
Abstract
Titanates and zirconates of light rare earth elements (REE): REE2TiO5, REE2Ti2O7, REE4Ti9O24, and REE2Zr2O7, are of interest as matrices for isolating the REE actinide fraction of high-level waste from the reprocessing of irradiated nuclear fuel. Data on the incorporation of impurities (Zr, U, Ca) into Nd and La titanates are examined. They display limited isomorphism toward these elements, including by the reaction 2REE3+ ↔ Ca2+ + U4+, which is common for minerals and their synthetic analogues. The reasons for the low solubility of Zr and U in Nd titanates and the role of the crystal chemical factor in the choice of crystalline matrices for the immobilization of the REE actinide fraction are considered.
Similar content being viewed by others

INTRODUCTION
The strategy of two-component nuclear power with slow and fast neutron reactors is implemented in Russia in a closed cycle mode, i.e., with reprocessing of irradiated nuclear fuel (SNF) [1, 2]. As a result, radioactive waste (RW), including high-level radioactive waste (HLW), is generated. The key task of improving the nuclear power safety is to find ways to handle the most dangerous HLW radionuclides—long lived fission products and transuranium actinides. The latter are represented by Pu and minor actinides (MA: Np, Am, and Cm), the amount of which in SNF, as follows from their name, is significantly inferior to the plutonium content (Table 1).
Minor actinides are proposed to be recovered from HLW for transmutation in thermal neutron or fast neutron reactors in homogeneous (Pu, Np) or heterogeneous (Am) modes. Curium is proposed to be stored for 70 to 100 years to decay into plutonium and then utilize in the nuclear fuel production. It has been calculated [1, 2, 4] that in this case, after 300–500 years, the activity of the radionuclides remaining in the waste will be equal to the value of uranium ore (the principle of radiation equivalence); even faster, in 100 years, there will come a radiological (onco) equivalence, taking into account their potential harm to public health [5].
This approach is based on the use of very complex technologies for the separation of transplutonium elements (TPEs), including minor actinides of liquid HLW from spent nuclear fuel reprocessing, Am and Cm separation, and fuel fabrication. Methods for recovery of americium and curium from rare-earth elements close to them in properties, separating Am and Cm, and preparing fuel with Am are at the early stages of development [3, 6–8], far from implementation. The closing of the fuel cycle with fractionation of MA (Am, Cm) by fabrication and irradiation of the fuel and its subsequent processing is expected by 2050 [9], which is 30 years more than previously estimated.
Comparison of harm to human health from radioactive waste and uranium ore is based on the assumption of their dissolution in groundwater. However, there are no grounds for this assumption, since uranium-ore objects are known with an age of many millions of years and reserves of hundreds of thousands of tons of U at a concentration in ores of up to 20 wt %. Reliably established reserves of uranium in the deposits are approaching 6 million tons, its estimated resources are 7.5 million tons, and several million tons have already been recovered from the bowels for use in the nuclear industry. The number of known deposits exceeds 1800, the oldest of which are more than 2 billion years old [10–12]. Uranium ore formation is still taking place due to the concentrating the element in a limited volume of rocks. Therefore, in our opinion, a comparison of the radiation and radiological characteristics of HLW and uranium ores makes no sense, since the solubility of both in groundwater is very low. The use of the same methods for assessing the hazard of elements through the water volume to dissolve them to a safe level showed [13, 14] that Hg, Se, Pb, Cd, As are of a much greater threat to human health at contents in ores from tiny parts to several weight percents. Note that the danger of these elements does not decrease with time unlike that of radionuclides whose content decreases as they decay.
The primary industrial uranium minerals are oxides (pitchblende), phosphate (ningyoite), silicate (coffinite), and titanate (brannerite) of tetravalent uranium. The development of oxide and phosphate ores is carried out by underground leaching using oxidizing agents and aggressive acidic or alkaline solutions. The stability of uranium, thorium, and REE minerals, such as pyrochlore, zirconolite, perovskite, monazite, britholite, etc., is even higher [15, 16]. Their isotope systems are closed for hundreds of millions of years [17], which makes it possible to use minerals for dating the age of ores and rocks. The existence in nature of environments in which there is no migration of radionuclides underlies the method alternative to transmutation for handling long-lived isotopes, including actinides. It consists in introducing them in stable matrices and placing them at depths of several hundred meters in a special repository of highly active materials [13, 18–22]. The reports of the International Atomic Energy Agency and the Nuclear Energy Agency argue for the safe disposal of SNF and solidified HLW [23–25]. Many countries, including Russia, are already implementing programs to select sites for the construction of such SNF and HLW storage facilities [21–29].
