wozniak et al. showed Аβ deposition in the brain of HSV-1 infected mice, with high titers of HSV-1 in the amyloid plaques [68]. In addition, HSV-1 promotes the induction of neural excitotoxicity and the activation of intracellular calcium signaling, potentiating also the intracellular accumulation of Аβ and hyperphosphorylation of the τ protein [72]. And though the mechanisms of amyloidogenesis in herpes-virus infection are not completely known, it is supposed that HSP-1 can induce accumulation of the amyloid protein. W. A. Eimer’s study has shown that Аβ oligomers inhibit HSP-1 replication in vitro and prevent the development of acute viral encephalitis in transgenic mice [25].
Serological studies confirm the importance of enteroviruses and herpes-group viruses for the manifestation of amyotrophic lateral sclerosis, in the case of which fatal damage to motor neurons is observed. The flavivirus (the Japanese encephalitis pathogen) was considered for a long time in the early to mid 20th century (and later the influenza virus) as an etiological factor of PD. The herpes-group viruses, mainly cytomegalovirus, are associated with the development of multiple sclerosis. At present, the interrelationship between the direct invasion of an infectious agent and the development of a neurodegenerative disease has not been definitively proven; however, the presented data confirm the hypothesis that all manner of pathogens can trigger a cascade of pathological processes resulting in neurodegeneration.
Considerable progress in studying the role of infections as a possible cause of PD was achieved with the advent of the “dual-hit” hypothesis proposed by C.H. Hawkes et al. It is supposed that a pathological process with the deposition of Lewy bodies occurring at the early stages of the disease involves the olfactory bulb and preganglionic parasympathetic fibers of the vagus nerve [30]. Firstly, perineural invasion of the CNS by viruses, e.g., the influenza virus, can contribute to microglia activation, a considerable increase in phosphorylation and the aggregation of alpha-synuclein (α-syn), which results in dopaminergic-neuron degeneration of the substantia nigra 60 days after infection resolution [33]. Secondly, gut dysbiosis is supposed to play a key role [4]. A biopsy taken from the duodenal and small intestinal mucosa of children with norovirus infection shows the accumulation of α‑synuclein (α-syn). The level of the latter correlates with the degree of lymphoid infiltration and is maintained for more than 6 months after clinical reconvalescence [36]. Simultaneously, the impaired removal of excess aggregates of pathological forms of α-syn may be very important for the development of PD; however, the role of any single specific causative agent is doubtful [6]. The subsequent trans-synaptic invasion of the CNS by infectious agents (among them, viruses are considered more often) via n. vagus and n. olfactorius is accompanied by the hyperproduction of inflammatory mediators and subsequent formation of α-syn deposits. The spread of synucleinopathy can partially decrease after vagotomy, and the risk of PD development in persons who underwent vagotomy within a 20-year period of observation is almost two times lower (OR = 0.53; 95% CI 0.28–0.99) [61].
It should be noted that the role of neuroinflammation in the development of PD has been studied to an even greater extent than for AD. Microglial activation was shown in autopsy material as early as in 1988 [47]; the development of radioligands for positron emission tomography (11C-BU99088) provided the possibility of intravital assessment of the activation of astrocytic glia in PD [67]. By analogy, the activation of microglia assessed by positron emission tomography using [11C]PK11195 has also been shown to play a role in prediction of the development of progressive supranuclear palsy. At the same time, the dynamics of structural changes, according to magnetic resonance imaging (MRI) data, does not correspond with the rate of disease development, in contrast to the intensity of progressive neuroinflammation [45].
POSSIBILITY OF SARS-COV-2-INDUCED NEURODEGENERATION
The infectious hypothesis becomes especially relevant in our time: in the epoch of the novel coronavirus (COVID-19) pandemic, when the problem of studying the etiology and pathogenesis of this disease, as well as immediate and long-term outcomes, has become obvious.
The data obtained over the past year demonstrate that the brain can also be a target organ for SARS-CoV-2. This is confirmed by more and more often reported neurological manifestations of COVID-19, from nonspecific symptoms (headache, anosmia and disorder of consciousness) to life-threatening states (disorder of cerebral circulation, acute necrotizing hemorrhagic encephalopathy and encephalitis) [46]. This hypothesis is supported by the fact that a high level of tropism of β-coronaviruses to nervous tissue has been known for already 70 years [21].
Angiotensin-converting enzyme 2 (ACE2) [11], serine protease TMPRSS2 [31], as well as extracellular matrix metalloproteinase inducer (EMMPRIN) CD147 [22], which are the “site of entry” for the pathogen, are also present in the CNS under physiological conditions [15, 16]. ACE2 is expressed by astrocytes, neurons and endotheliocytes, making the brain susceptible to COVID-19 [28].
