


Abstract
The dopamine D2 receptor (D2R) family is upregulated in many cancers and tied to stemness. Reduced cancer risk has been correlated with disorders such as schizophrenia and Parkinson’s disease, in which dopaminergic drugs are used. D2R antagonists are reported to have anticancer efficacy in cell culture and animal models where they have reduced tumor growth, induced autophagy, affected lipid metabolism, and caused apoptosis, among other effects. This has led to several hypotheses, the most prevalent being that D2R ligands may be a novel approach to cancer chemotherapy. This hypothesis is appealing because of the large number of approved and experimental drugs of this class that could be repurposed. We review the current state of the literature and the evidence for and against this hypothesis. When the existing literature is evaluated from a pharmacological context, one of the striking findings is that the concentrations needed for cytotoxic effects of D2R antagonists are orders of magnitude higher than their affinity for this receptor. Although additional definitive studies will provide further clarity, our hypothesis is that targeting D2-like dopamine receptors may only yield useful ligands for cancer chemotherapy in rare cases.
The Concatenation of Cancer and Neuropharmacology
The serendipitous discovery of chlorpromazine (Delay et al., 1952; Delay and Deniker, 1955) over 60 years ago may be considered a landmark in several ways. Besides offering the first effective treatment of some of the symptoms of schizophrenia, it opened new doors to an understanding of the chemoarchitecture of the brain, especially the role of dopamine (Carlsson et al., 1958; Carlsson and Lindqvist, 1963). This led to millions of people being treated with drugs that targeted dopamine receptors. In psychiatry, this complicated a decades-long debate about whether schizophrenia itself affected cancer risk. For a review, see Gulbinat et al. (1992), who noted that pharmacological mechanisms were of particular interest, especially because some phenothiazine-based drugs had antitumor activity in murine leukemia and melanoma, and high concentrations of the antipsychotics or their metabolites were found in the lung (Driscoll et al., 1978). These latter findings might explain a lower occurrence of malignancies sometimes reported in schizophrenics. Conversely, because classic antipsychotics markedly increased serum prolactin resulting from antagonism of inhibitory dopamine receptors on anterior pituitary lactotrophs, this also might explain an increased risk of breast cancer in females (Gulbinat et al., 1992). These early observations led to the hypotheses, first suggested in 1972, that dopamine agonists (then all of the D2 type) might be a potential therapeutic approach in cancer (Csatary, 1972), as will be discussed later.
Dopamine Receptors
Dopamine receptors are members of the heptahelical G protein-coupled receptor (GPCR) superfamily and are divided pharmacologically into two subfamilies (Fig. 1): “D1-like” and “D2-like” (Garau et al., 1978; Kebabian and Calne, 1979). The molecular biology and pharmacology of these receptors have been the subject of numerous reviews and books (Neve and Neve, 1997; Mailman and Huang, 2007). Dopamine receptors are encoded by five genes, with DRD1 and DRD5 encoding the two D1-like receptors (D1 and D5), and DRD2, DRD3, and DRD4 encoding four expressed mammalian proteins (D2long, D2short, D3, and D4). D2long and D2short are splice variants from DRD2 and together are the most highly expressed of the D2-like receptors (Dal Toso et al., 1989; Giros et al., 1989; Monsma et al., 1989b; Chio et al., 1990). As noted earlier, the first drugs that were shown to bind to dopamine receptors (e.g., chlorpromazine) were discovered serendipitously because of effects in controlling positive symptoms of schizophrenia. The target of early antipsychotic drugs was soon identified, then validated, via radioreceptor studies and receptor cloning (Burt et al., 1976; Seeman et al., 1976; Dal Toso et al., 1989; Giros et al., 1989; Monsma et al., 1989a, 1990). When using drugs as research tools, it is imperative to understand the relative effects of a molecule on both primary and secondary targets; antipsychotics in particular have many off-target actions. In addition, although they may have selectivity for one subfamily of dopamine receptor, there is often much less selectivity for an individual member (e.g., D2 vs. D3 vs. D4). Thus, when we discuss clinical findings, reference to “D2” will be a reference to D2-like affinity unless otherwise specified.

Dopamine receptors are G protein-coupled receptors, which are divided into the D1- and D2-like families. Some tissues of interest where these receptors are expressed are included here.
There is a rich literature on both agonist and antagonist effects on dopamine receptors, but it has largely been focused on central nervous system modulation of dopamine function in the context of schizophrenia and other brain disorders (Neve and Neve, 1997). On the periphery, dopamine is known to play an important role in cardiovascular control and kidney function. The notion that dopamine receptor ligands might affect the biology of neoplastic cells independent of their actions on neurotransmission is provocative, and offers both a novel mechanism and the ability to both purpose and repurpose the huge libraries of dopaminergic ligands and drugs that have resulted from neuropharmacological drug discovery and development (Schalop and Allen, 2016). Thus, an examination of this arena is timely.
Clinical Studies of Dopaminergic Drugs and Cancer
Correlative Studies and Case Reports Support a Role for the D2 Receptor in Cancer Development and Treatment Response.
To date, all antipsychotic drugs engage D2 receptors, usually as antagonists (Creese et al., 1976; Mailman, 2007; Boyd and Mailman, 2012), whereas therapy for Parkinson disease (PD) relies primarily on activation of dopamine receptors indirectly via levodopa, or directly by direct agonists (Mailman and Huang, 2007). The accepted targets of current dopamine agonists in PD have been the D2 and D3 receptors. Although some findings suggest a greater role for D1 receptors (Taylor et al., 1991; Mailman et al., 2001), the clinical data of relevance to this topic deals with D2R-targeted therapeutics.
Investigations into the relationship between D2R antagonists and cancer began almost as soon as these drugs were approved for psychiatric indications (Table 1), starting with isolated case reports of increased treatment response from cancer patients treated concurrently with antipsychotics (Osterman, 1961; Csatary, 1972; Eicke, 1973; Hercbergs, 1988). Correlative studies of cancer risk in the context of other diseases strengthened this anecdotal association (Fig. 2), (Fig. 3), Table 2). By the 1980s, population-based correlative studies to determine cancer risk within groups of patients with schizophrenia and PD were underway. Many studies showed clear, significant differences in cancer development, yet methodologies were quite variable, and cohorts often small. Some studies were prospective and followed matched cohorts, whereas others mined national healthcare databases. These differences complicate arriving at a unitary hypothesis.