Crystalline phases for the immobilization of the REE-actinide fraction. There are various methods for separating HLW into fractions of elements [30–36], mainly for nitric acid wastes from the Purex process. The type of extractant is determined by the composition of the elements to be recovered. For the partitioning of Cs and Sr, CSEX and DDC processes are proposed; of REE and TPE, TRUEX, UREX, TRPO, TODGA, DIAMEX; for recovery of all actinides, GANEX; The UNEX process is proposed for the Cs + Sr + REE + TPE group with its subsequent separation into Cs–Sr and REE–TPE fractions. Some of the techniques have been tested on real active solutions and have a technology readiness level of 6 or higher [35]. The presence of stable phases of actinides and the existence in nature of media in which there is no their migration [13, 18–20], underlie handling the REE-MA fraction by including them in matrices and burials. The localization of radionuclides will be ensured by the engineering barriers of the repository and the protective properties of the host rocks. The main engineering barrier is the matrix, which is highly stable in groundwater [37]. The REE–MA fraction is dominated by light lanthanides (La, Ce, Pr, Nd, Sm), minor actinides (MA = Am, Cm) account for up to 10 wt % (Table 1). Crystal matrices (ceramics) with a high isomorphic capacity and stability in water have been proposed for the isolation of the REE–MA fraction [38–41]. In their synthesis and study of their properties, stable rare-earth simulants are used. Most often, this is Nd [41, 42], primarily because of the close radii of the Nd3+, Am3+, and Cm3+ ions and also because of the predominance of Nd in the hypothetical rare-earth–MA fraction composition.
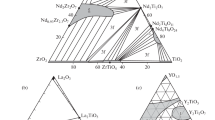
REE compounds (Nd) are potential matrices for minor actinides. Promising actinide matrices are REE titanates and zirconates [38–49]. Among them, much attention was paid to phases with pyrochlore and fluorite structures [38, 39, 44]. Data on REE titanates are scarce and concern mainly the influence of the composition (REE type) on the structure of REE2TiO5 and REE2Ti2O7 phases and their behavior upon ion irradiation [40, 45–49]. In view of the fact that Nd3+ serves as an analog of Am3+ and Cm3+, neodymium compounds are of great interest in the search for REE–MA fraction matrices. According to [49], the NdO1.5–TiO2–ZrO2 system contains the Nd2(Ti,Zr)2O7 phase with the pyrochlore structure (hereinafter NTZ), TiO2 (T, rutile), ZrTiO4 (ZT, srilankite), and tetragonal ZrO2 (Zt). Nd titanates are represented by Nd2TiO5 (NT), Nd2Ti2O7 (NT2), Nd2Ti4O11 (NT4), Nd4Ti9O24 (N2T9) (Fig. 1). The NTi2 and NT4 structures contain blocks of the perovskite type; N2T9 has no natural analogues. Other titanate and zirconate systems with REEs have also been studied: La2O3–TiO2–ZrO2 [50], Y2O3–TiO2–ZrO2 [51, 52], Nd2O3–TiO2 [53], La2O3–TiO2 [54], Nd2O3–ZrO2 [55]. The study of the Nd2O3–TiO2 system [53] proved the similarity of the structures of Nd2Ti4O11 and Nd4Ti9O24 and revealed the Nd2Ti3O9 (NT3) phase.

System NdO1.5–TiO2–ZrO2. (P) Phase field with pyrochlore structure [49].
In the La2O3–TiO2–ZrO2 system, the following phases were identified (Fig. 2a): La2TiO5 (LT), La4Ti3O12 (L2T3), La2Ti2O7 (LT2), La4Ti9О24 (L2T9), La2Zr2О7 (LZ2), ZrTiО4 (ZT), ZrO2 (Z), TiO2 (T). The systems with Nd2O3 and La2O3 are similar in the set of phases, but the phase associations in them are different, mainly due to the smaller pyrochlore region (LZ2). A characteristic feature of REE titanates is weak compositional variations; the Ti : Nd(La) ratios in them are close to the values in the formulas, which is illustrated in the narrow phase fields in the diagrams. At 1350°С, LT2, L2T3, and LT contain less than 2 mol % ZrО2 [50], and the ZrО2 content in L2T9 reaches 4 mol % (Table 2). Pyrochlore La2Zr2O7 (LZ2) contains up to 35 mol % La2O3 and 69 mol % ZrO2 with values in the formula equal to 33 and 67 mol %. The isomorphism of La3+ in ZrO2 and ZrTiO4 is limited to 1 mol % La2O3. High contents of ZrO2 or TiO2 (12–14 mol %) are observed in the oxides (Ti,Zr)O2 and (Zr,Ti)O2, respectively, and the Zr : Ti ratio in the ZrTiO4 compound varies over a wide range, from 1.4 to 0.9.

The replacement of large La3+ and Nd3+ cations by Y3+ leads to noticeable changes in the structure of the diagram (Fig. 2b). The Y2Ti2O7 titanate structure becomes cubic (pyrochlore) and a large (Zr,Y)O2–x field with a fluorite structure appears. TiO2, ZrO2, and ZrTiO4 oxides remain along the TiO2–ZrO2 line; there is a Y2TiO5 phase with a cubic structure (space group Fm3–m), while Nd2TiO5 and La2TiO5 have orthorhombic crystal symmetry (space group Pnma).