By analogy with the viral infectious agents described above (in particular, HSV-1) and other coronaviruses, the SARS-CoV-2 neuroinvasion occurs according to several pathways: in a retrograde manner through nerves (olfactory, vagus), in a transsynaptic manner through infected neurons, in a transendothelial manner via the damaged endothelium of cerebral vessels, or by the migration of leukocytes through the blood–brain barrier [34, 51]. The glymphatic system of the brain, where glial cells play an important role in communication between the blood and the nervous system, can also promote invasion of the brain by the virus [55].
The experimental models of J. Netland et al. with transgenic mice showed that SARS-CoV-2 penetrated the brain through the epithelium of the nasal cavity, spreading further via brain tissue: at the initial stage (the first 4 days), the virus was found in the piriform cortex, the basal ganglia, the mesencephalon and the hypothalamus (i.e., the areas that are in some way related to the olfactory system); later it affected the substantia nigra, the amygdaloid body, the hippocampus and the cerebellum. This process caused the development of marked neurological disorders (often resulting in death of animals), which in the first 24 h were associated with the intensive death of neurons [34]. In subsequent days, the hyperproduction of proinflammatory cytokines in brain tissue was recorded, often causing high lethality [52]. The impaired integrity of the blood–brain barrier resulted in its enhanced permeability to cytokines (IL-1β) and the activation of microglia, GFAP+ astrocytes in the olfactory bulb and the brainstem. 310 patients with PCR+ SARS-Cov-2 having neurological symptoms had higher levels of total τ protein, NfL, GFAP, pTau-181, pTau/Aβ-42 in the acute period. These values correlated with the levels of С-RP in blood plasma, which could be evidence of injury to the blood–brain barrier, rapid development of neuroinflammation, astroglia and neuron impairment. In addition, the moderate and severe courses of the disease were accompanied by elevated NfL in blood plasma, indicating marked neuroaxonal damage after COVID-19 [13].
Although at the moment there are not enough data, it is obvious that neurological manifestations of COVID-19 can be a result of both the direct cytopathic effect of the pathogen and the activation of neuroinflammation accompanied by impaired integrity of the blood–brain barrier (which makes a substantial contribution to penetration of the virus into the CNS and to the maintenance of neuroinflammatory reactions). Neuroglia are directly involved in the inflammatory changes in nervous tissue and (as in the case of already studied neuroinfections) it can be part of a universal trigger mechanism of neurodegenerative process. Further investigation of the molecular and cellular mechanisms of neuroinflammation and neurodegeneration in COVID-19 will be a basis for the development of methods for treating neurological complications.
ROLE OF GENETIC FACTORS IN THE DEVELOPMENT OF INFECTION-INDUCED NEUROINFLAMMATION
The reactivation of HSV-1-associated latent infection in APOE4-carrying AD patients significantly increases the risk of development of this disease. The higher risk of AD is typical of people carrying HSP-1 with the APOE4 genotype. In addition, the anti-HSP-1 antibodies in the cerebrospinal fluid are detected much more often in AD patients. A prospective study with the involvement of more than 3 thousand patients has shown that the detection of anti-HSV IgM doubles the risk of AD development [32]. According to the latest data, the carriage of the pathological isoform APOE4/4 considerably increases the risk of a severe course of COVID-19 and the possibility of pulmonary edema and a cytokine storm, and the necessity of artificial ventilation of the lungs (AVL) increases 2.31-fold [95% CI: 1.65–3.24] [35].
Different polymorphisms of the loci of human leukocyte antigens (HLA) or the major histocompatibility complex (MHC) are associated with the development of quite a number of diseases, including multiple sclerosis, schizophrenia, psoriasis, etc. The emergence and progression of neurodegenerative diseases can be largely related to class-II genes of the HLA locus (HLA II), which are expressed in the CNS mainly by microglial cells.
For example, in patients with multiple sclerosis, high titers of IgG against the Epstein–Barr virus (EBV) were associated with the presence of the DRB1*1501 allele in the HLA II genotype [64]. The study of the mechanisms of interaction between the virus and the HLA II genotype in MS patients has shown a close relationship between the active HHV-6 infection and the expression of MHC class II transactivator (MHC2TA/CIITA). The rarer allele of the MHC2TA gene, rs4774C, was found in patients with multiple sclerosis and HHV-6A reactivation symptoms in almost 50% of cases. HHV-6A-positive patients with the minor allele rs4774C of the MHC2TA gene were predisposed to progressive multiple sclerosis within 2 years to the absence of a response to β-interferon (IFN-β) therapy [12]. In such cases, the increased titers of anti-EBV antibodies in multiple sclerosis patients (compared to healthy carriers of the virus) are evidence of continuous reactivation of EBV, because the virus can disturb the tolerance to myelin antigens by the mechanism of molecular mimicry [58].