Timeline of D2 receptor pharmacology and early cancer findings
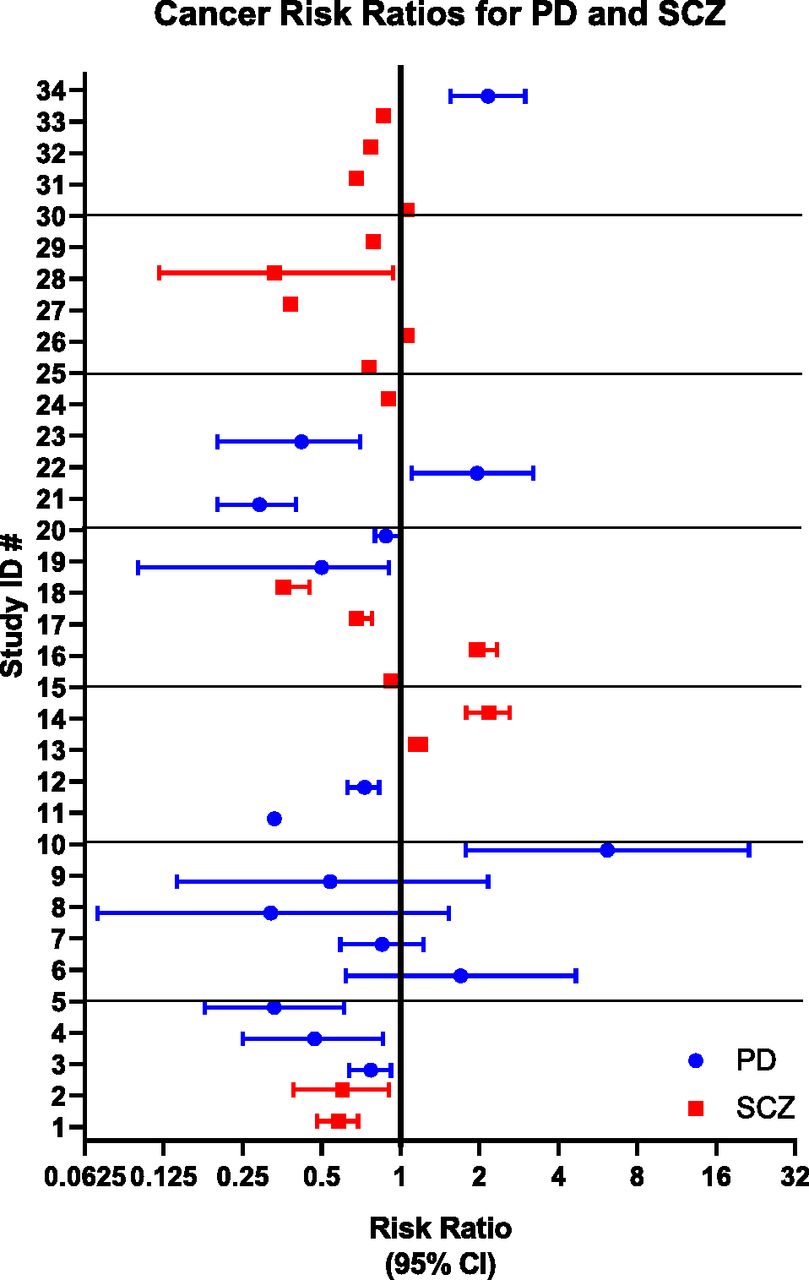
Forest plot of risk ratios from Table 2, by ID number. Bars represent 95% confidence intervals. Studies of PD patients are shown in blue, and studies of SCZ patients are in red.

Treatment with D2 antagonists affects many vital metabolic processes within cancer cells and tumors. Cancer stem cell-like activities, survival signaling, and proliferation are reduced by treatment. However, intracellular calcium levels, autophagy, and apoptosis are increased. Additionally, lipid synthesis and trafficking are disrupted. The direct mechanisms by which these alterations occur is not currently known, but these compounds may ultimately lead to cell death through these or other pathways.
Cancer risk in schizophrenia and Parkinson’s disease patients
Of particular note was a study of more than 100,000 age- and gender-matched, primarily Han Chinese schizophrenia patients in which both male and female subjects showed a strong inverse correlation for age and development of cancers (Wu et al., 2013). One possible explanation for this trend is that older patient populations had undergone long-term treatment with neuroleptic agents that might have attenuated the increased risk inherent in schizophrenics. This study was limited, however, by the lack of ethnic diversity, as well as the lack of stratification for other risk factors, such as smoking status.
The D2 Receptor Is Expressed in a Number of Cancer Cell Lines and in Patient Samples.
D2 receptor expression has been reported at both the mRNA and protein levels in a variety of cancers. Increased immunohistochemical staining has been reported in cervical, esophageal, and lung cancers, often correlating with tumor grade or survival (Li et al., 2006; Hoeppner et al., 2015; Kanakis et al., 2015; Mao et al., 2015; Cherubini et al., 2016). In acute myeloid leukemia (AML), D2R protein is also highly expressed. DRD2 mRNA levels are elevated in breast cancer (Pornour et al., 2014), ovarian cancer (Moreno-Smith et al., 2011), glioma (Li et al., 2014), and neuroblastoma (Deslauriers et al., 2011). Peripheral blood mononuclear cells of breast cancer patients express DRD1, DRD2, DRD3, and DRD4 mRNA (Pornour et al., 2014). Because the D3R and D4R have significant homology to the D2R and often recognize the same drugs, these D2-like receptors may also be relevant.
It is a reasonable hypothesis that tumors derived from cells in which dopamine plays a clear D2-mediated inhibitory role (e.g., from the pituitary, etc.) would be inhibited by D2 agonists. Indeed, the early suggestions to this effect (Csatary, 1972; Jacobs and Franks, 1975) have led to the use of dopamine agonists as one mechanism for controlling such tumors, in which a clear role of dopamine receptors can be demonstrated (Hoeppner et al., 2015), sometimes involving effects on angiogenesis (Chauvet et al., 2017). In clinical, and many of laboratory in vivo, studies of such uses of dopamine agonists, the doses used, after allometric adjustment, are consistent with mediation via the D2 receptor rather than off-target effects. Yet whereas targeting certain types of tumors with dopamine agonists has a sound physiologic rationale, many of the studies ascribing roles for dopamine receptors have important limitations: the use of small numbers of patient samples, lack of blinding, and use of antibodies with poor specificity (Stojanovic et al., 2017). Few studies have ascertained both protein and mRNA levels of the D2R, and no histochemical studies have published replicate data with other probes to verify selectivity. Importantly, reported mRNA levels have typically been quite low, so large fold-changes in mRNA presence may have little functional impact. Thus, although many studies have reported potential anticancer efficacy of dopamine receptor ligands, a large number have failed to show definitive presence of D2R protein or message, especially when the drugs being studied were antagonists. We shall explore these important issues below.