The variation in the REE3+ radius in the REE2O3–TiO2 and REE2O3–ZrO2 systems is accompanied by polymorphism—transformation of the phase structure. The REE2Ti2O7 and REE2Zr2O7 compounds can have a structure like fluorite, pyrochlore, and perovskite. The stability regions of pyrochlore correspond to the ratio rA/rB (А = REE3+, B = Ti4+ or Zr4+) from 1.46 to 1.82 [57, 58], rA, rB are ionic radii [59]. At rA/rB > 1.82, a perovskite-like structure is formed, while at rA/rB < 1.46, an anion-deficient structure is generated, similar to fluorite. The titanates of medium and heavy REE (Ln2Ti2O7, Ln = Sm–Lu, Y) and zirconates of medium and light REE (Ln2Zr2O7, Ln = La–Gd) have the pyrochlore structure. The REE2TiO5 phases, where REE = La–Sm, have an orthorhombic structure, from Er to Lu and for Sc they have cubic one; the titanates of elements from Eu to Ho, as well as Y zirconate, have a hexagonal lattice symmetry [46, 56]. As can be seen, the crystal chemical features of the matrix phases have a significant effect on the incorporation of HLW elements therein.
Crystal chemical features of REE titanates and zirconates. Crystal chemical aspects of potential matrices of radionuclides, including actinides, are the subject of a large number of reports and reviews, for example, [39–44, 60–65]. From structural positions, they consider the features of the isomorphism of radionuclides and their simulants in matrices, the element solubility limits in the phase lattice, and the possibility of their increase. The crystal chemical approach has predictive properties and helps to find the compositions of phases of a specific structural type to include the maximum amount of waste therein. In particular, it was shown [62] that calcium, zirconium, and REE ferrites have the highest capacity with respect to tetravalent actinides among phases with a garnet structure. The preservation of the structure with variations in composition, i.e., wide ranges of solid solution stability, is an important characteristic of matrices. The formation of compounds with higher solubility in water during the synthesis of the target phases will worsen the insulating properties of the matrix. As noted, the main interest in the study of REE titanates and zirconates as potential matrices for the REE–MA fraction is associated with Nd phases. The compositions studied in the NdO1.5–TiO2–ZrO2 system [49] lie mainly along the Nd2Zr2O7–Nd2Ti2O7 line; therefore, the phase fields in Fig. 1 are shown with some degree of conventionality.
We obtained data on the content of impurities in REE titanates and zirconates, their behavior upon irradiation, and interaction with solutions [66–72]. The samples were prepared by cold pressing the oxide blend and sintering it (CPS), as well as by cold crucible induction melting (CCIM) or in glassy carbon crucibles in electric furnaces (EF). The experimental conditions and the results of studying the samples are summarized in Tables 3–5 and Fig. 3.

SEM images of samples: (а) LT2 (1—LnT2, 2—Ln2T9); (b) 3b (1—LnT2; 2—UT2, brannerite; 3— (Ln,U)O2–x; 4— REE titanosilicate); (c) 4 (1—Ln2T9; 2—UT2, brannerite; 3— (Ln,U)O2–x; 4— REE titanosilicate); (d) MPM-2 (1—T, rutile; 2—Ln2T9), (e) NTC-2 (1— pyrochlore, 2— (Ln,U)O2–x); (f) NTU-4 (1—LnT2, 2—Ln2T9, 3— (Ln,U)O2–x). (Black) pores. The mark is (a, d–f) 50, (b) 20, or (c) 10 µm. The phase compositions are given in Tables 3–5.
Let us note the variations in the phase compositions with respect to trivalent REE (Table 4). This is due to the fact that lanthanide titanates (La, Ce, Nd) of the same stoichiometry (LnT, LnT2, Ln2T9) are isostructural. Similar behavior can be expected from MA3+ which are close to them in size [59]. Unlike other REE compounds (pyrochlore, zirconolite, monazite, brannerite, etc.), titanates have low solubility in relation to tetra- (U, Zr) and bivalent (Ca) elements. The ZrO2 concentration in the Ln2T9 phase (sample MPM-2) is 2.0 wt %; in rutile it increases to 8.5 wt %. The uranium content in Ln2T9 was determined as 1.3 wt % (Table 4), while in LnT and LnT2 it is below the detection limit of 0.3–0.5 wt % (Tables 4, 5). The highest contents of U and Zr (10 wt % and more) are detected in pyrochlore (Table 5).
REE titanates and zirconates have several types of structures (Table 6, Figs. 4–7). The coordination numbers (CN) of large light REEs (La–Sm) are 7 (polyhedron: truncated cubic prism), 8 (cube, distorted cube, becapped antiprism), 9 (tricapped trigonal prism). As the REE3+ radius decreases, the role of CN 6 (octahedron) increases. Ti and Zr atoms are usually surrounded by six oxygen atoms forming an octahedron.