Monogenic mutations have been identified in more than 20 genes during PD development. Different variants of mutations in the PARK2, PARK7, PINK1, SNCA, LRRK2 and other genes can lead to the prevalence of ameboid microgliocytes and astrocyte function impairments such as disturbance of the trapping of glutamate and liposomal homeostasis, lysosomal and mitochondrial dysfunction, the release of IL-1β, TNF-α, and nitrogen oxide. In addition, we should take a fresh look at the role of peripheral immune cells, especially T cells, in disease development. The CD8+ and CD4+ T cells were found in postmortem examinations of brain tissue, and the degree of their expression correlated with the accumulation of α-syn [44]. It has been shown that the enhanced T-cell activity against α-syn appears before the motor stage of PD, reaches its maximum at the onset of motor symptoms, and then drastically decreases [40]. Such data, firstly, confirm the possibility of the early immunological diagnosis of PD and, secondly, open up new prospects for immunotherapeutic intervention at the initial stages of the disease.
The pathomorphological comparison of materials from deceased people with AD and elderly persons without AD showed a specific increase in the level of expression of the HLA-DRA gene in AD patients. At the same time, PD proved to be associated with quite a number of single-nucleotide polymorphisms in the HLA II locus [1]. The involvement of the DRB1_15:01 and DRB5_01:01 МНС alleles was highly specific [60].
Thus, all of the above-mentioned pathogens not only potentiate the synthesis of pathological protein isoforms (in this case, Аβ-amyloid, α-syn, τ) but also impair phagocytosis, intensify oxidant stress, impair synaptic neurotransmission and induce apoptosis. However, neuronal inflammation and subsequent apoptosis is certainly induced not only by damaging agents, but also by the state of the immune system, as well as by genetic peculiarities such as the HLA or APOE genotype.
OXIDANT STRESS AND ACCUMULATION OF REDOX-ACTIVE METALS
What can the resultant essence of infection-induced neuroinflammation consist in? One of the mechanisms underlying the pathway of neuronal degeneration due to glial activation is the oxidation of metals and their compounds. Elevated concentrations of Fe2+ and reduced levels of ferritin are detected in the cerebrospinal fluid (CSF) of PD patients with progression of the disease. The Fe2+ concentration in CSF correlates with the levels of α-syn (r = 0.78; p < 0.0001) and phosphorylated τ-protein (r = 0.72; p < 0.001) [43]. The data on the metabolism of iron in microglia are currently insufficient; however, it is known that microglial activation results in increased iron consumption [37]. Neuroinflammation, in turn, results in the activation of glial cells, impairing iron homeostasis.
An increase in the iron concentration in CNS related to physiological aging can be caused by several factors: increased vascular permeability in the brain, inflammation, iron redistribution, and altered iron homeostasis. Aging slows down the work of the system for the maintenance of iron homeostasis described above, which leads to iron accumulation as a result of ineffective chelation [26]. An increase in the level of iron in glia can be induced by inflammation due to the increased release of proinflammatory cytokines, which leads to neurodegeneration.
An age-related increase in the iron concentration occurs in the substantia nigra, the putamen, the globus pallidus, the caudate nucleus, and the cortex [69]. The regional heterogeneity of iron distribution in the brain, as well as its age-related change, has been confirmed in vivo my MRI [7]. Histochemistry demonstrated the presence of ferritin deposits in the microglia and astrocytes of the cerebral cortex, the cerebellum, the hippocampus and basal ganglia and in amygdaloid bodies, and their number usually increases with age. Oligodendrocytes also contain ferritin and transferrin; however, their concentrations remain constant with aging [18]. The subpopulations of ferritin-positive cells of the microglia can be detected in elderly people; most of them are aberrant and have dystrophic changes. The iron phagocyted by this type of cells probably leads to intoxication and causes cell degeneration.
Functionally altered ferritin-positive microglia can be involved in the pathogenesis of AD and PD. Excess iron can cause oxidative stress through the generation of ROS, in particular, the hydroxyl radical [23]. ROS can induce the release of iron from mitochondrial iron–sulfur clusters and other iron-storage proteins, which results in the triggering of Fenton’s reaction. Impaired iron homeostasis can influence mitochondrial functions, leading to the acceleration of mechanisms of neurodegeneration [54]. Moreover, it has been shown in vitro that the aggregation of proteins involved in the pathogenesis of neurodegenerative diseases (α-syn, hyperphosphorylated τ-protein) is caused by an elevated level of iron [24]. Thus, neurodegeneration developing as a result of the toxic effect of iron can lead to apoptosis and ferroptosis: programmed oxidative necrotic cell death with iron-dependent LPO [71]. Figure 2 schematically shows the major molecular mechanisms involved in ferroptosis.

Main mechanisms of ferroptosis. CP is cerruloplasmin; TF is transferrin; TFR is transferrin receptor; DMT-1 is divalent metal transporter-1; O2-R is reactive oxygen species; Fer is ferritin.