Cancer and the “Non-Neuropharmacology” of Dopamine Receptor Ligands
Some of the earliest indications of anticancer activity for D2R ligands were from Driscoll et al. (1978) and Akiyama et al. (1986). Micromolar concentrations of phenothiazine antipsychotics reversed KB-cell resistance to doxorubicin, vinblastine, dactinomycin, and daunorubicin in a noncalmodulin-dependent manner (Akiyama et al., 1986). In contrast, another study concluded that a reduced proliferative effect of the D2-like antagonists thioridazine and pimozide in Michigan Cancer Foundation (MCF)-7 cells was attributable to calmodulin antagonism (Strobl et al., 1990). Yet, Iishi et al. (1992) soon reported that the D2-like agonist bromocriptine promoted gastric carcinogenesis in a rat model, shortly followed by the suggestion of genetic linkage between the DRD2 gene and BRCA1-sufficient breast cancer (Cortessis et al., 1993). Although these early studies hinted at a potential role for D2R antagonism in cancer development and treatment, there are some issues that should be considered in interpreting these data. In particular, the effects of the four antipsychotic drugs noted above required concentrations two or more orders-of-magnitude higher than their KD (Table 3).
Ligand affinities of select D2 antagonists, agonists, and functionally selective ligands (nM)1
Large-Scale Screens Have Identified D2R as a Potential Target for Anticancer Therapies.
Since 2003, several screening studies identified D2R antagonists as potential therapeutics for cancer treatment on the basis of their biologic activity and/or presence in cancer cells. Like calmodulin inhibitors, phenothiazines selectively increased Forkhead box (FOX)O transcription factor nuclear localization in 786-O renal cell adenocarcinoma cells (Kau et al., 2003), yet FOXO localization remained unchanged when treated with D2R antagonists of different chemotypes (i.e., clozapine and haloperidol) to control for off-target effects. Although this suggests that the D2R is not involved, it contrasts with previous reports noting that D2R agonist treatment increases phospho-Akt levels in neurons, an effect that would be expected to exclude FOXO from the nucleus (Brami-Cherrier et al., 2002; Kihara et al., 2002). Nuclear localization and transcriptional activity of FOXO3 in the human breast cancer BT549 cell line, however, was increased by 5 μM-concentrations of the calcium channel blocker bepridil or the antipsychotic trifluoperazine (Park et al., 2016).
An in silico screening approach suggested thioridazine may inhibit the Akt/ phosphoinositide 3-kinase (PI3K) pathway as well (Rho et al., 2011). Experimentally, thioridazine (20 μM) decreased PI3K pathway activation, inhibited cell cycle progression at G1, reduced cell viability, and induced apoptosis via caspase-3 cleavage over 24 hours of treatment in a manner that was additive with paclitaxel and cisplatin. This suggested that phenothiazines could impact Akt/PI3K signaling in a cell type–specific manner, but target engagement was not verified and may not involve the D2R (Rho et al., 2011). More recently, Gutierrez et al. (2014) did dual screening seeking compounds that were toxic toward zebrafish thymocytes that overexpress MYC and synergized with Notch inhibitors in human T-cell acute lymphoblastic leukemia (T-ALL) cells. They identified several phenothiazines (including perphenazine and chlorpromazine) as potential anti-T-ALL treatments that bound protein phosphatase 2A (PP2A) (Gutierrez et al., 2014).
Two large-scale screens identified the D2R protein itself as a potential target that is upregulated in pancreatic cancer and glioblastoma multiforme. The D2R and its associated G protein Gαi2 were highly upregulated in pancreatic ductal adenocarcinoma tissue samples (Jandaghi et al., 2016). In an short-hairpin (sh)RNA screen to identify genes necessary for glioblastoma (GBM) cell line survival, the D2R was also identified (Li et al., 2014). Inhibition of D2R signaling with shRNA, short-interfering (si)RNA, and several antagonists (i.e., spiperone, haloperidol, risperidone, and L-741,626) reduced cell viability, proliferation, and clonogenicity in U87MG glioblastoma cells. To our knowledge, this was the only study to show that DRD2 knockdown reduces cell viability and tumor growth.
D2R Antagonists Reduce Cell Proliferation and Induce Apoptosis In Vitro.
During the past 20 years, other studies also have identified D2R antagonists as potential anticancer therapeutics through in vitro studies utilizing cell lines and patient samples (Table 4). Phenothiazines, most notably thioridazine, have been suggested as anticancer therapeutics more often than other chemotypes, but haloperidol, pimozide, and olanzapine also have been studied. These compounds have been shown to reduce cell viability, induce apoptosis, cause necrotic cell death, induce cell-cycle arrest, and alter protease activity (Fig. 1). This anticancer activity is apparent in a broad range of cancer types, including gender-specific (Kang et al., 2012; Mao et al., 2015; Park et al., 2016; Ranjan and Srivastava, 2016; Ranjan et al., 2016; Zhou et al., 2016), pancreatic (Ranjan and Srivastava, 2016), nervous system (Gil-Ad et al., 2004; Daley et al., 2005; Levkovitz et al., 2005; Shin et al., 2012, 2013; Li et al., 2014; Karpel-Massler et al., 2015), blood (Zhelev et al., 2004), oral (Choi et al., 2014), lung (Yue et al., 2016), gastric (Mu et al., 2014), and renal (Min et al., 2014) cancers, among others (Levkovitz et al., 2005; Nagel et al., 2012). Typical in vitro cell viability assay IC50 values for D2R antagonists range from 5 to 20 μM, yet D2R antagonists appear to be only modestly selective for cancer cells. Fibroblasts were less sensitive to pimozide treatment than five different pancreatic cancer cell lines, but there was a trivial difference in IC50 (2-fold selectivity, 10 vs. 20 μM) (Jandaghi et al., 2016). Astrocytic cell lines were also less sensitive to haloperidol compared with GBM cells (Li et al., 2014). These concentrations exceed the known maximum tolerated plasma concentrations in humans (Table 5) and suggest a narrow therapeutic window or even dose-limiting toxicity if applied to clinical use. In most cases, cytotoxic concentrations of these compounds are much higher (>100-fold) than would be expected for a D2R-based mechanism, as determined from D2R receptor affinity (Table 3). It is possible that this is owing to differences in receptor environment or functional partners, but it is also important to consider other mechanisms, especially because of the multiple targets that high concentrations of these drugs might engage (Besnard et al., 2012).