Polyhedral structure of (а) Nd2Zr2O7 and (b) Nd2TiO5.

Structure of Nd2Ti2O7: (а) perovskite blocks from TiO6 octahedra. (b) Nd distribution in perovskite blocks, yellow balls center the oxygen nine vertex polyhedron, blue balls are seven vertex polyhedron, purple ones are in the center of oxygen nine vertex polyhedron.

Structure of Nd4Ti9O24: (a) In the [11] direction, (b) 3D framework of TiO6 octahedra, (c) layers of Nd(1) (brown) and Nd(3) (violet) polyhedra.

Structure of Nd2Ti3O9: green, TiO6 octahedra; red, O atoms; white, Nd atoms.
Phase Nd2Zr2O7 is of cubic symmetry (Fig. 4a), space group Fd3–m. There are eight formula units in the unit cell, the Nd polyhedron is a scalenohedron (distorted cube), it is formed with 8 O atoms: six equidistant and two at a greater distance. The Zr cations are surrounded by 6 O atoms located at the vertices of a trigonal antiprism (distorted octahedron). The pyrochlore structure can be described in terms of interpenetrating frameworks of BO6 and A2X octahedra. This structure is derived from the lattice of oxides of the AO2 fluorite type (space group Fm3m). The Nd2TiO5 structure (Fig. 4b) consists of a 3D net of seven-vertex NdO7 connected by edges and of chains of square TiO5 pyramids connected by vertices in the [010] direction. The structure of Nd2Ti2O7 is derived from the structure of perovskite (Fig. 5): TiO6 octahedrons are connected at their vertices and form blocks having 4 octahedron thick (about 12 Å) in the a and b directions, with monocapped NdO7 trigonal prisms located between them. Tricapped NdO9 trigonal prisms occupy the cavities of octahedral blocks. Bicapped NdO8 prisms are present inside and between TiO6 octahedral blocks. The structure of Nd4Ti9O24 is the most complex among REE titanates (Fig. 6). Three types of Nd polyhedra are located in the Ti–O cavities of the framework: Nd(1) polyhedron is a distorted square antiprism, Nd(2) is an octahedron, and Nd(3) is a distorted square prism. Nd(1)O8 polyhedra, connected by edges and vertices, form layers in parallel (110). Nd(3) polyhedra are connected by edges to Nd(1) layers; they form layers containing Nd(2) octahedra. The structure of Nd2Ti3O9 (Fig. 7) is derived from the perovskite structure and contains perovskite layers three octahedra thick, connected at the vertices. Nd1 atoms are located in cuboctahedral voids, while Nd2 and O4 atoms are statistically distributed in interlayer spaces of the crystal lattice.
In all phases under study, the polyhedra of the Zr4+ and Ti4+ cations are represented by octahedra (CN 6), except for Nd2TiO5, in which the Ti4+ CN is 5 and the polyhedron is a shape of a square pyramid. The Nd3+ ions in them, as a rule, are characterized by odd CN values (7, 9). This, apparently, explains the low content of impurity elements (Ca, Zr, U) in them and weak variations in the composition, which are close to ideal formulas. Their maximum content and wide compositional field of the solid solution are characteristic of compounds with the pyrochlore structure. The same can be expected from the Nd2Ti3O9 phase with the perovskite structure, in which the CN of Nd3+ is 12, as is typical of other compounds of similar structure [39, 41, 60, 61, 64], including titanates, but this assumption requires additional experimental testing.
The narrow stability fields of La and Nd titanates (Figs. 1, 2a) contrast with large variations in the pyrochlore, zirconolite, and brannerite composition, which are also considered as matrices for the REE–actinide fraction. Zirconate and titanium–zirconate pyrochlore are characterized by a high structural capacity for actinides in oxidation states from 3+ to 6+ [40, 44, 77, 78]. The Zr4+ position of Nd2Zr2O7 pyrochlore can contain up to 20 at % U, while the Nd3+ position contains from 10 to 20 at % U and Th. The wide ranges of zirconolite and brannerite compositions are due to the peculiarities of their crystal structure [40, 43, 79]. Zirconolite CaZrTi2O7 has monoclinic symmetry (C2/m), Ca and Zr coordination numbers are 8 and 7 (Fig. 8a). Cations of different valence and size can replace Ca, Zr, and Ti. Three main substitution mechanisms are known for REE and actinides. The first of them is heterovalent: Ca2+ + Zr4+ → 2 M13+ (where M1 are lanthanides and actinides). At high concentrations of M1, the zirconolite structure transforms into pyrochlore of composition (M1)2Ti2O7. The second type of heterovalent isomorphism: Ca2+ + Ti4+ → M13+ + M23+ (M1 = Ln, An = Pu, Am, Cm; M2 = Al, Fe). An isovalent substitution of Zr4+ for M34+ cations (M3 = Ce, An) is also possible. In zirconolite, actinides (U, Np, Pu) enter the Ca and Zr positions in an amount of up to ~0.3 atoms in the formula, and when Zr4+ is completely replaced by actinides, pyrochlore CaAn4+Ti2O7 is formed. Brunnerite (ATi2O6) has monoclinic symmetry (space group C2/m). The layers of TiO6 octahedra are connected by vertices and edges, they are parallel to the (001) plane and are held together by UO6 octahedra columns extended along the b axis (Fig. 8b). Positions A are occupied by four-charged cations (Ce, Th, U, Np, Pu). Cations of a higher charge (Np5+, U5+) can also located therein at simultaneous entering di- and trivalent cations, for example, Ca2+ and REE3+, to maintain electrical neutrality of the lattice, according to the isomorphic substitution reaction: REE3+ + U5+ → 2U4+ [43].

