
Molecular packing analyses were carried out on 15 crystal data sets of chloro-substituted Schiff bases, including that of the title compound, C
15H
15ClN
2. C—H

π and π–π interactions play a major role in the molecular self-assembly in the crystal. The former interactions favor molecules assembling into a screw, with a non-centrosymmetric crystal structure. When the molecular dipole is small, π–π interactions favor a parallel, but not usually antiparallel, mode of packing. Weak C—H
X hydrogen bonds (
X = Cl or Br) and
XX interactions seem to be a secondary driving force in packing. The title molecule takes the
trans form and the two benzene rings are twisted around the central linkage in opposite directions. In the crystal structure, molecules interact through C—H
π and π–π interactions, forming a `dimer' and further forming double chains along [001]. The double chains are extended along [10
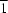
] through C—H
Cl hydrogen bonds, forming double layers in (010). In the third direction, there are only ordinary, weaker, van der Waals interactions, which explains the crystal habit (
i.e. thin plate).
Supporting information
CCDC reference: 251337
To a solution of 4-(N,N-dimethyl)benzaldehyde (10 mmol) in ethanol (10 ml), 4-chlor-aniline (11 mmol) was added. The solution was refluxed for 30 min at 363 K and then cooled to ambient temperature, yielding a pale-yellow product. The product was recrystallized three times from 85% ethanol, and colorless plate-like crystals were obtained from acetone solution by slow evaporation at ambient temperature for a week. Analysis calculated for C15H15N2Cl: C 69.63, H 5.84, N 10.82%; found: C 69.73, H 5.79, N 10.84%. IR (KBr pellets, cm−1): 1649, 1582, 1519, 1420, 765,726. 1H NMR (CDCl3, 399.97 MHz): δ 3.08 [s, 6H, –N(CH3)2], 6.73–6.75 (d, 2H, Ph), 7.15 (s, 2H, Ph), 7.32–7.34 (d, 2H, Ph), 7.76 (s, 2H, Ph), 8.28 (s, 1H, –CH=N–).
H atoms were positioned geometrically and treated as riding, with C—H distances of 0.98 Å (methyl H atoms) and 0.95 Å (other H atoms), and with Uiso(H) values of 1.5Ueq(C) (methyl) and 1.2Ueq(C) (other H atoms).
Data collection: CrystalClear (Rigaku/MSC, 2001); cell refinement: CrystalClear; data reduction: CrystalStructure (Rigaku/MSC & Rigaku Corporation, 2001); program(s) used to solve structure: SHELXS97 (Sheldrick, 1990); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: reference?; software used to prepare material for publication: reference?.
4-chloro-
N-[4-(dimethylamino)benzylidene]aniline
top
Crystal data top
| C15H15ClN2 | F(000) = 544 |
| Mr = 258.74 | Dx = 1.301 Mg m−3 |
| Monoclinic, P21/c | Melting point: 378 K |
| Hall symbol: -P 2ybc | Mo Kα radiation, λ = 0.71070 Å |
| a = 9.852 (6) Å | Cell parameters from 3935 reflections |
| b = 16.268 (9) Å | θ = 3.5–27.5° |
| c = 9.512 (6) Å | µ = 0.27 mm−1 |
| β = 119.904 (6)° | T = 193 K |
| V = 1321.5 (14) Å3 | Thin plate, colorless |
| Z = 4 | 0.70 × 0.70 × 0.30 mm |
Data collection top