D2 ligand IC50 values in cell culture
Tolerated human plasma levels of selected D2 antagonists
In Vivo Models of Cancer Suggest Efficacy of D2R Antagonism.
Animal models of cancer have suggested that D2R antagonists might have chemotherapeutic utility (Table 6). Authors have reported significant reductions in tumor growth with D2R antagonist treatment in gastric, glial, ovarian, medulloblastoma, oral, lung, pancreatic, prostate, and breast cancer xenograft models. Many of these studies observed evidence of Akt-signaling inhibition and/or alterations in autophagic flux in vivo. In an OVCAR-3 murine xenograft model, 10 mg/kg of thioridazine, trifluoperazine, or chlorpromazine reduced tumor growth, but an equivalent dose of fluphenazine was found to be toxic to the animals (Choi et al., 2008), again suggesting a narrow therapeutic window. A dose of 300 μg/day thioridazine or 400 μg/day mepazine reduced tumor size by half in OCI-Ly10 but not in Su-DHL-6 xenograft models (Nagel et al., 2012). These doses led to compound plasma levels of 200 ng/ml, well below the achievable plasma level of 2000 ng/ml in humans.
D2 antagonist efficacy in animal studies
In summary, many animal studies have suggested that D2R antagonists are efficacious in reducing tumor size and prolonging survival in xenograft models. In general, plasma and tumor drug concentrations were not quantified, but they may be expected to be well above selective concentrations. When measured in one study, plasma levels were, however, less than those achievable in human patients (Table 5) (Nagel et al., 2012). Unfortunately, toxicity of some compounds was observed. Therefore, D2 receptor involvement is difficult to ascertain solely on the basis of pharmacological data. Ideally, such findings would be corroborated by studies that employed genetic methods to identify a target. To our knowledge, only one study probed the role of DRD2 in a xenograft model in this way. In this study, a doxycycline-inducible DRD2 knockout in U87MG intracranial xenografts prevented tumor growth in Nu/Nu mice, providing strong support for a role of D2R in cancer growth (Li et al., 2014). Although most of these studies were carried out in the context of immunodeficient mice, it is tempting to speculate on the effects that D2R modulators may have on the immune system through both indirect and direct means (i.e., through psychoactive effects or through direct interaction with immune cells).
D2R Antagonists Are Associated with Anti–Cancer Stem Cell Activity.
D2R expression is also implicated in stem-like cells [cancer stem cells (CSCs)], hypothesized slow-cycling cells that promote tumor growth, chemoresistance, and metastasis. One in silico study using the Connectivity Map identified phenothiazines, notably trifluoperazine, as potential therapeutic agents capable of reversing stem-like gene expression profiles (Yeh et al., 2012). Trifluoperazine concentration-dependently induced apoptosis in a patient-derived, gefitinib-resistant lung cancer cell line, and reduced clonogenicity in a number of other patient-derived lines, regardless of epidermal growth factor receptor (EGFR) status. In a green fluorescent protein (GFP) reporter-based screen for Oct4 and Sox2 in human neoplastic pluripotent stem cells (hnPSCs), thioridazine appeared to target CSCs with an EC50 of 7 μM; prochlorperazine and fluphenazine were also identified, but not further characterized in this work (Sachlos et al., 2012). D2R antagonists, including thioridazine (10 μM), reduced cell number and colony-forming units in AML samples and hPSCs (Sachlos et al., 2012). This work was the first to conclude that D2R activity contributes to the survival and function of CSCs and employed both agonists and antagonists to examine this possibility. In glioblastoma CSCs, similar results were seen for the D2R functionally selective, partial agonist aripiprazole (10 μM) (Suzuki et al., 2016) as well as the D2R antagonists thioridazine and trifluoperazine and the selective D4 antagonists PNU 96415E and L-741,742 (Dolma et al., 2016). Taken together, all of these results suggest that D2R is expressed in CSCs and may impact stemness.
D2R Receptor Signaling Mechanisms and Cancer Cell Growth
STAT and RTK Signaling.
Signal transducer and activator of transcription (STAT) proteins are attractive therapeutic targets because of their role in cellular proliferation and angiogenesis. In a screen for potential STAT5 inhibitors using chronic myelogenous leukemia cell lines, the D2R antagonist pimozide (5–10 μM) decreased STAT5 phosphorylation and function, even downstream of potent oncogenic activation (Nelson et al., 2011). Moreover, pimozide inhibited IL-6-induced growth and migration via inhibition of STAT3 in prostate cancer cells (Zhou et al., 2016). It is unknown if other D2R antagonists inhibit STAT directly, but they reduce proneoplastic receptor tyrosine kinase (RTK) signaling upstream of Janus kinase (JAK)/STAT. The D2R agonists quinpirole (10 μM) and pramipexole (10 μM) both increased phosphorylation of extracellular signal-regulated kinases (ERK) and RTK EGFR in a D2R-dependent manner (Yoon and Baik, 2013). Antagonism with thioridazine reduced vascular EGFR (VEGFR) phosphorylation and VEGF availability (Park et al., 2014). These studies suggest an RTK/JAK/STAT mechanism or downstream effect of D2R antagonists and a possible role for D2R in proneoplastic EGFR signaling.
Wnt.