Structure of zirconolite CaZrTi2O7 (a: gray—layers of Ti octahedra, blue and green—Ca and Zr atoms) and brannerite UTi2O6 (b: yellow U octahedra and gray Ti octahedra), according to [43].
CONCLUSIONS
Variations in the composition and crystal chemistry of the Nd2O3–TiO2–ZrO2 system phases are considered based on Nd as an analog of the REE-actinide fraction. The Nd2O3–TiO2–ZrO2 system contains Nd2(Zr,Ti)2O7, Nd4Ti9O24, Nd2Ti3O9, Nd2Ti2O7, Nd2TiO5, ZrTiO4, (Zr,Ti)O2, (Ti,Zr)O2 phases. Nd2(Zr,Ti)2O7–x (pyrochlore structure), Nd2Ti3O9, and Nd2Ti2O7 (perovskite), Nd4Ti9O24, and Nd2TiO5 can serve as matrices for the REE–MA fraction. The highest concentrations of Ca, Zr, and U are characteristic of pyrochlore Nd2(Ti,Zr)2O7 (10 wt % and more) and Nd4Ti9O24 (2–3 wt %) with even Nd coordination numbers in the structural polyhedra. The capacity of Nd2TiO5 and Nd2Ti2O7 toward impurities is below the detection limit in a scanning electron microscope (0.3–0.5 wt %), which is related to the features of their structure. In such matrices, at high U and Zr contents, brannerite and zirconolite will appear. These phases are stable in solutions; their appearance will not lead to a decrease in the insulating properties of the matrix.
Changes in the REE3+ size in the REE2O3–TiO2 and REE2O3–ZrO2 systems are accompanied by polymorphism [80, 81]. As the REE3+ radius decreases (from La3+ to Yb3+), the REE2Ti2O7 structure changes from monoclinic perovskite-like to cubic pyrochlore: the boundary passes between Nd and Sm. REE2Zr2O7 phases crystallize in two types of related structures, pyrochlore and fluorite. The first is formed at a significant difference between the REE3+ and Zr4+ radii (rREE : rZr > 1.46). As the difference in cation sizes diminishes, the pyrochlore structure transforms into an anion-deficient fluorite structure (REE,Zr)4O7, the boundary between them passes through Gd3+. Zirconates of light REE of the Ce group crystallize in the pyrochlore structure, while the phases of smaller cations of the Y group have the fluorite structure. For REE–2TiO5 compounds, the situation is more complicated: REE phases from La to Sm have an orthorhombic structure; those from Er to Lu, and Sc titanate have a cubic structure, while REE phases from Eu to Ho and Y have a hexagonal crystal symmetry [46, 48, 56]. As the temperature increases, the boundaries between these phases shift with the expansion of the fields of the cubic and hexagonal phases.
Awareness on the crystal chemical features of the phases (solubility limits of elements) makes it possible to control the phase composition of matrices. Impurities (Zr, residual amounts of Pu, U, Np) can initially be in the composition of the REE–MA fraction [30, 36, 82, 83] or introduced into the batch in the form of inactive additives (CaO, Fe2O3, Al2O3) before the matrix synthesis. Thus, the addition of Ca with Al or Fe promotes the appearance of a phase with the zirconolite structure [66]. There is a connection between the features of the crystal chemistry of compounds and the possibility of their use for the isolation of radioactive waste. Therefore, when searching for optimal materials for the isolation of long lived radionuclides, along with the conditions of corrosion and radiation resistance, it is necessary to take into account the structural features of the phases, as shown in [61–64, etc.].
REFERENCES
Adamov, E.O., Lopatkin, A.V., Murav’ev, E.V., Rachkov, V.I., Khomyakov, Yu.S., Izv. RAN. Energetika, 2019, no. 1, p. 3.
Adamov, E.O., Mochalov, Yu.S., Rachkov, V.I., Khomyakov, Yu.S., Shadrin, A.Yu., Kascheev, V.A., and Khaperskaya, A.V., At. Energy, 2021, vol. 130, no. 1, p. 29.
Spent Nuclear Fuel Reprocessing Flowsheet, Paris: OECD NEA, 2012.