An increase in the level of iron with a simultaneous decrease in the level of ferritin has been shown in the substantia nigra in the case of PD, and iron in Lewy bodies is represented by redox ions. It is precisely the level of ferritin that controls the number of redox ions [20]. The level of redox ions of iron correlates with the severity of neuronal death [27]. In spite of a PD-related increase in iron content in the brain structures described above, the enhanced concentration of serum iron is an anti-risk factor of PD, and vice versa [48]. The higher risk of PD in people with a low content of serum iron is probably explained by the need for its adequate supply for the normal synthesis of dopamine, because iron is a cofactor of tyrosine hydroxylase, the key enzyme of dopamine synthesis [63].
The impaired homeostasis of redox-active metals, first of all iron and copper, is probably a component of AD pathogenesis. At present, it has been shown that amyloid plaques and neurofibrillary tangles have high concentrations of zinc, copper and iron. The impaired homeostasis of these metals is involved in the synthesis of Аβ and hyperphosphorylated τ protein and the oxidative stress of neurons [57]. The accumulation of τ protein in neurofibrillary tangles results in the induction of heme oxygenase (HO-1) catalyzing heme destruction. This leads to the further release of iron, which in turn can trigger Fenton’s reaction [59].
PROMISING TRENDS FOR PREVENTING INFECTION-INDUCED NEURODEGENERATION
Undoubtedly, the main objective in the early, preclinical prevention of neurodegenerative diseases is sanitation of the foci of chronic infection. In addition, it would be reasonable to assume that the application of antiviral drugs in persons with manifested forms of HSV-1 infection, or vaccination against it, could prevent the further progress of neurodegeneration.
The analysis of available health-insurance data on the state of health of more than 99.9% of Taiwan residents indicate that AD in elderly and senile people was observed less frequently in persons who have been administered with anti-herpes drugs at some point in their lives [62]. At present, a 78-week randomized study of Valacyclovir administration in patients with prodromal AD is being carried out in the United States; however, its preliminary results will be known no earlier than in mid–late 2022 [46]. The second phase of the clinical trial of a Pleconaril/Ribavirin combination was completed, where 69 AD patients (MMSEcp 23.4 ± 1.8) received this combination for 9 months, followed by an observation period of 12 months. In the group administered with the preparation, the level of Аβ-42 in the cerebrospinal fluid decreased by 70.4 pg/mL (p < 0.03). The findings probably represent a decrease in the synthesis of Аβ as a protein with antiviral activity and a decrease in the viral load [39]. Now a trial of fecal microbiome transplantation is being performed, and its preliminary results will allow us to count on the success of PD therapy [70].
CONCLUSIONS
At present it is obvious that there is a universal trigger mechanism for the neurodegenerative process. A specific bacterial or viral pathogen (including those persisting in nervous tissue in the latent state for a long time), reactivating, penetrates particular cerebral structures where it is exposed to the effects of either Аβ or CNS-resident macrophages. The latter, in turn, are activated and induce the release of proinflammatory cytokines, resulting in the development of neuronal inflammation, autophagy and neurodegeneration. Neuroinflammation intensifies the processes of lipid peroxidation and metal oxidation, which supplements neurodegeneration-related apoptosis with one more mechanism: ferroptosis.
Undoubtedly, we should not consider the infection factor as the only one in the induction of neuronal inflammation, because the roles of other endogenous and exogenous factors in the development of this process are also quite significant. However, together with chronic stress, circadian-rhythm disorders leading to dysfunction of the glymphatic system and the impaired elimination of pathological metabolic products from the cerebral parenchyma, low physical activity, “western diet,” metabolic disorders and diabetes mellitus, against the background of a genetic predisposition, infectious agents play a significant role in the development of neurodegeneration.
REFERENCES
Aliseichik, M.P., Andreeva, T.V., and Rogaev, E.I., Immunogenetic factors of neurodegenerative diseases: role of class HLA II, Biokhimiya, 2018, vol. 83, no. 9, pp. 1385–1398.
Emelin, A.Yu., Litvinenko, I.V., and Lobzin, V.Yu., Erroneous management of patients with Alzheimer’s disease: analysis of problems and pathways of their solution, Nevrol., Neiropsikhiatr., Psikhosom., 2019, vol. 11, no. 4, pp. 141–146.
Emelin, A.Yu., Lobzin, V.Yu., and Vorob’ev, S.V., Kognitivnye narusheniya: Ruk. dlya vrachei (Cognitive Disorders: Manual for Physicians), Moscow, 2019.
Krasakov, I.V., Litvinenko, I.V., Rodionov, G.G., et al., Assessment of intestinal microbiota in patients with Parkinson’s disease using the method of gas chromatography–mass spectrometry, Annaly Klin. Exp. Nevrol., 2018, vol. 12, no. 4, pp. 23–29.
Litvinenko, I.V., Emelin, A.Yu., Lobzin, V.Yu., et al., Amyloid hypothesis of Alzheimer’s disease: past and present, hopes and disappointments, Nevrol., Neiropsikhiatr., Psikhosom., 2019, vol. 11, no. 3, pp. 4–10.