The wingless/integrated (Wnt) pathway affects development, carcinogenesis, and stem-like behavior, and is reportedly inhibited by D2R antagonists. In a patient-derived lung cancer cell line, trifluoperazine concentration-dependently inhibited T-cell factor–mediated transcription (Yeh et al., 2012), with the decreases in Wnt signaling being concomitant with the induction of cytotoxicity. Such findings are supported by an in silico docking and network analysis study identifying the Wnt pathway protein glycogen synthase kinase (GSK)3β as potentially affected by phenothiazine treatment (Qi and Ding, 2013). Spiperone (10 μM) had similar effects, but these were not mediated by D2R, serotonin, or σ1/2 receptor activity by comparison with selective receptor ligands but may have involved intracellular calcium signaling and protein kinase C (PKC) (Lu and Carson, 2009). Furthermore, D2R and Wnt5a coimmunoprecipitated from HEK293T cells with a KI of 165 nM for competition with [3H]-spiperone, suggesting a possible direct interaction (Yoon et al., 2011). The quinpirole-induced upregulation of Wnt pathway protein Dvl-3 induces ERK activation in mesencephalic neuronal culture but did not occur using cells from D2R−/− mice (Yoon et al., 2011). These data suggest that the D2R may interact with the Wnt pathway in neuronal cells and that D2R antagonists can decrease Wnt signaling, but further studies are needed to see if this is more broadly applicable to the malignant phenotype.
PI3K.
The PI3K/Akt pathway, a critical regulator of the cell cycle, has been suggested as a target pathway for D2R antagonists in cancer-related cell lines. In Chinese hamster ovary (CHO) cells expressing the human D2R, dopamine and quinelorane activated the PI3K pathway by increasing phospho-Akt (at both Ser-473 and Thr-308) and GSK-3β (at Ser-9) levels, with maximal effects at 10 μM (Mannoury la Cour et al., 2011). Pertussis toxin, as well as D2R antagonists, blocked this, suggesting a dependence on D2R G protein signaling. When receptor internalization was blocked with phenylarsine oxide, phosphorylation levels were reduced by half. Likewise, disruption of cholesterol-rich lipid rafts with methyl-β-cyclodextrin inhibited phosphorylation. These latter data suggest that both G-protein and β-arrestin signaling are important. Increased Akt phosphorylation was PKC- and calmodulin-dependent, and GSK-3β phosphorylation was attributable, at least in part, to Akt activity. Thus, there is the potential for these mechanisms to affect cancer cell growth, proliferation, and metabolism via Akt downstream effectors, including transcription factors (like FOXO). In vivo, 25 mg/kg thioridazine given every third day to 2774-xenografted (ovarian cancer) nude mice reduced phosphorylation levels of PI3K, Akt, pyruvate dehydrogenase kinase 1 (PDK1), and mammalian target of rapamycin (mTOR) (Park et al., 2014). In normal rat brain, however, D2R antagonist raclopride (3 mg/kg per day) enhanced phosphorylation at both Thr308 and Ser473 of Akt, which indicates activation, but did not alter total Akt protein levels. In the same model, agonist quinpirole reduced phosphorylation (Sutton and Rushlow, 2012). In normal brain, Akt phosphorylation is reduced by D2-receptor activation in a β-arrestin-2 (βArr2)-mediated manner involving a complex with PP2A (Sotnikova et al., 2005). Antagonism may increase the overall level of Akt phosphorylation or block cell sensitivity to βArr2-mediated Akt regulation (Beaulieu et al., 2004). D2R−/− mouse striatal lysates have increased Akt phosphorylation at Thr-308 both basally and in response to amphetamine (3 mg/kg) challenge (Beaulieu et al., 2007). Overall, it appears PI3K signaling is increased by D2R agonists but reduced by D2R antagonists in malignant tissues, whereas the opposite may be true in normal tissues.
Thioridazine (15 μM) induced apoptosis and inhibited the phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) pathway in endometrial and cervical cancer cell lines (Kang et al., 2012), and at similar concentrations had effects resembling PI3K/Akt inhibition (Rho et al., 2011), decreasing PI3K activity by 60%, inducing G1 arrest after 24-hour treatment, reducing cell viability by half at 48 hours, and inducing apoptosis. Phosphorylation of Akt, mTOR, and GSK-3β were also reduced by several antidopaminergic phenothiazine drugs at low micromolar concentrations in EGF-stimulated OVCAR-3 ovarian cancer cells, although the concentration-response relationship did not parallel D2R affinity (Choi et al., 2008). PI3K activation was unaffected by these phenothiazines.
MAPK/ERK.
The mitogen-activated protein kinase (MAPK)/ERK pathway, known to be involved in cancer cell survival and proliferation, was inhibited in U87MG and A172 glioma cell lines by four different D2R antagonists, albeit at relatively high concentrations (spiperone and haloperidol at 5 μM, risperidone and L-741,626 at 10 μM) (Li et al., 2014). MAPK8 and MAPK10 were also identified as potential targets by a correlational in silico docking and network analysis study of phenothiazines, including chlorpromazine, fluphenazine, and trifluoperazine (Qi and Ding, 2013). This may involve a cascade wherein interaction with peroxisome proliferator-activated receptors γ affects MAPK8 status, leading to a protein kinase-modulated alteration of activity in downstream effectors cyclin-dependent kinase 2 and GSK3β (see section on Wnt signaling). In normal rat and mouse brain slices, the D2 agonist quinpirole (60 μM) increased MAPK and cAMP response element-binding protein phosphorylation, with effects blocked by the D2 antagonist eticlopride (40 μM), the calcium chelator BAPTA-AM, or the PKC antagonist Go6976 (Yan et al., 1999). Although these investigators did not directly assay G protein activity, they hypothesized a role for Gαq activation (Yan et al., 1999), although the D2-like receptors normally are not considered to couple readily to this α-subunit. Owing to the heterogeneous nature of the system and use of healthy tissue, these findings may or may not have any relationship to the behavior of cancer cells exposed to ligands that modulate D2R function.
Calcium Signaling.
D2R signaling and antagonist treatments both alter calcium signaling. Wolfe and Morris (1999) found that both the long and short D2R isoforms interacted with Gαo to reduce high-voltage-activated calcium channel activity. In wild-type astroglia, dopamine signaling is capable of both increasing and reducing intracellular calcium levels in a manner dependent on local neural type in brain slices (Jennings et al., 2017). Dopamine D2/D3 receptors were involved in the negative regulation of Ca2+ in this study.
The calcium channel blocker bepridil and the D2R antagonist triflupromazine had similar effects on PI3K signaling through FOXO3 in MDA-MB-231 breast cancer cells (Park et al., 2016). FOXO3 activity was required to reduce colony formation with both trifluoperazine and bepridil, and FOXO3-regulated proteins D2R, KLF-5, and c-Myc were downregulated by treatment with either drug. In vivo, 10 mg/kg trifluoperazine or bepridil three times a week significantly reduced tumor volume of MDA-MB-231 xenografts in female athymic (nu/nu) mice (Park et al., 2016). A calmodulin mechanism was posited for both compounds but not explored experimentally.