Implications of Partitioning and Transmutation in Radioactive Waste Management, Vienna: IAEA, 2004.
Ivanov, V.K., Chekin, S.Yu., Menyailo, A.N., Maksyutov, M.A., Tumanov, K.A., Kashcheeva, P.V., Lovachev, S.S., Spirin, E.V., and Solomatin, V.M., Radiatsiya i risk, 2019, vol. 28, no. 2, p. 8.
State-of-the-Art Report on Innovative Fuels for Advanced Nuclear Systems, Paris: NEA, 2014.
Skupov, M.V., Glushenkov, A.E., Tarasov, B.A., Abramov, S.V., Kuzin, M.A., Nikitin, O.N., Zabudko, L.M., Grachev, A.F., Zherebtsov, A.A., Mochalov, Yu.S., Nucl. Eng. Des., 2021, vol. 382, 111379.
Kuzin, M.A., Abramov, C.V., Grachev, A.F., Zherebtsov, A.A., Zabud’ko, L.M., Nikitin, O.N., Kuz’min, S.V., Khim. Tekhnologiya, 2021, vol. 22, no. 1, p. 36.
2020 NEA Annual Report, Paris: NEA, 2021.
Uranium 2016: Resources, Production and Demand, Paris: NEA, 2016.
Geological Classification of Uranium Deposits and Description of Selected Examples, Vienna: IAEA, 2018.
World Uranium Geology, Exploration, Resources and Production, Vienna: IAEA, 2020.
Brookins, D.G., Geochemical Aspects of Radioactive Waste Disposal, New York: Springer, 1984.
Plutonium Separation in Nuclear Power Programs. Status, Problems, and Prospects of Civilian Reprocessing Around the World, Princeton, USA: Princeton Univ., 2015.
Omel’yanenko, B.I., Livshits, T.S., Yudintsev, S.V., and Nikonov, B.S., Geologiya rud. Mestorozhdenii, 2007, vol. 49, no. 3, p. 173.
Lumpkin, G.R. and Geisler-Wierwille, T., Comprehensive Nuclear Materials, Konings, R.J.M., Ed., Amsterdam: Elsevier, 2012.
Wu, F.-Y., Yang, Yu., and Mitchell, R.H., Chem. Geol., 2010, vol. 277, p. 178.
Laverov, N.P., Velichkin, V.I., Omel’yanenko, B.I., and Yudintsev, S.V., Geologiya rud. Mestorozhdenii, 2003, vol. 45, no. 1, p. 3.
Laverov, N.P., Velichkin, V.I., Omel’yanenko, B.I., Yudintsev, S.V., Petrov, V.A., Bychkov, A.V., Izolyatsiya otrabotavshikh yadernykh materialov: geologo-geokhimicheskie osnovy (Isolation of Spent Nuclear Materials: Geological and Geochemical Fundamentals), Moscow: IFZ RAN, 2008.
Laverov, N.P., Omel’yanenko, B.I., and Yudintsev, S.V., Russ. J. Gen. Chem., 2011, vol. 8, no. 9, p. 1980.
Geological Repository Systems for Safe Disposal of Spent Nuclear Fuels and Radioactive Waste, Apted, M.J. and Ahn, J., New York: Woodhead Publishing Ser. in Energy, Elsevier, 2017.
Amiard, J.-C., Management of Radioactive Waste, New York: Wiley, 2021.
Scientific and Technical Basis for Geological Disposal of Radioactive Wastes, Vienna: IAEA, 2003.
Disposal of Radioactive Waste no. SSR-5. Specific Safety Requirements, Vienna: IAEA, 2011.
Management and Disposal of High-Level Radioactive Waste: Global Progress and Solutions, Paris: NEA, 2020. 45 p.
Tsebakovskaya, N.S., Utkin, S.S., Kapyrin, I.V., Medyantsev, N.V., and Shamina, A.V., Obzor zarubezhnykh praktik zakhoroneniya OYaT i RAO (Review of Foreign Practices of SNF and RW Disposal), Moscow: Komtekhprint, 2015.
Laverov, N.P., Yudintsev, S.V., Kochkin, B.T., Malkovsky, V.I., Elements, 2016, vol. 12, no. 4, p. 253.
Sorokin, V.T., Pavlov, D.I., Radioaktivnye otkhody, 2018, no. 4 (5), p. 24.
Strategies and Considerations for the Back End of the Fuel Cycle, Paris: NEA, 2021. 67 p.
Kopyrin, A.A., Karelin, A.I., and Karelin, V.A., Tekhnologiya proizvodstva i radiokhimicheskoi pererabotki yadernogo topliva (Technology of Production and Radiochemical Processing of Nuclear Fuel), Moscow: Atomenergoizdat, 2006.
Zilberman B.Ya., Puzikov, E.A., Ryabkov, D.V., Makarychev-Mikhailov, M.N., Shadrin A.Yu., Fedorov, Yu.S., and Simonenko, V.A., At. Energy, 2009, vol. 107, no. 5, p. 333.