Litvinenko, I.V., Krasakov, I.V., Bisaga, G.N., et al., Modern concept of pathogenesis of neurodegenerative diseases and therapeutic strategy, Zh. Nevrol. Psikhiatr. im. S.S. Korsakova, 2017, vol. 117, no. 6–2, pp. 3–10.
Litvinenko, I.V., Krasakov, I.V., and Trufanov, A.G., Cerebral disorders of iron metabolism as a basis of development and progression of neurodegenerative diseases, Vestn. Ros. VMA, 2018, no. S3, pp. 68–78.
Litvinenko, I.V., Lobzin, V.Yu., and Pushkarev, V.A., Role of infectious agents in the development of neurodegenerative diseases, Izv. Ros. VMA, 2021, vol. 40, no. 4, pp. 25–32.
Lobzin, V.Yu., Vascular-neurodegenerative cognitive disorders (pathogenesis, clinical manifestations, early and differential diagnostics), D. Sci. (Med.) Dissertation, St. Petersburg, 2016.
Lobzin, V.Yu., Kolmakova, K.A., and Emelin, A.Yu., Novel view of pathogenesis of Alzheimer’s disease: modern ideas of amyloid clearance, Obozrenie Psikhiatr. Med. Psikhol., 2018, no. 2, pp. 22–28.
Alenina, N. and Bader, M., ACE2 in brain physiology and pathophysiology: evidence from transgenic animal models, Neurochem. Res., 2019, vol. 44, no. 6, pp. 1323–1329. https://doi.org/10.1007/s11064-018-2679-4
Alvarez-Lafuente, R., De las Heras, V., and Bartolomè, M., Relapsing-remitting multiple sclerosis and human herpesvirus 6 active infection, Arch. Neurol., 2004, vol. 61, no. 10, pp. 1523–1527.https://doi.org/10.1001/archneur.61.10.1523
Ameres, M., Brandstetter, S., Toncheva, A.A., et al., Association of neuronal injury blood marker neurofilament light chain with mild-to-moderate COVID-19, J. Neurol., 2020, vol. 267, no. 12, pp. 3476–3478. https://doi.org/10.1007/s00415-020-10050-y
Atwood, C.S., Bowen, R.L., Smith, M.A., and Perry, G., Cerebrovascular requirement for sealant, anti-coagulant and remodeling molecules that allow for the maintenance of vascular integrity and blood supply, Brain Res. Rev., 2003, vol. 43, no. 1, pp. 164–178. https://doi.org/10.1016/s0165-0173(03)00206-6
Baig, A.M., Khaleeq, A., Ali, U., et al., Evidence of the COVID-19 virus targeting the CNS: tissue distribution, host-virus interaction, and proposed neurotropic mechanisms, ACS Chem. Neurosci., 2020, vol. 11, no. 7, pp. 995–998. https://doi.org/10.1021/acschemneuro.0c00122
Bender, S.J., Phillips, J.M., Scott, E.P., and Weiss, S.R., Murine coronavirus receptors are differentially expressed in the central nervous system and play virus strain-dependent roles in neuronal spread, J. Virol., 2010, vol. 84, no. 21, pp. 11030–11044. https://doi.org/10.1128/jvi.02688-09
Benedet, A.L., Milà-Alomà, M., Vrillon, A., et al., Alzheimer’s and families (ALFA) study, and BioCogBank Paris Lariboisiére cohort. Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum, J.A.M.A. Neurol., 2021, vol. 78, no. 12, pp. 1471–1483. https://doi.org/10.1001/jamaneurol.2021.3671
Block, M.L., Zecca, L., and Hong, J.S., Microglia-mediated neurotoxicity: uncovering the molecular mechanisms, Nat. Rev. Neurosci., 2007, vol. 8, no. 1, pp. 57–69. https://doi.org/10.1038/nrn2038
Bourgade, K., Le Page, A.Y., Bocti, C., et al., Protective effect of amyloid-β peptides against herpes simplex virus-1 infection in a neuronal cell culture model, J. Alzheimers Dis., 2016, vol. 50, no. 4, pp. 1227–1241. https://doi.org/10.3233/JAD-150652
Castellani, R.J., Siedlak, S.L., Perry, G., et al., Sequestration of iron by Lewy bodies in Parkinson’s disease, Acta Neuropathol., 2000, vol. 100, no. 2, pp. 111–114. https://doi.org/10.1007/s004010050001