Mercury CCD
diffractometer | 2956 independent reflections |
| Radiation source: fine-focus sealed tube | 2773 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.029 |
| Detector resolution: 7.31 pixels mm-1 | θmax = 27.5°, θmin = 3.5° |
| ω scans | h = −12→12 |
Absorption correction: multi-scan
(Jacobson, 1998) | k = −21→17 |
| Tmin = 0.832, Tmax = 0.923 | l = −11→12 |
| 10180 measured reflections | |
Refinement top
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.056 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.143 | H-atom parameters constrained |
| S = 1.16 | w = 1/[σ2(Fo2) + (0.0677P)2 + 0.3993P]
where P = (Fo2 + 2Fc2)/3 |
| 2956 reflections | (Δ/σ)max < 0.001 |
| 166 parameters | Δρmax = 0.22 e Å−3 |
| 0 restraints | Δρmin = −0.23 e Å−3 |
Crystal data top
| C15H15ClN2 | V = 1321.5 (14) Å3 |
| Mr = 258.74 | Z = 4 |
| Monoclinic, P21/c | Mo Kα radiation |
| a = 9.852 (6) Å | µ = 0.27 mm−1 |
| b = 16.268 (9) Å | T = 193 K |
| c = 9.512 (6) Å | 0.70 × 0.70 × 0.30 mm |
| β = 119.904 (6)° | |
Data collection top
Mercury CCD
diffractometer | 2956 independent reflections |
Absorption correction: multi-scan
(Jacobson, 1998) | 2773 reflections with I > 2σ(I) |
| Tmin = 0.832, Tmax = 0.923 | Rint = 0.029 |
| 10180 measured reflections | |
Refinement top
| R[F2 > 2σ(F2)] = 0.056 | 0 restraints |
| wR(F2) = 0.143 | H-atom parameters constrained |
| S = 1.16 | Δρmax = 0.22 e Å−3 |
| 2956 reflections | Δρmin = −0.23 e Å−3 |
| 166 parameters | |
Special details top
Experimental. Elemental analysis (Perkin-Elmer 240 C elemental analyzer) IR (FT—IR spectrometer with KBr pellets, cm−1) 1H NMR (Bruker AV-400 NMR spectrometer, CDCl3 as solvent, 1H (399.97 MHz) NMR, Ambient temperature) |
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Refinement. Refinement using reflections with F2 > 3.0 σ(F2). The weighted R-factor (wR) and goodness of fit (S) are based on F2. R-factor (gt) are based on F. The threshold expression of F2 > 2.0 σ(F2) is used only for calculating R-factor (gt). |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2) top| | x | y | z | Uiso*/Ueq | |
| Cl1 | 0.89087 (6) | 0.37831 (4) | 0.21784 (6) | 0.0580 (2) | |
| N1 | 0.60073 (17) | 0.38891 (8) | 0.63361 (17) | 0.0362 (3) | |
| N2 | 0.18129 (18) | 0.37376 (10) | 0.97868 (17) | 0.0417 (4) | |
| C1 | 0.40283 (18) | 0.35404 (9) | 0.69945 (19) | 0.0340 (4) | |
| C2 | 0.47823 (19) | 0.38991 (10) | 0.8534 (2) | 0.0356 (4) | |
| H2 | 0.5813 | 0.4108 | 0.8951 | 0.043* | |
| C3 | 0.40763 (19) | 0.39577 (10) | 0.94605 (19) | 0.0351 (4) | |
| H3 | 0.4632 | 0.4197 | 1.0509 | 0.042* | |
| C4 | 0.25288 (19) | 0.36664 (9) | 0.88783 (19) | 0.0335 (3) | |
| C5 | 0.17790 (19) | 0.32941 (10) | 0.7333 (2) | 0.0385 (4) | |
| H5 | 0.0748 | 0.3084 | 0.6905 | 0.046* | |
| C6 | 0.2521 (2) | 0.32317 (10) | 0.6435 (2) | 0.0389 (4) | |
| H6 | 0.1993 | 0.2971 | 0.5405 | 0.047* | |
| C7 | 0.47312 (19) | 0.35135 (10) | 0.5968 (2) | 0.0356 (4) | |
| H7 | 0.4226 | 0.3206 | 0.4989 | 0.043* | |