Modolo, G., Geist, A., and Miguirditchian, M., Reprocessing and Recycling of Spent Nuclear Fuel, Elsevier, 2015. Ch. 10, p. 245.
Veliscek-Carolan, J., J. Hazard. Mater., 2016, vol. 318, p. 266.
State-of-the-Art Report on the Progress of Nuclear Fuel Cycle Chemistry, Paris: NEA, 2018.
Baron, P., Cornet, S.M., Collins, E.D., DeAngelis, G., Del Cul, G., Fedorov, Yu., Glatz, J.P., Ignatiev, V., Inoue, T., Khaperskaya, A., Kim, I.T., Kormilitsyn, M., Koyama, T., Law, J.D., Lee, H.S., Minato, K., Morita, Y., Uhlíř, J., Warin, D., Taylor, R.J.A, Prog. Nucl. Energy, 2019, vol. 117, 103091.
Mamchich, M.V., Goletsky, N.D., Tkachenko, L.I., Viznyi, A.N., Naumov, A.A., Belova, E.V., Puzikov, E.A., and Zil’berman, B.Ya., Radiochemistry, 2021, vol. 63, no. 4, p. 462
Frankel, G.S., Vienna, J.D., Lian, J., Guo, X., Gin, S., Kim, S.H., Du, J., Ryan, J.V., Wang, J., Windl, W., Taylor, C.D., and Scully, J.R., Chem. Rev., 2021, vol. 121, no. 20, p. 12327
Ewing, R.C., Earth Planet. Sci. Lett., 2005, vol. 229, p. 165.
Stefanovsky, S.V. and Yudintsev, S.V., Russ. Chem. Rev., 2016, vol. 85, p. 962.
Lumpkin, G.R., Experimental and Theoretical Approaches to Actinide Chemistry, Gibson, J.K. and de Jong, W.A., Eds., New York: Wiley, 2018.
Yudintsev, S.V., Radiochemistry, 2021, vol. 63, no. 5, p. 527.
Yudintsev, S.V., Dokl. Earth Sci., 2015, vol. 460, p. 130.
Zhang, Y., Kong, L., Ionescu, M., Gregg, D.J., J. Eur. Ceram. Soc., 2022, vol. 42, pp. 1852.
Ewing, R.C., Weber, W.J., and Lian, J., J. Appl. Phys., 2004, vol. 95, no. 11, p. 5949.
Smith, K.L., Blackford, M.G., Lumpkin, G.R., Zaluzec, N.J., Microsc. Microanal., 2006, vol. 12, p. 1094.
Aughterson, R.D., Lumpkin, G.R., Ionescu, M., de los Reyes, M., Gault, B., Whittle, K.R., Smith, K.L., and Cairney, J.M., J. Nucl. Mater., 2015, vol. 467, p. 683.
Aughterson, R.D., A Thesis Submitted in Fulfilment of the Requirements for the Degree of Doctor of Philosophy, Sydney: Univ. of Sydney, 2018.
Aughterson, R.D., Lumpkin, G.R., Smith, K.L., and Cairney, J.M., J. Am. Ceram. Soc., 2020, vol. 103, p. 5536.
Shoup, S.S., Bamberger, C.E., Tyree, J.L., and Anovitz, L., J. Solid State Chem., 1996, vol. 127, p. 231.
Skapin, S.D., Kolar, D., and Suvorov, D., Solid State Sci., 1999, vol. 1, p. 245.
Schaedler, T.A., Fabrichnaya, O., Levi, C.G., J. Eur. Ceram. Soc., 2008, vol. 28, p. 2509.
He, S., Liu, X., Feng, Q., Chen, G., Zou, X., Wu, Z., Li, C., and Lu, X., Ceram. Int., 2021, vol. 47, p. 23991.
Gong, W. and Zhang, R., J. Alloys Compd., 2013, vol. 548, p. 216.
Gong, W. and Zhang, R., Thermochim. Acta, 2012, vol. 534, p. 28.
Fabrichnaya, O., Savinykh, G., Schreiber, G., and Sei-fert, H.J., J. Phase Equil. Diffus., 2011, vol. 32, no. 4, p. 284.
Petrova, M.A., Novikova, A.S., and Grebenshchikov, R.G., Inorg. Mater., 1982, vol. 18, no. 2, p. 287.
Subramanian, M.A., Aravamudan, G., and Rao, G.S., Prog. Solid State Chem., 1983, vol. 15, p. 55.
Nickolsky, M.S., Acta Crystallogr., Sect. B, 2015, vol. 71, no. 2, p. 235.
Shannon, R.D., Acta Crystallogr., Sect. A, 1976, vol. 32, no. 5, p. 751.
Ringwood, A.E., Kesson, S.E., Ware, N.G., Hibberson, W.O., and Major, A., Geochem. J., 1979, vol. 13, p. 141.
Fielding, P.E. and White, T.J., J. Mater. Res., 1987, vol. 2, p. 387.