In pancreatic cancer lines MiaPaCa-2 and Panc-1, 10 μM pimozide, or L-741,626 increased intracellular calcium levels sharply within seconds of treatment and concentration-dependently increased phospho–protein kinase R–like endoplasmic reticulum kinase (PERK), suggesting an increase in endoplasmic reticulum stress (Jandaghi et al., 2016). PKA phosphorylation activity was also modestly increased. Caspase activity upon treatment with pimozide was reduced by around 25% when ATF4 was silenced with shRNA, further supporting the involvement of the unfolded protein response. Similar results were found for haloperidol, except IC50 values were increased and fibroblasts seemed even more resistant. Overall, it appears that multiple chemotypes of D2R antagonists can alter intracellular calcium levels and initiate cellular stress in cancer cells.
Autophagy May Be Affected by D2 Antagonists.
Numerous studies have suggested that D2R antagonists are able to induce autophagic cell death in the context of in vitro and in vivo studies of cancer. One trifluoperazine derivative, A4, increased reactive oxygen species (ROS), DNA damage, and autophagic cell death, while also causing apoptosis and activating AMP-activated protein kinase K (AMPK) (Wu et al., 2016). AMPK phosphorylation increases were also seen in D2R antagonist-treated GBM stem cell cultures (Cheng et al., 2015). In SH-SY-5Y neuroblastoma cells, sertindole, pimozide, and trifluoperazine were identified as autophagy-inducing agents by a large-scale fluorescence-based screen (Shin et al., 2012). Increases in GFP-LC3 puncta were sertindole concentration- and time- dependent; autophagosome formation was also verified by electron microscopy. LC3 cleavage was responsive to GFP 3-methyladenine, suggesting autophagic induction was partially regulated by the PI3K pathway. Conditional siRNA knockdown of the essential autophagic protein, ATG5, reduced autophagosome formation, enhanced cell viability, and reduced LC3 cleavage under treatment with 10 μM sertindole. A fluorescence assay that included ROS scavengers indicated a partial role for ROS in the cytotoxicity of sertindole. Similar results have been reported in glioma cell lines (Shin et al., 2013; Cheng et al., 2015). Although autophagy can contribute to D2 antagonist-mediated cell death, D2 activity does not appear to be involved in this mechanism since thioridazine reduced D2R protein levels and increased autophagy, whereas trifluoperazine reduced D2R protein levels and did not increase autophagy at the same concentrations.
Lipid Synthesis and Trafficking Is Altered by D2R Antagonist Treatment.
An early study reported that chlorpromazine (10 μM) inhibited both sphingomyelinase activity and esterification of cholesterol in human fibroblasts in a manner comparable to 10 μM W-7, a known calmodulin antagonist (Masson et al., 1992). Chlorpromazine treatment resulted in accumulation of unesterified cholesterol in lysosomal vacuoles reminiscent of a Niemann-Pick type C (NPC) lipidosis phenotype (Masson et al., 1992). Similar results were seen with 10–50 μM haloperidol, and concomitant insulin receptor signaling inhibition was reversed by cholesterol addback, suggesting lipid raft disruption (Sanchez-Wandelmer et al., 2010).
Other antipsychotics like haloperidol (10 μM) and clozapine (30 μM) increased cholesterol and fatty acid synthesis enzyme mRNA by 2- to 4-fold in GaMg glioma cells at 5–10 hours (Ferno et al., 2006). Sterol regulatory element-binding proteins (SREBP)-1 and SREBP-2, sterol-responsive transcription factors that regulate these genes, were upregulated at the protein level, supporting the idea that antipsychotic treatment may upregulate lipogenesis via SREBP signaling. Cholesterol-related mRNAs, including 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR), apolipoprotein E (APOE), ATP binding cassette subfamily A member 1 (ABCA1), liver X receptors (LXR)α/β, and NPC1/2, were increased after 24- to 48-hour treatment with clozapine (25 μM), haloperidol (10 μM), olanzapine (10 μM), or imipramine in GaMg cells (Vik-Mo et al., 2009). Protein levels of apoE also increased in GaMg and HepG2 human hepatocellular carcinoma cells. Message-level increases were more striking in glial cell cultures, suggesting the activation of LXR and its downstream targets may occur as an effect of earlier SREBP-modulated lipogenesis within the cell (Ferno et al., 2006). Lipogenesis and adequate cholesterol stores are essential for cancer cell survival, particularly in the case of gliomas, which are highly sensitive to exogenous cholesterol levels and LXR activity (Villa et al., 2016).
Although haloperidol and pimozide treatment (10 μM) slightly increased the expression of some SREBP-responsive genes, they also disrupted cholesterol trafficking, causing intracellular accumulation of unesterified cholesterol in intracellular puncta in CHO-7 cells (Kristiana et al., 2010). Despite increases in active SREBP-2, cholesterol synthesis was ablated under treatment with these compounds. Aripiprazole, clozapine, quetiapine (all 10 μM), olanzapine, risperidone, and ziprasidone (25 μM) showed similar behavior, suggesting that the effect may be mediated by D2R or another common target of these compounds. Kristiana et al. (2010) posited that the intracellular trafficking of cholesterol was disrupted by these drugs, inhibiting SREBP cleavage-activating protein (SCAP) activation of SREBP and sterol O-acyltransferase 1 (SOAT-1) esterification of cholesterol. Likewise, 10–50 μM haloperidol reduces biosynthesis of cholesterol in SH-SY-5Y cells while generating a buildup of sterol precursors (Sanchez-Wandelmer et al., 2010). Risperidone, ziprasidone, and clozapine (5–25 μM) also induced buildup of sterol intermediates in HepG2 cells (Canfran-Duque et al., 2013).