| C8 | 0.66290 (18) | 0.38361 (9) | 0.52806 (19) | 0.0324 (3) | |
| C9 | 0.66878 (19) | 0.31082 (10) | 0.4542 (2) | 0.0382 (4) | |
| H9 | 0.6249 | 0.2620 | 0.4696 | 0.046* | |
| C10 | 0.7380 (2) | 0.30911 (11) | 0.3584 (2) | 0.0416 (4) | |
| H10 | 0.7420 | 0.2593 | 0.3083 | 0.050* | |
| C11 | 0.80115 (19) | 0.38026 (11) | 0.3362 (2) | 0.0389 (4) | |
| C12 | 0.7964 (2) | 0.45362 (10) | 0.4078 (2) | 0.0403 (4) | |
| H12 | 0.8390 | 0.5025 | 0.3906 | 0.048* | |
| C13 | 0.7288 (2) | 0.45433 (10) | 0.5045 (2) | 0.0381 (4) | |
| H13 | 0.7272 | 0.5040 | 0.5561 | 0.046* | |
| C14 | 0.0142 (2) | 0.36234 (16) | 0.9064 (3) | 0.0590 (6) | |
| H14A | −0.0407 | 0.4069 | 0.8292 | 0.089* | |
| H14B | −0.0143 | 0.3629 | 0.9914 | 0.089* | |
| H14C | −0.0157 | 0.3095 | 0.8495 | 0.089* | |
| C15 | 0.2598 (3) | 0.41126 (16) | 1.1376 (2) | 0.0572 (5) | |
| H15A | 0.3671 | 0.3903 | 1.1992 | 0.086* | |
| H15B | 0.2032 | 0.3979 | 1.1951 | 0.086* | |
| H15C | 0.2621 | 0.4710 | 1.1263 | 0.086* | |
Atomic displacement parameters (Å2) top| | U11 | U22 | U33 | U12 | U13 | U23 |
| Cl1 | 0.0501 (3) | 0.0794 (4) | 0.0518 (3) | 0.0009 (2) | 0.0309 (2) | −0.0057 (2) |
| N1 | 0.0375 (7) | 0.0308 (6) | 0.0378 (7) | 0.0027 (5) | 0.0169 (6) | 0.0006 (5) |
| N2 | 0.0350 (7) | 0.0525 (9) | 0.0339 (7) | −0.0057 (6) | 0.0145 (6) | 0.0014 (6) |
| C1 | 0.0342 (8) | 0.0264 (7) | 0.0355 (8) | 0.0023 (6) | 0.0128 (7) | 0.0027 (6) |
| C2 | 0.0288 (7) | 0.0332 (8) | 0.0364 (8) | −0.0020 (6) | 0.0099 (6) | 0.0007 (6) |
| C3 | 0.0326 (8) | 0.0339 (8) | 0.0299 (7) | −0.0028 (6) | 0.0089 (6) | −0.0004 (6) |
| C4 | 0.0327 (8) | 0.0287 (7) | 0.0318 (8) | −0.0012 (6) | 0.0105 (6) | 0.0058 (6) |
| C5 | 0.0332 (8) | 0.0379 (8) | 0.0370 (8) | −0.0082 (6) | 0.0119 (7) | −0.0009 (7) |
| C6 | 0.0374 (8) | 0.0357 (8) | 0.0353 (8) | −0.0053 (6) | 0.0119 (7) | −0.0050 (6) |
| C7 | 0.0358 (8) | 0.0304 (7) | 0.0351 (8) | 0.0043 (6) | 0.0134 (7) | 0.0012 (6) |
| C8 | 0.0290 (7) | 0.0305 (7) | 0.0308 (7) | 0.0049 (6) | 0.0097 (6) | 0.0021 (6) |
| C9 | 0.0358 (8) | 0.0311 (8) | 0.0418 (9) | −0.0017 (6) | 0.0147 (7) | −0.0045 (6) |
| C10 | 0.0368 (8) | 0.0388 (8) | 0.0430 (9) | 0.0016 (7) | 0.0154 (7) | −0.0100 (7) |
| C11 | 0.0288 (7) | 0.0488 (9) | 0.0323 (8) | 0.0056 (7) | 0.0100 (6) | −0.0003 (7) |
| C12 | 0.0401 (9) | 0.0348 (8) | 0.0419 (9) | 0.0020 (7) | 0.0174 (7) | 0.0041 (7) |
| C13 | 0.0435 (9) | 0.0277 (7) | 0.0405 (8) | 0.0040 (6) | 0.0191 (7) | 0.0002 (6) |
| C14 | 0.0356 (10) | 0.0907 (16) | 0.0478 (11) | −0.0047 (10) | 0.0186 (8) | −0.0038 (10) |
| C15 | 0.0519 (11) | 0.0780 (14) | 0.0446 (10) | −0.0163 (10) | 0.0263 (9) | −0.0139 (10) |
Geometric parameters (Å, º) top
| Cl1—C11 | 1.744 (2) | C7—H7 | 0.9500 |
| N1—C7 | 1.280 (2) | C8—C13 | 1.393 (2) |
| N1—C8 | 1.415 (2) | C8—C9 | 1.393 (2) |
| N2—C4 | 1.366 (2) | C9—C10 | 1.384 (3) |
| N2—C14 | 1.445 (3) | C9—H9 | 0.9500 |
| N2—C15 | 1.445 (3) | C10—C11 | 1.379 (3) |
| C1—C6 | 1.397 (2) | C10—H10 | 0.9500 |
| C1—C2 | 1.397 (2) | C11—C12 | 1.387 (2) |