Cheever, F.S., Daniels, J.B., Pappenheimer, A.M., et al., A murine virus (JHM) causing disseminated encephalomyelitis with extensive destruction of myelin, J. Exp. Med., 1949, vol. 90, no. 3, pp. 181–210. https://doi.org/10.1084/jem.90.3.181
Chen, Z., Mi, L., Xu, J., et al., Function of HAb18G/ CD147 in invasion of host cells by severe acute respiratory syndrome coronavirus, J. Infect. Dis., 2005, vol. 191, no. 5, pp. 755–760. https://doi.org/10.1086/427811
Connor, J.R., Menzies, S.L., St Martin, S.M., et al., Cellular distribution of transferrin, ferritin, and iron in normal and aged human brains, J. Neurosci. Res., 1990, vol. 27, no. 4, pp. 595–611. https://doi.org/10.1002/jnr.490270421
Di Monte, D.A., Schipper, H.M., Hetts, S., et al., Ironmediated bioactivation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in glial cultures, Glia, 1995, vol. 15, no. 2, pp. 203–206. https://doi.org/10.1002/glia.440150213
Eimer, W.A. and Vijaya Kumar, D.K., Navalpur Shanmugam, N.K., et al., Alzheimer’s disease-associated β-amyloid is rapidly seeded by Herpes viridae to protect against brain infection, Neuron, 2018, vol. 99, no. 1, pp. 56–63. https://doi.org/10.1016/j.neuron.2018.06.030
Farrall, A.J. and Wardlaw, J.M., Blood-brain barrier: ageing and microvascular disease—systematic review and meta-analysis, Neurobiol. Aging, 2009, vol. 30, no. 3, pp. 337–352. https://doi.org/10.1016/j.neurobiolaging.2007.07.015
Faucheux, B.A., Martin, M.E., Beaumont, C., et al., Lack of up-regulation of ferritin is associated with sustained iron regulatory protein-1 binding activity in the substantia nigra of patients with Parkinson’s disease, J. Neurochem., 2002, vol. 83, no. 2, pp. 320–330. https://doi.org/10.1046/j.1471-4159.2002.01118.x
Finsterer, J. and Stollberger, C., Update on the neurology of COVID-19, J. Med. Virol., 2020, vol. 92, no. 11, pp. 2316–2318. https://doi.org/10.1002/jmv.26000
Gosztyla, M.L., Brothers, H.M., and Robinson, S.R., Alzheimer’s amyloid-β is an antimicrobial peptide: a review of the evidence, J. Alzheimers Dis., 2018, vol. 62, no. 4, pp. 1495–1506. https://doi.org/10.3233/JAD-171133
Hawkes, C.H., Del Tredici, K., and Braak, H., Parkinson’s disease: a dual-hit hypothesis, Neuropathol. Appl. Neurobiol., 2007, vol. 33, no. 6, pp. 599–614. https://doi.org/10.1111/j.1365-2990.2007.00874.x
Heurich, A., Hofmann-Winkler, H., Gierer, S., et al., TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein, J. Virol., 2014, vol. 88, no. 2, pp. 1293–1307. https://doi.org/10.1128/jvi.02202-13
Itzhaki, R.F., Golde, T.E., Heneka, M.T., et al., Do infections have a role in the pathogenesis of Alzheimer disease?, Nat. Rev. Neurol., 2020, vol. 16, no. 4, pp. 193–197. https://doi.org/10.1038/s41582-020-0323-9
Jang, H., Boltz, D., Sturm-Ramirez, K., et al., Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration, Proc. Nat. Acad. Sci. U.S.A., 2009, vol. 106, no. 33, pp. 14063–14068. https://doi.org/10.1073/pnas.0900096106
Kumar, A., Pareek, V., Prasoon, P., et al., Possible routes of SARS-CoV-2 invasion in brain: in context of neurological symptoms in COVID-19 patients, J. Neurosci. Res., 2020, vol. 98, no. 12, pp. 2376–2383. https://doi.org/10.1002/jnr.24717
Kuo, C.L., Pilling, L.C., Atkins, J.L., et al., APOE e4 genotype predicts severe COVID-19 in the UK biobank community cohort, J. Gerontol. A Biol. Sci. Med. Sci., 2020, vol. 75, no. 11, pp. 2231–2232. https://doi.org/10.1093/gerona/glaa131
Labrie, V. and Brundin, P., Alpha-synuclein to the rescue: immune cell recruitment by alpha-synuclein during gastrointestinal infection, J. Innate Immunol., 2017, vol. 9, no. 5, pp. 437–440. https://doi.org/10.1159/000479653