Yudintsev, S.V., Geol. Ore Depos., 2003, vol. 45, no. 2, p. 151.
Orlova, A.I., Vopr. Radiats. Bezopasnosti, 2015, no. 3, p. 67.
Orlova, A.I. and Ojovan, M.I., Materials, 2019, vol. 12, 2638.
Blackburn, L.R., Bailey, D.J., Sun, S.-K., Gardner, L.J., Stennett, M.C., and Corkhill, C.L., Adv. Appl. Ceram., 2021, vol. 120, no. 2, p. 69.
Yudintsev, S.V., Stefanovsky, S.V., Kalenova, M.Yu., Nikonov, B.S., Nikol’skii, M.S., Koshcheev, A.M., and Shchepin, A.S., Radiochemistry, 2015, vol. 57, no. 3, p. 321.
Yudintsev, S.V., Stefanovsky, S.V., Nikonov, B.S., Nikol’skii, M.S., and Livshits, T.S., Radiochemistry, 2015, vol. 57, no. 2, p. 187.
Yudintsev, S.V., Livshits, T.S., Zhang, J., and Ewing, R.C., Dokl. Earth Sci., 2015, vol. 461, p. 247.
Yudintsev, S.V., Lizin, A.A., Livshits, T.S., Stefanovsky, S.V., Tomilin, S.V., and Ewing, R.C., J. Mater. Res., 2015, vol. 30, no. 9, p. 1516.
Yudintsev, S.V., Radiochemistry, 2018, vol. 60, no. 3, p. 315.
Yudintsev, S.V., Nikolskii, M.S., Nikonov, B.S., and Malkovskii, V.I., Dokl. Earth Sci., 2018, vol. 480, p. 631.
Yudintsev, S.V., Malkovsky, V.I., Nikolsky, M.S., Nikonov, B.S., Dokl. Earth Sci., 2019, vol. 485, p. 303.
Harvey, E.J., Whittle, K.R., Lumpkin, G.R., Smith, R.I., and Redfern, S.A.T., J. Solid State Chem., 2005, vol. 178, no. 3, p. 800.
Mueller-Buschbaum, H. and Scheunemann, K., J. Inorg. Nucl. Chem., 1973, vol. 35, p. 1091.
Hübner, N. and Gruehn, R., Z. Anorg. Allg. Chem., 1992, vol. 616, no. 10, p. 86.
Richard, M., Brohan, L., and Tournoux, M.J., Solid State Chem., 1994, vol. 112, p. 345.
Wang, L., Li, J., Xie, H., Chen, Q., and Xie, Y., Prog. Nucl. Energy, 2021, vol. 137, 103774.
Sun, J., Zhou, J., Hu, Z., Chan T.-S., Liu, R., Yu, H., Zhang, L., and Wang, J.-Q., J. Synchr. Radiat., 2022, vol. 29, p. 37.
Blackburn, L.R., Bailey, D.J., Sun, S.-K., Gardner, L.J., Stennett, M.C., and Corkhill, C.L., Adv. Appl. Ceram., 2021, vol. 120, no. 2, p. 69.
Yang, K., Lei, P., Yao, T., Gong, B., Wang, Y., Li, M., Wang, J., and Lian, J., Corros. Sci., 2021, vol. 185, 109394.
Yang, K., Wang, Y., Lei, P., Yao, T., Zhao, D., and Lian, J., J. Eur. Ceram. Soc., 2021, vol. 41, p. 6018.
Demine, A.V., Krylova, N.V., Polyektov, P.P., Shestoperov, I.N., Smelova, T.V., Gorn, V.F., and Medvedev, G.M., Mat. Res. Soc. Symp. Proc., 2001, vol. 663, p. 27.
Vandegrift, G.F., Regalbuto, M.C., Aase, S.B., Arafat, H.A., Bakel, A.J., Bowers, D.L., Byrnes, J.P., Clark, M.A., Emery, J.W., Falkenberg, J.R., Lohman, A.V.G., Hafen-richter, D., Leonard, R.A., Pereira, C., Quigley, K.J., Tsai, Y., Vander Pol, M.H., and Laidler, J.J., Proc. WM’04 Conf. Tucson, AZ, Feb. 29–March 4, 2004, WM-4323.
Funding
The work was carried out within the framework of the research topic of the state assignment for the Institute of Geology of Ore Deposits, Petrography, Mineralogy and Geochemistry of the Russian Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare no conflicts of interest.
Additional information
Translated from Radiokhimiya, No. 6, pp. 503–514, December, 2022 https://doi.org/10.31857/S0033831122060016
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yudintsev, S.V., Nickolsky, M.S., Stefanovskaya, O.I. et al. Crystal Chemistry of Titanates and Zirconates of Rare Earths—Possible Matrices for Actinide Isolation. Radiochemistry 64, 667–679 (2022). https://doi.org/10.1134/S1066362222060017
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1066362222060017