| C1—C7 | 1.452 (2) | C12—C13 | 1.379 (3) |
| C2—C3 | 1.371 (2) | C12—H12 | 0.9500 |
| C2—H2 | 0.9500 | C13—H13 | 0.9500 |
| C3—C4 | 1.419 (2) | C14—H14A | 0.9800 |
| C3—H3 | 0.9500 | C14—H14B | 0.9800 |
| C4—C5 | 1.411 (2) | C14—H14C | 0.9800 |
| C5—C6 | 1.377 (3) | C15—H15A | 0.9800 |
| C5—H5 | 0.9500 | C15—H15B | 0.9800 |
| C6—H6 | 0.9500 | C15—H15C | 0.9800 |
| | | |
| C7—N1—C8 | 119.36 (14) | C10—C9—C8 | 120.50 (16) |
| C4—N2—C14 | 120.87 (16) | C10—C9—H9 | 119.8 |
| C4—N2—C15 | 121.45 (15) | C8—C9—H9 | 119.8 |
| C14—N2—C15 | 116.13 (17) | C11—C10—C9 | 119.46 (16) |
| C6—C1—C2 | 117.26 (16) | C11—C10—H10 | 120.3 |
| C6—C1—C7 | 120.36 (15) | C9—C10—H10 | 120.3 |
| C2—C1—C7 | 122.33 (15) | C10—C11—C12 | 121.24 (17) |
| C3—C2—C1 | 121.85 (15) | C10—C11—Cl1 | 119.74 (14) |
| C3—C2—H2 | 119.1 | C12—C11—Cl1 | 119.01 (14) |
| C1—C2—H2 | 119.1 | C13—C12—C11 | 118.75 (16) |
| C2—C3—C4 | 121.10 (15) | C13—C12—H12 | 120.6 |
| C2—C3—H3 | 119.5 | C11—C12—H12 | 120.6 |
| C4—C3—H3 | 119.5 | C12—C13—C8 | 121.28 (15) |
| N2—C4—C5 | 121.90 (15) | C12—C13—H13 | 119.4 |
| N2—C4—C3 | 121.19 (15) | C8—C13—H13 | 119.4 |
| C5—C4—C3 | 116.90 (16) | N2—C14—H14A | 109.5 |
| C6—C5—C4 | 120.90 (16) | N2—C14—H14B | 109.5 |
| C6—C5—H5 | 119.5 | H14A—C14—H14B | 109.5 |
| C4—C5—H5 | 119.5 | N2—C14—H14C | 109.5 |
| C5—C6—C1 | 121.95 (15) | H14A—C14—H14C | 109.5 |
| C5—C6—H6 | 119.0 | H14B—C14—H14C | 109.5 |
| C1—C6—H6 | 119.0 | N2—C15—H15A | 109.5 |
| N1—C7—C1 | 122.31 (15) | N2—C15—H15B | 109.5 |
| N1—C7—H7 | 118.8 | H15A—C15—H15B | 109.5 |
| C1—C7—H7 | 118.8 | N2—C15—H15C | 109.5 |
| C13—C8—C9 | 118.75 (16) | H15A—C15—H15C | 109.5 |
| C13—C8—N1 | 117.70 (14) | H15B—C15—H15C | 109.5 |
| C9—C8—N1 | 123.46 (14) | | |
| | | |
| C6—C1—C2—C3 | −0.8 (2) | C6—C1—C7—N1 | 170.00 (16) |
| C7—C1—C2—C3 | 176.70 (14) | C2—C1—C7—N1 | −7.4 (2) |
| C1—C2—C3—C4 | −1.1 (2) | C7—N1—C8—C13 | 140.53 (16) |
| C14—N2—C4—C5 | −15.4 (3) | C7—N1—C8—C9 | −43.0 (2) |
| C15—N2—C4—C5 | 179.36 (18) | C13—C8—C9—C10 | −0.4 (2) |
| C14—N2—C4—C3 | 165.29 (18) | N1—C8—C9—C10 | −176.86 (15) |
| C15—N2—C4—C3 | 0.1 (3) | C8—C9—C10—C11 | −0.1 (2) |
| C2—C3—C4—N2 | −178.80 (15) | C9—C10—C11—C12 | −0.1 (3) |
| C2—C3—C4—C5 | 1.9 (2) | C9—C10—C11—Cl1 | 179.16 (12) |
| N2—C4—C5—C6 | 179.76 (15) | C10—C11—C12—C13 | 0.9 (3) |
| C3—C4—C5—C6 | −0.9 (2) | Cl1—C11—C12—C13 | −178.39 (12) |
| C4—C5—C6—C1 | −0.9 (3) | C11—C12—C13—C8 | −1.4 (2) |
| C2—C1—C6—C5 | 1.7 (2) | C9—C8—C13—C12 | 1.2 (2) |
| C7—C1—C6—C5 | −175.78 (15) | N1—C8—C13—C12 | 177.87 (15) |
| C8—N1—C7—C1 | 179.93 (13) | | |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| C14—H14B···Cl1i | 0.98 | 2.75 | 3.729 (3) | 175 |
| C2—H2···Cl1ii | 0.95 | 3.11 | 3.812 (3) | 132 |
| C6—H6···N2iii | 0.95 | 2.83 | 3.481 (3) | 127 |
| C14—H14A···N1iv | 0.98 | 3.08 | 3.602 (4) | 115 |
| C10—H10···N1iii | 0.95 | 2.87 | 3.728 (3) | 152 |
| Symmetry codes: (i) x−1, y, z+1; (ii) x, y, z+1; (iii) x, −y+1/2, z−1/2; (iv) x−1, y, z. |
Experimental details
| Crystal data |
| Chemical formula | C15H15ClN2 |
| Mr | 258.74 |
| Crystal system, space group | Monoclinic, P21/c |
| Temperature (K) | 193 |
| a, b, c (Å) | 9.852 (6), 16.268 (9), 9.512 (6) |
| β (°) | 119.904 (6) |
| V (Å3) | 1321.5 (14) |
| Z | 4 |
| Radiation type | Mo Kα |
| µ (mm−1) | 0.27 |
| Crystal size (mm) | 0.70 × 0.70 × 0.30 |
| |
| Data collection |
| Diffractometer | Mercury CCD
diffractometer |
| Absorption correction | Multi-scan
(Jacobson, 1998) |
| Tmin, Tmax | 0.832, 0.923 |
No. of measured, independent and
observed [I > 2σ(I)] reflections | 10180, 2956, 2773 |
| Rint | 0.029 |
| (sin θ/λ)max (Å−1) | 0.649 |
| |
| Refinement |
| R[F2 > 2σ(F2)], wR(F2), S | 0.056, 0.143, 1.16 |
| No. of reflections | 2956 |
| No. of parameters | 166 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.22, −0.23 |
Selected geometric parameters (Å, º) top| Cl1—C11 | 1.744 (2) | N2—C14 | 1.445 (3) |
| N1—C7 | 1.280 (2) | N2—C15 | 1.445 (3) |
| N1—C8 | 1.415 (2) | C1—C7 | 1.452 (2) |
| N2—C4 | 1.366 (2) | | |
| | | |
| C7—N1—C8 | 119.36 (14) | C5—C4—C3 | 116.90 (16) |
| C4—N2—C14 | 120.87 (16) | N1—C7—C1 | 122.31 (15) |
| C4—N2—C15 | 121.45 (15) | C13—C8—C9 | 118.75 (16) |
| C14—N2—C15 | 116.13 (17) | C13—C8—N1 | 117.70 (14) |
| C6—C1—C2 | 117.26 (16) | C9—C8—N1 | 123.46 (14) |
| C6—C1—C7 | 120.36 (15) | C10—C11—C12 | 121.24 (17) |
| C2—C1—C7 | 122.33 (15) | C10—C11—Cl1 | 119.74 (14) |
| N2—C4—C5 | 121.90 (15) | C12—C11—Cl1 | 119.01 (14) |
| N2—C4—C3 | 121.19 (15) | | |
| | | |
| C7—C1—C2—C3 | 176.70 (14) | C8—N1—C7—C1 | 179.93 (13) |
| C14—N2—C4—C5 | −15.4 (3) | C6—C1—C7—N1 | 170.00 (16) |
| C15—N2—C4—C5 | 179.36 (18) | C2—C1—C7—N1 | −7.4 (2) |
| C14—N2—C4—C3 | 165.29 (18) | C7—N1—C8—C13 | 140.53 (16) |
| C15—N2—C4—C3 | 0.1 (3) | C7—N1—C8—C9 | −43.0 (2) |
| C7—C1—C6—C5 | −175.78 (15) | N1—C8—C9—C10 | −176.86 (15) |
Hydrogen-bond geometry (Å, º) top
| D—H···A | D—H | H···A | D···A | D—H···A |
| C14—H14B···Cl1i | 0.979 | 2.753 | 3.729 (3) | 174.5 |
| C2—H2···Cl1ii | 0.950 | 3.110 | 3.812 (3) | 132.0 |
| C6—H6···N2iii | 0.950 | 2.828 | 3.481 (3) | 126.8 |
| C14—H14A···N1iv | 0.981 | 3.078 | 3.602 (4) | 114.9 |
| C10—H10···N1iii | 0.951 | 2.865 | 3.728 (3) | 151.5 |
| Symmetry codes: (i) x−1, y, z+1; (ii) x, y, z+1; (iii) x, −y+1/2, z−1/2; (iv) x−1, y, z. |
The chloro-substituted Schiff bases in this study top | Refcode | Space group | S | S' | µ(D) |
| 1 | BADDAL10 | P21/c | 2-OH | 4'-Cl | 2.20 |
| 2 | BEYQEB | P212121 | 2-OH | 5'-Cl,2'-Me | 3.03 |
| 3 | CBZYAN | P212121 | 2,4-Cl | | 1.10 |
| 4 | CHLSAN | P212121 | 2-OH | 2'-Cl | 3.73 |
| 5 | CSALAN02 | Pca21 | 2-OH, 5-Cl | | 2.35 |
| 6 | FAKDIE | P21/n | 4-Cl | 2'-OH | 2.76 |
| 7 | RONKEK | P21 | 4-Cl | 3'-Cl | 0.52 |
| 8 | RONKOU | P21 | 4-Br | 3'-Cl | 0.57 |
| 9 | RONKUA | P21 | 4-Cl | 2'-Br | 0.48 |
| 10 | RONLAH | P21/c | 3-Cl | 4'-Br | 2.78 |
| 11 | WEMJIH | P212121 | 3-Br | 3'-Cl | 1.46 |
| 12 | YICNON | P21/c | 3,5-Cl, 2-OH | 4'-NEt2 | 6.22 |
| 13 | YICPAB | P21/c | 3,5-Cl, 2-OH | 4'-NMe2 | 6.16 |