Lee, P., Peng, H., Gelbart, T., et al., The IL-6- and lipopolysaccharide-induced transcription of hepcidin in HFE-, transferrin receptor 2-, and beta 2-microglobulin-deficient hepatocytes, Proc. Nat. Acad. Sci. U.S.A., 2004, vol. 101, no. 25, pp. 9263–9265. https://doi.org/10.1073/pnas.0403108101
Lewandowski, G., Zimmerman, M.N., Denk, L.L., et al., Herpes simplex type 1 infects and establishes latency in the brain and trigeminal ganglia during primary infection of the lip in cotton rats and mice, Arch. Virol., 2002, vol. 147, pp. 167–179. https://doi.org/10.1007/s705-002-8309-9
Lindblom, N., Lindquist, L., Westman, J., et al., Potential virus involvement in Alzheimer’s disease: results from a phase IIa trial evaluating Apovir, an antiviral drug combination, J. Alzheimers Dis. Rep., 2021, vol. 5, no. 1, pp. 413–431. https://doi.org/10.3233/ADR-210301
Lindestam Arlehamn, C.S., Dhanwani, R., Pham, J., et al., α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease, Nat. Commun., 2020, vol. 11, no. 1, p. 1875. https://doi.org/10.1038/s41467-020-15626-w
Luna, S., Cameron, D.J., and Ethell, D.W., Amyloid-β and APP deficiencies cause severe cerebrovascular defects: important work for an old villain, PLoS One, 2013, vol. 8, no. 9. e75052. https://doi.org/10.1371/journal.pone.0075052
Lustig, R.H., Ultraprocessed food: addictive, toxic, and ready for regulation, Nutrients, 2020, vol. 12, p. 3401. https://doi.org/10.3390/nu12113401
Maass, F., Michalke, B., Willkommen, D., et al., Cerebrospinal fluid iron–ferritin ratio as a potential progression marker for Parkinson’s disease, Mov. Disord., 2021, vol. 36, no. 12, pp. 2967–2969. https://doi.org/10.1002/mds.28790
MacMahon Copas, A.N., McComish, S.F., Fletcher, J.M., et al., The pathogenesis of Parkinson’s disease: a complex interplay between astrocytes, microglia, and T lymphocytes?, Front. Neurol., 2021, vol. 12, p. 666737. https://doi.org/10.3389/fneur.2021.666737
Malpetti, M., Passamonti, L., Jones, P.S., et al., Neuroinflammation predicts disease progression in progressive supranuclear palsy, J. Neurol. Neurosurg. Psychiatr., 2021, vol. 92, pp. 769–775.
Mao, L., Jin, H., Wang, M., et al., Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China, J.A.M.A. Neurol., 2020, vol. 77, no. 6, pp. 683–690. https://doi.org/10.1001/jamaneurol.2020.1127
McGeer, P.L., Itagaki, S., Boyes, B.E., et al., Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains, Neurology, 1988, vol. 38, pp. 1285–1291. https://doi.org/10.1212/WNL.38.8.1285
Miyake, Y., Tanaka, K., Fukushima, W., et al., Dietary intake of metals and risk of Parkinson’s disease: a case-control study in Japan, J. Neurol. Sci., 2011, vol. 306, nos. 1–2, pp. 98–102. https://doi.org/10.1016/j.jns.2011.03.035
Mori, I., Goshima, F., Ito, H., et al., The vomeronasal chemosensory system as a route of neuroinvasion by herpes simplex virus, Virology, 2005, vol. 334, pp. 51–58.
Morley, J.E. and Farr, S.A., The role of amyloid-beta in the regulation of memory, Biochem. Pharmacol., 2014, vol. 88, no. 4, pp. 479–485. https://doi.org/10.1016/j.bcp.2013.12.018
Najjar, S., Najjar, A., Chong, D.J., et al., Central nervous system complications associated with SARS-CoV-2 infection: integrative concepts of pathophysiologyand case reports, J. Neuroinflammat., 2020, vol. 17, no. 1, p. 231. https://doi.org/10.1186/s12974-020-01896-0
Netland, J., Meyerholz, D.K., Moore, S., et al., Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2, J. Virol., 2008, vol. 82, no. 15, pp. 7264–7275. https://doi.org/10.1128/jvi.00737-08
Perez-Nievas, G.B. and Serrano-Pozo, A., Deciphering the astrocyte reaction in Alzheimer’s disease, Front. Aging Neurosci., 2018, vol. 10, p. 114.