Clearly, numerous chemotypes of D2R antagonists can reduce cellular cholesterol levels, disrupt lipid rafts, and alter lipid trafficking. These effects, however, have not been shown to be the cause of D2R antagonist-induced cytotoxicity; it is possible that lipid alterations result from cellular coping mechanisms for other types of stress, such as ROS or autophagic stress. Indeed, these lipid phenotypes indicate that cancer cells treated with these compounds behave as though they are lipid-starved and frustrated in their attempts to synthesize more. One final point should be noted—many (but not all) of the D2-like antagonists used clinically can cause a metabolic syndrome that can include hyperlipidemia (Hirsch et al., 2017; Hoffman, 2017), yet the latter effects require chronic use of the antipsychotics, and the drug concentrations in human tissue are far lower than those causing anticancer effects in vitro or in animals. These factors suggest that different mechanisms probably are involved.
D2R Antagonists May Interact Positively with Other Compounds to Increase Their Anticancer Efficacy.
Studies also indicate that D2R antagonists can be additive with common chemotherapeutics. Aripriprazole sensitized CSC-enriched cultures to gemcitabine, 5-FU, and cisplatin treatment in an additive manner (Suzuki et al., 2016). Likewise, the proapoptotic effects of trifluoperazine were synergistic with cisplatin (10 μM) and gefitinib (2.5–10 μM) in a patient-derived lung cancer cell line (Yeh et al., 2012). Tumor volume and weight of G362 GBM xenografts were decreased in mice treated with 20 mg/kg of either PNU 96415E or L-741,742 over control, though the difference in size was not large (Dolma et al., 2016). L-741,742 treatment on its own failed to improve survival of xenografted mice, but survival increased under cotreatment with temozolomide over treatment with temozolomide alone. Likewise, thioridazine increased the efficacy of AraC in leukemia (Dolma et al., 2016), and cisplatin or paclitaxel in ovarian cancer (Rho et al., 2011). In treatment-resistant endometrial cancer cell lines ISK and KLE, combination treatment with 20 μM medroxyprogesterone acetate and 10 μM thioridazine reduced cell viability by half after 4 days (Meng et al., 2016). Such observations could potentially be explained by inhibition of P-glycoprotein or other efflux pumps associated with drug resistance, as suggested by the fact that thioridazine sensitizes chemoresistant oral squamous cancer cells (KBV20C) to vinblastine owing to inhibited P-glycoprotein efflux (Choi et al., 2014). Likewise, ABCG2-mediated chemoresistance in MDR cells is reduced by 10 μM D3 antagonists PG01037, NGB 2904, SB27 7011A, and U99194 (Hussein et al., 2017). Hussein et al. (2018) later reported that cariprazine (which they termed a D2/D3 partial agonist) had similar effects and suggested it might be repurposed for cancer chemotherapy. The concentrations required, however, were ≥1 μM, much higher than found clinically with maximal doses of the cariprazine, a drug that also has active metabolites that accumulate at even higher levels (Nakamura et al., 2016). These facts suggest that repurposing of the parent molecule might be problematic, and that the reported actions might not be via D2 or D3 receptors.
Critical Interpretation and Future Directions
As the literature currently stands, evidence is suggestive, but by no means conclusive, of an anticancer role for D2R antagonists. Correlative studies of patients with schizophrenia and PD, case studies of cancer patients under concomitant antipsychotic therapy, and repeated hits by unbiased screens support the notion that D2R may have a significant role in cancer development and may be a reasonable therapeutic target. Also, D2R antagonists of varying chemotypes have anticancer activity both in vitro and in vivo, where they induce apoptosis, autophagic cell death, and cell cycle arrest (Fig. 1). In some studies, they also induce CSC differentiation and/or disrupt cholesterol trafficking and synthesis. Such effects are favorable for anticancer therapies, especially since these compounds are modestly selective for cancer cells over normal cell type controls of various lineages.
Yet, although these compounds have effects and can affect many signaling pathways (Figure 3), the role of the D2R itself is still unclear. One major factor is that invariably the concentrations required to induce cytotoxicity are many orders of magnitude higher than the KD for this receptor. At these concentrations, this class of drug has many off-target actions. As approved drugs, there is a great deal of data regarding pharmacokinetics, pharmacodynamics, and toxicity profiles, which when considered in the light of the modest selectivity in cell culture studies, suggests that it may be difficult to achieve circulating plasma levels sufficient for meaningful anticancer activity (Table 5). Maximal circulating levels are reported as concentrations of parent compound, although some of these compounds would also be present as active or inactive metabolites that may or may not have anticancer activities. Many D2R antagonists also have profound side effects that included marked increase in serum prolactin, large increases in body weight and metabolic syndrome, neurologic side effects, and potentially fatal cardiac complications like torsades de pointes that results from QT prolongation. Although some of these are quite serious, they may be tolerable in patients with cancers that are unresponsive to other therapies, especially if the side effects are reversible. The question is whether there is an adequate therapeutic window and an adequate degree of efficacy.
Another issue arising from the high concentrations necessary for anticancer effects is that of target determination; it is far from clear that the D2R is a valid anticancer target on the basis of pharmacological studies alone. Aside from the studies of Li et al. (2014) with GBM, there is little in vitro or in vivo data to suggest that alteration of D2R levels can affect cell growth, viability, or response to D2R antagonist treatment. Studies to determine the role of the D2R will require both understanding of basic principles of pharmacology and the use of orthogonal approaches to decrease the likelihood of erroneous conclusions. Thus, if the D2R is hypothesized to be the target by which an antipsychotic drug kills or inhibits cancer cell growth, then rigorous evidence must be provided to demonstrate that the receptor is both expressed on the cell type of interest and the principal target that needs to be engaged. Ideally a combination of approaches such as receptor binding assays, Western blot, immunosorting analysis, mRNA quantification, molecular ablation, and the like are needed to provide a rigorous test of the underlying hypothesis. Without these types of data, assigning activity to a specific target is risky.