| 14 | ZAMMEF | P21/c | 2,3-OH | 2'-Cl | 3.73 |
| 15 | (I) | P21/c | 4-NMe2 | 4'-Cl | 6.20 |
| Notes: plane 1 is the plane directly attached to the central C atom; plane 2 is the plane directly attached to the central N atom; S and S' refer to the substituent groups in planes 1 and 2, respectively; µ is the molecular dipole moment calculated by MOPAC (Stewert, 1989). |
C—H···π and π–π interactions in (I) (Å, °) top| E(SM)% | Interaction | H···P | C—H···P | Symmetry code |
| 15.3 | C—H12···P1 | 3.47 | 126.9 | -x + 1,-y + 1, −z + 1 |
| 15.3 | C—H13···P3 | 3.65 | 121.7 | -x + 1,-y + 1, −z + 1 |
| 15.3 | Plane 3–3 | 4.24 | 4.66 | -x + 1,-y + 1, −z + 1 |
| 12.6 | C—H10···P3 | 3.63 | 138.8 | x, −y + 1/2, z + 1/2 |
| Notes: plane 3–3 for the π–π interaction between the two planes 3 in the interacting molecules. The parameters followed are the distances between the planes and between their centers, respectively; E(SM)% is the percentage interaction energy between the two interacting molecules in the total packing energy, calculated by OPEC (Gavezzotti, 1983) |


 journal menu
journal menu organic compounds
organic compounds











![[Figure 1]](http://journals.iucr.org/c/issues/2004/09/00/de1245/de1245fig1thm.gif)
![[Figure 2]](http://journals.iucr.org/c/issues/2004/09/00/de1245/de1245fig2thm.gif)




Benzylideneanilines are an important class of Schiff bases, which have been widely used in coordinate, medical and biological chemistry for some time (Metzler, 1980; Tarafder, 2002). Recently, the thermochromism (Pistolis, 1996), photochromism (Jalali-Heravi, 2000) and non-linear optical properties of these compounds have found applications in modern technologies (Sekikawa, 1997). In the design of solid materials, one of the key steps is to understand how the constituent molecules are packed, what kinds of interactions are playing a role in crystal packing and how they are interplaying (Desiraju, 1989). Since any centrosymmetric crystal has no odd rank tensor property, such as SHG and piezoelecticity, the design of non-centrosymmetric crystals is one of the important and also difficult problems in crystal engineering. As an example, molecular packing analyses were carried out for 14 chloro-substituted Schiff bases retrieved from the Cambridge Structural Database (CSD; Allen, 2002); this class of compounds was chosen because the percentage of non-centrosymmetric space groups for these compounds is 57.1%, much higher than that for general organic compounds (about 25%). The crystal data of these 14 compounds are listed in Table 3.
These crystals contain no strong hydrogen-bond-forming groups, such as-COOH, –NH2CO– or –NO2; various weak interactions, such as CH/π (Dmezawa, 1998), π···π (Sharma, 1993) and weak hydrogen bonds, and the interplay between these interactions, must therefore play an important role in determining the crystal structures.
The crystal packing analysis was carried out using OPEC (Gavezzotti, 1983), which was locally modified with additional calculation routines. Given a reference molecule (FM), and with the distance of interacting molecules limited to within 15 Å, the program calculated approximate 150 surrounding molecules (SM), which form the crystal model.
The main results are listed in Table 1S (supplementary material). From Table 1S, we can see the following:
(i) The CH/π interactions play a major role in controlling the molecular packing. 15 CH/π interactions, out of 18 listed for the most important intermolecular interactions, assemble molecules into a screw, which might explain why the crystals studied are more likely to crystallize in non-centrosymmetric groups.
2. The π···π interactions almost equally play the same role. Out of 19 listed, half of the interactions assemble the molecules in a translation-related mode, favouring an acentric crystal. This mode appears to occur when the molecular dipole is relatively small. When the molecular dipole is relatively large, the π···π interactions cause the molecules to packed in an antiparallel fashion into centric crystals.
3. Weak interactions, such as C—H···Cl (Thallapally & Nangia, 2001), also favour the assembling of molecules with the first kind of symmetric operators (screw 10 and traslation 13 in the 27 important interactions listed), but the percentage of packing energy is relatively small.
4. To our suprize, the role played by X atoms (X = Cl or Br) through so-called X···X interactions (Desiraju, 1994) seems to be only a secondary one, perhaps because the atomic fractions are not large enough. However, the? replacement of substituents like NO2 by X atoms? can also modify the properties of crystals designed for similar purposes.
During our study on the design of organic functional materials, the title compound, (I), was obtained, and its crystal structure is reported here. The selected geometric parameters are listed in Table 1.
The title molecule has a trans configuration and the C1—C7—N1—C8 (plane 3) torsion angle is 179.9°. A dihedral angle of 50.4° exists between planes 1 and 2 (plane 1 is the plane directly attached to the central C atom and plane 2 is the plane directly attached to the central N atom), which are twisted ?in opposite directions? around the central linkage by 9.6 and 41.3° for planes 1 and 2, respectively. For the title crystal, there are no strong hydrogen-bond-forming groups, such as –COOH, –NH2CO– or –OH, –NO2, so weak interactions must play determining roles in the crystal packing. The molecules that interacte strongly with the FM are listed in Tables 2 and 4. As shown by these tables, molecules interact through CH/π and π···π interactions, forming a `dimer' and further forming double chains along [001]. The double chains are connected by a C14—H14B···Cl1 hydrogen bond (3.729 Å and 174.5 °), extending along [10–1], forming double layers (010). In the third direction, there are only ordinary, weaker, van der Waals interactions, consistent with the formation of thin plates.