Perluigi, M., Coccia, R., and Butterfield, D.A., 4-Hydroxy-2-nonenal, a reactive product of lipid peroxidation, and neurodegenerative diseases: a toxic combination illuminated by redox proteomics studies, Antioxid. Redox. Signal., 2012, vol. 17, no. 11, pp. 1590–1609. https://doi.org/10.1089/ars.2011.4406
Plog, B.A. and Nedergaard, M., The glymphatic system in central nervous system health and disease: past, present, and future, Ann. Rev. Pathol., 2018, vol. 13, no. 1, pp. 379–394. https://doi.org/10.1146/annurev-pathol-051217-111018
Prokop, S., Lee, V.M.Y., and Trojanowski, J.Q., Neuroimmune interactions in Alzheimer’s disease—new frontier with old challenges?, Prog. Molec. Biol. Transl. Sci., 2019, vol. 168, pp. 183–201. https://doi.org/10.1016/bs.pmbts.2019.10.002
Roberts, B.R., Ryan, T.M., Bush, A.I., et al., The role of metallobiology and amyloid-β peptides in Alzheimer’s disease, J. Neurochem., 2012, vol. 120, pp. 149–166. https://doi.org/10.1111/j.1471-4159.2011.07500.x
Salvetti, M., Giovannoni, G., and Aloisi, F., Epstein–Barr virus and multiple sclerosis, Curr. Opin. Neurol., 2009, vol. 22, no. 3, pp. 201–206. https://doi.org/10.1097/WCO.0b013e32832b4c8d
Sayre, L.M., Perry, G., Harris, P.L., et al., In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer’s disease: a central role for bound transition metals, J. Neurochem., 2000, vol. 74, no. 1, pp. 270–279. https://doi.org/10.1046/j.1471-4159.2000.0740270.x
Sulzer, D., Alcalay, R.N., Garretti, F., et al., T cells from patients with Parkinson’s disease recognize alpha-synuclein peptides, Nature, 2017, vol. 546, pp. 656–661. https://doi.org/10.1038/nature22815
Svensson, E., Horváth-Puhó, E., Thomsen, R.W., et al., Vagotomy and subsequent risk of Parkinson’s disease, Ann. Neurol., 2015, vol. 78, no. 4, pp. 522–529. https://doi.org/10.1002/ana.24448
Tzeng, N.S., Chung, C.H., Lin, F.H., et al., Anti-herpetic medications and reduced risk of dementia in patients with herpes simplex virus infections—a nationwide, population-based cohort study in Taiwan, Neurotherapeutics, 2018, vol. 15, no. 2, pp. 417–429. https://doi.org/10.1007/s13311-018-0611-x
Uversky, V.N., Li, J., and Fink, A.L., Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure, J. Biol. Chem., 2001, vol. 276, no. 47, pp. 44284–44296. https://doi.org/10.1074/jbc.M105343200
Waubant, E., Mowry, E.M., and Krupp, L., Common viruses associated with lower pediatric multiple sclerosis risk, Neurology, 2011, vol. 76, no. 23, pp. 1989–1995. https://doi.org/10.1212/WNL.0b013e31821e552a
Więckowska-Gacek, A., Mietelska-Porowska, A., Wydrych, M., et al., Western diet as a trigger of Alzheimer’s disease: from metabolic syndrome and systemic inflammation to neuroinflammation and neurodegeneration, Ageing Res. Rev., 2021, vol. 70, p. 101397. https://doi.org/10.1016/j.arr.2021.101397
Więckowska-Gacek, A., Mietelska-Porowska, A., Chutoránski, D., et al., Western diet induces impairment of liver–brain axis accelerating neuroinflammation and amyloid pathology in Alzheimer’s disease, Front. Aging Neurosci., 2021, vol. 13, p. 654509. https://doi.org/10.3389/fnagi.2021.654509
Wilson, H., Dervenoulas, G., Pagano, G., et al., Imidazoline 2 binding sites reflecting astroglia pathology in Parkinson’s disease: an in vivo 11C-BU99008 PET study, Brain, 2019, vol. 142, pp. 3116–3128. https://doi.org/10.1093/brain/awz260
Wozniak, M.A., Itzhaki, R.F., Shipley, S.J., et al., Herpes simplex virus infection causes cellular-amyloid accumulation and secretase upregulation, Neurosci. Let., 2007, vol. 429, nos. 2–3, pp. 95–100. https://doi.org/10.1016/j.neulet.2007.09.077
Xu, J., Jia, Z., Knutson, M.D., et al., Impaired iron status in aging research, Int. J. Molec. Sci., 2012, vol. 13, no. 2, pp. 2368–2386. https://doi.org/10.3390/ijms13022368
Xue, L.J., Yang, X.Z., Tong, Q., et al., Fecal microbiota transplantation therapy for Parkinson’s disease: a preliminary study, Medicine, 2020, vol. 99. e22035. https://doi.org/10.1097/MD.0000000000022035
Yamamoto, A., Shin, R.W., Hasegawa, K., et al., Iron(III) induces aggregation of hyperphosphorylated tau and its reduction to iron(II) reverses the aggregation: implications in the formation of neurofibrillary tangles of Alzheimer’s disease, J. Neurochem., 2002, vol. 82, no. 5, pp. 1137–1147. https://doi.org/10.1046/j.1471-4159.2002.t01-1-01061.x
Zambrano, A., Solis, L., Salvadores, N., et al., Neuronal cytoskeletal dynamic modification and neurodegeneration induced by infection with herpes simplex virus type 1, J. Alzheimers Dis., 2008, vol. 14, no. 3, pp. 259–269. https://doi.org/10.3233/jad-2008-14301
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest.
Additional information
Translated by E. Makeeva
Rights and permissions
About this article

Cite this article
Litvinenko, I.V., Lobzin, V.Y. On a New Paradigm of the Development of Neurodegenerative Diseases by the Example of Alzheimer’s Disease and Parkinson’s Disease. Adv Gerontol 12, 386–395 (2022). https://doi.org/10.1134/S2079057022040117
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S2079057022040117