As an example, ONC201, a small-molecule inhibitor of TNF-related apoptosis-inducing ligand (TRAIL), reduced proliferation and viability in HCT116 gastric cancer cells (Allen et al., 2016) with antagonism of the D2R as a major part of its activity. The mechanism was defined further by the suggestion by Leng et al. (2017), who hypothesized a D5R modulation of the D2R by direct experiments using the nanomolar affinity D1/D5 agonist SKF83959 (Lee et al., 2014a) and the nanomolar affinity D2-like agonist cabergoline (Newman-Tancredi et al., 2002). They found that ligand concentrations ≥5 μM or higher were required to cause effects for these drugs. Despite the good selectivity of SKF83959, and to a lesser extent cabergoline, micromolar concentrations will engage many off-target effects as known for SKF83939 (Lee et al., 2014b). On the basis of the pharmacological principles underlying the current analysis, there should be significant skepticism about this proposed D5R/D2R mechanism. Shortly thereafter, it was shown that the cytotoxicity of ONC201 was not eliminated by D2R knockdown or knockout (Kline et al., 2018). Moreover, in a preliminary clinical study against glioblastoma multiformae, ONC201 increased circulating prolactin by only 20% (Arrillaga-Romany et al., 2017), whereas known D2 antagonists cause multifold increases, inconsistent with effects via the D2R. Although they had shown that D2R knockdown or knockout was not the primary mechanism for ONC201 (Kline et al., 2018), these investigators (Prabhu et al., 2019) use associations and correlations of expression data (without any direct assessment of pharmacology or signaling) to elaborate further on the D5R-D2R modulation suggested by Leng et al. (2017). A pharmacological analysis suggests that neither receptor is of primary importance.
In summary, we were attracted to this topic because it seemed like an excellent example of the potential for drug repurposing with a known target (i.e., D2R) for which dozens of drugs are approved, and for which there are probably thousands of experimental compounds that already exist. If the D2R is a viable target, such a wealth of compounds and data would be a very fertile field for study. Yet, our attempt at a critical view of the literature has altered our initial opinion, such that we believe it is probable that the actions of D2R antagonists both in vivo and in vitro will not in most cases involve effects mediated primarily by the D2 receptor. Indeed, novel phenothiazine derivatives have been shown to have many potential anticancer activities aside from the established activities with respect to calmodulin, dopamine receptors, and other known psychiatrically relevant targets. These include antioxidant ability, inhibition of tubulin polymerization, and inhibition of farnesyl transferase (Prinz et al., 2011; Baciu-Atudosie et al., 2012; Engwa et al., 2016; Ghinet et al., 2016). We recognize how our hypothesis, which runs counter to a voluminous literature, could be interpreted, but we believe it would be useful if this generates controversy that leads to hypothesis-driven studies using orthogonal approaches and varying structural series of D2R antagonists. Such rigorous pharmacological evidence could help clarify many of the intrinsic issues. Whether our supposition is correct or not, the field will benefit from a clear resolution of these questions, and the knowledge might impact on the development of new therapeutic paradigms.
Acknowledgments
This work was funded by the Penn State Cancer Institute, the Pritchard Distinguished Graduate Fellowship, and the National Cancer Institute (T32CA060395-21A1). The authors declare no conflicts of interest in this work.
Authorship Contributions
Performed data analysis: Weissenrieder, Mailman.
Wrote or contributed to the writing of the manuscript: Weissenrieder, Mailman, Neighbors, Hohl.
Note Added in Proof—Some references and footnote were accidentally not included in Table 3 in the Fast Forward version published April 18, 2019. Table 3 has now been corrected.
Footnotes
- Received January 25, 2019.
- Accepted April 16, 2019.
This work was supported by Public Health grants, the Penn State Cancer Institute, the Pritchard Distinguished Graduate Fellowship (Penn State College of Medicine, Hershey, PA), and the National Institutes of Health National Cancer Institute [2T32CA060395-21A1].
Abbreviations
- Akt
- protein kinase B
- AML
- acute myeloid leukemia
- AraC
- Cytosine arabinoside
- BAPTA-AM
- 1,2-bis(2-aminophenoxy)ethane-N,N,N,N-tetraacetic acid acetoxymethyl ester
- CHO
- Chinese hamster ovary
- CSCs
- cancer stem cells
- EGFR
- epidermal growth factor receptor
- ERK
- extracellular signal-regulated kinases
- FOX
- Forkhead box
- GFP
- green fluorescent protein
- GBM
- glioblastoma
- 10 GPCR
- G protein-coupled receptor
- Go6976
- 5,6,7,13-Tetrahydro-13-methyl-5-oxo-12H-indolo[2,3-a]pyrrolo[3,4-c]carbazole-12-propanenitrile
- HMGCR
- 3-hydroxy-3-methylglutaryl coenzyme A reductase
- L-741,626
- 3-[4-(4-chlorophenyl)-4-hydroxypiperidinyl]methylindole
- LXR
- liver X receptors
- MAPK
- mitogen-activated protein kinase
- NGB2904
- N-[4-[4-(2,3-dichlorophenyl)-1-piperazinyl]butyl]-9H-fluorene-2-carboxamide
- ONC201
- 7-benzyl-4-(2-methylbenzyl)-1,2,6,7,8,9-hexahydroimidazo[1,2-a]pyrido[3,4-e]pyrimidin-5(4H)-one
- PD
- Parkinson disease
- PG01037
- N-[(2E)-4-[4-(2,3-dichlorophenyl)-1-piperazinyl]-2-buten-1-yl]-4-(2-pyridyl)-benzamide
- PI3K
- phosphoinositide 3-kinase
- PKC
- protein kinase C
- PNU 96415E
- 1-[2-(3,4-dihydro-1H-2-benzopyran-1-yl)ethyl]-4-(4-fluorophenyl)piperazine
- PPAR
- Peroxisome proliferator-activated receptor
- ROS
- reactive oxygen species
- RTK
- receptor tyrosine kinase
- shRNA
- short-hairpin RNA
- siRNA
- short-interfering RNA
- ROS
- reactive oxygen species
- SB-277011A
- N-{trans-4-[2-(6-cyano-3,4-dihydroisoquinolin-2(1H)-yl)ethyl]cyclohexyl}quinoline-4-carboxamide
- SCZ
- Schizophrenia
- SKF-83,959
- 6-Chloro-2,3,4,5-tetrahydro-3-methyl-1-(3-methylphenyl)-1H-3-benzazepine-7,8-diol
- SREBP
- sterol regulatory element-binding proteins
- STAT
- signal transducer and activator of transcription
- TRAIL
- TNF-related apoptosis-inducing ligand
- U99194
- 2,3-dihydro-5,6-dimethoxy-N, N-dipropyl-1H-inden-2-amine
- VEGFR
- vascular EGFR
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics
References
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- The Concatenation of Cancer and Neuropharmacology
- Dopamine Receptors
- Clinical Studies of Dopaminergic Drugs and Cancer
- Cancer and the “Non-Neuropharmacology” of Dopamine Receptor Ligands
- D2R Receptor Signaling Mechanisms and Cancer Cell Growth
- Critical Interpretation and Future Directions
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters