





Abstract
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) is an extremely potent environmental contaminant that produces a wide range of adverse biological effects, including the induction of cytochrome P450 1A1(CYP1A1) that may enhance the toxic effects of TCDD. Several studies indicated that concurrent supplementation of vitamin A could reduce the toxicity, and potentially inhibit CYP1A1 activity (measured as ethoxyresorufin-O-deethylase [EROD] activity). In the present study, we investigated the in vivo effects of vitamin A on EROD activities and the expression of CYP1A1 in the liver of TCDD-treated mice. In Experiment I, the mice were given a single oral dose of 40 μg TCDD/kg body weight with or without the continuous administration of 2500 IU vitamin A/kg body weight/day, and were killed on day 1, 3, 7, 14, or 28. In Experiment II, the mice were given daily an oral dose of 0.1 μg TCDD/kg body weight with or without supplement of 2000 IU vitamin A/kg body weight, and were killed on day 14, 28, or 42. In both experiments, TCDD caused liver damage and increase in relative liver weights, augmented the EROD activities and CYP1A1 expression, and increased the aryl hydrocarbon receptor (AhR) mRNA expression, but did not alter the AhR nuclear translocator (ARNT) mRNA expression. CYP1A1 mRNA expression and AhR mRNA expression showed a similar time course. The liver damage in TCDD + vitamin A–treated mice was less severe than that in TCDD-treated mice. EROD activities, CYP1A1 expression, and AhR mRNA expression in vitamin A + TCDD–treated mice were lower than those in TCDD-treated mice, indicating that supplementation of vitamin A might attenuate the liver damage caused by TCDD.
Cytochrome P450s belong to the superfamily of enzymes that are involved in the metabolism of many endogenous and exogenous compounds, such as steroid hormones, drugs, and environmental pollutants (Honkakoski and Negishi, 2000; Gonzalez and Gelboin, 1994). These enzymes can be modulated by endogenous and exogenous factors (Nelson et al., 1996), and in turn, they may influence the biological actions of the environmental compounds. Therefore, the effects of natural and synthetic compounds on cytochrome P450 enzymes have attracted considerable interest. Cytochrome P4501A1 (CYP1A1) is of major interest for its bioactivation of many procarcinogens including benzo(a)pyrene (B(a)P) (Shou et al., 1994; Whitlock, 1999). Its most potent known inducer is 2,3,7,8-tetrachlorodi-benzo-p-dioxin (TCDD).
TCDD is a ubiquitous environmental contaminant, which induces a variety of toxic responses via binding and activating the aryl hydrocarbon receptor (AhR). The TCDD–AhR complex forms a heterodimer with the AhR nuclear translocator (ARNT), and the TCDD–AhR–ARNT complex then binds to specific sequences in the DNA called dioxin responsive elements (DRE) and alters the expression of a wide variety of genes, including CYP1A1 (Schmidt and Bradfield, 1996).
Vitamin A (retinol) is an essential micronutrient that plays a central role in various physiological processes, such as reproduction, cell growth and differentiation, immunity, embryogenesis, and vision (Chambon, 1994; Maden et al., 1998). It exerts its effects by conversion to the biologically active derivatives, retinal and retinoic acid (RA) (Chambon, 1996). With the exception of vision, RA fulfills the majority of vitamin A actions via binding and activating two families of ligand-activated nuclear retinoid receptors, retinoic acid receptor (RAR) and retinoid X receptor (RXR) (Chambon, 1996).
Several studies indicated that some of the toxic effects of TCDD resembled retinoid deficiency. Low dietary intake of vitamin A impaired the tolerance to TCDD or decreased the survival time in experimental animals (Håkansson et al., 1991) and markedly enhanced the TCDD-induced liver foci development (Flodstrom et al., 1991). But concurrent supplementation of vitamin A could alleviate toxicity symptoms caused by TCDD (Zile, 1992). It was demonstrated that CYP1A1 activity was a well-characterized AhR-mediated response following exposure to TCDD (Okey et al., 1994). It was also reported that retinoids showed potent inhibitory effects on CYP1A1-dependent 7-ethoxycoumarin deethylation (Inouye et al., 1999). Huang et al. (1999) pointed out that the preventive effect of retinoids in B(a)P-induced carcinogenesis might be partially due to the specific inhibition of B(a)P activation catalyzed by CYP1A1. Therefore, inhibitory effects of vitamin A on some of the TCDD toxicity might be related to the reduction of CYP1A1 induction. This research was designed to investigate the in vivo effects of vitamin A on the activity and expression of CYP1A1 in the liver of TCDD-treated mice.
MATERIALS AND METHODS
Chemicals and reagents.
TCDD (>99%) was purchased from Supelco (Bellefonte, PA). The stock solutions of 10 μg TCDD per milliliter toluene were diluted with corn oil to prepare the dosing solutions, and the toluene was evaporated with a stream of nitrogen gas at 40°C. Vitamin A, as retinol palmitate, was bought from Xiamen Cod-liver Oil Company (Xiamen, China). Ethoxyresorufin, resorufin, glucose-6-phosphate, glucose-6-phosphate dehydrogenase, and NADPH were purchased from Sigma (St. Louis, MO). TRIzol reagents were purchased from Invitrogen Corp. (Carlsbad, CA). SYBR Green I was purchased from OPE Technology Development Co, Ltd (Shanghai, China). AMV Reverse Transcription System (A3500) was purchased from Promega (Madison, WI). Taq DNA polymerase and dNTP were purchased from MBI Fermentas (Lithuania). All the primers were designed using Primer Express Software v2.0 (Applied Biosystems, Foster City, CA) and synthesized by the Shanghai Sangon Biological Engineering Company, China. Other chemicals and solvents were of analytical grade or HPLC grade.
Animals.
Four-week-old male C57BL6 mice were obtained from Beijing Vital River Laboratory Animal Co, Ltd (Beijing, China). They were housed in plastic cages containing sawdust bedding in groups of five. The animals were kept at room temperature of 22 ± 2°C and fed standard rodent chow and water ad libitum. The feed, in which the vitamin A content was 11,000 IU/kg, was obtained from Laboratory Animal Center, Shantou University Medical College. All animal treatments were performed in accordance with the protocols approved by Medical Laboratory Administration Committee.
Treatment schedule.
In this study, single and repeated TCDD exposures were designed. The mice were randomly assigned to four groups in each experiment, A: control, B: vitamin A, C: TCDD, and D: TCDD with vitamin A. TCDD dose and duration of treatment in Experiment I or II are based on previous studies (Fletcher et al., 2001; Morris et al., 1998). With these doses of vitamin A it was possible to obtain healthy animals with a vitamin A status that varied within a physiological range.
In single TCDD exposure (Experiment I), mice received corn oil in A and B groups or 40 μg TCDD/kg body weight in C and D groups, and mice in A and C groups received corn oil daily, while those in B and D groups received 2500 IU vitamin A/kg body weight daily for one week prior to dosing of TCDD and throughout the experiment. Five mice per group were killed 1, 3, 7, 14, or 28 days after treatment with TCDD, respectively. In repeated TCDD exposure (Experiment II), mice were daily given corn oil in A and B groups or 0.1 μg TCDD/kg body weight in C and D groups, and the mice were daily administered corn oil in A and C groups or 2000 IU vitamin A/kg body weight in B and D groups for one week prior to the start of dosing of TCDD and throughout the experiment. Five mice per group were killed 14, 28, or 42 days after the start of dosing of TCDD, respectively. All the tested chemicals were delivered by gavage, and the dose volume was 0.2 ml solution per 20 g body weight. Animals were killed by cervical dislocation. Liver was excised and weighed. Representative samples of liver were fixed in buffered formalin for subsequent histological analyses. The remaining liver was snap frozen in liquid nitrogen and stored at −70°C for RNA extraction, enzyme assay, and Western blotting.
Histological examination.
Tissue samples were preserved in 10% neutral buffered formalin, embedded in paraffin wax, and cut to the thickness of 4 μm. The tissue sections were stained with hematoxylin and eosin (HE) and examined using light microscope independently by two pathologists. The degree of hepatoxicity was graded on a scale of 1 to 5, as categorized by Smith et al. (1998).
Hepatic CYP1A1 activity.
The hepatic microsomal fraction was isolated by differential centrifugation at 10,000 × g for 20 min, followed by a centrifugation at 105,000 × g for 1 h at 4°C (Wei et al., 1995). The resulting microsomal pellet was suspended in 0.1 M phosphate-buffered solution and stored in aliquots at −70°C. The protein content of the microsomes was measured by the Bradford method, using bovine serum albumin as the reference (Bradford, 1976).
Hepatic CYP1A1 activity was measured using ethoxyresorufin-O-deethylase (EROD) assay. EROD activity was determined by measuring the formation of resorufin at 37°C with an excitation wavelength of 530 nm and an emission wavelength of 585 nm using a spectrofluorometer (HITACHI, F-3000, Japan). Either 80 μg (in A and B groups) or 2 μg (in C and D groups) microsomal proteins were used in the reactions. These amounts of proteins had been confirmed to give linear enzymatic reactions for 15 min. The reaction mixture contained microsomal proteins, 50 mM potassium phosphate (pH 7.4), 1.5 μM ethoxyresorufin, and a NADPH regenerating system (4 mM MgCl2·6H2O, 5 mM glucose-6-phosphate, 1 unit/ml glucose-6-phosphate dehydrogenase, and 0.5 mM NADPH). These reactions were initiated by adding 10 μl NADPH and incubated at 37°C for 10 min, and then were stopped by adding 0.25 ml of a stop solution (20% trichloroacetic acid). The results are reported as nmol/min/mg protein.
RNA extraction and real-time PCR analysis.
Total RNA was extracted from the mouse liver tissue using TRIzol reagent according to the manufacturer's instructions. The yield and quality of the RNA samples were determined by absorbance at 260 nm and the ratio of absorbance at 260 nm to that at 280 nm (A260/280 ratio) using a spectrophotometer (DU650, Beckman). The isolated RNA samples with A260/280 ratio between 1.8 and 2.0 were used for subsequent analysis. First-strand cDNA was synthesized from 2 μg of total RNA using AMV Reverse Transcription System in a 20-μl reaction volume according to manufacturer's manual. The resultant cDNA was stored at −20°C and used for real-time PCR analysis later.
The double-stranded DNA binding dye method was used for quantitative PCR/RT-PCR. The quantitative PCR was performed in an ABI-Prism 7700 (Applied Biosystems, Foster City, CA) thermal cycler using a SYBR Green I. Primers of CYP1A1, AhR, ARNT, and β-actin are listed in Table 1. The synthesized cDNA products (1 μl) or plasmid DNA (2 μl) were subjected to real-time PCR in a reaction mixture (25 μl) containing 2 mM MgCl2, 200 nM dNTP, 1× Master SYBR Green I mix, 150 nM gene-specific forward and reverse primers, and 1.5 units Taq DNA polymerase. The thermal cycling conditions comprised an initial denaturation step at 95°C for 5 min, 40 cycles at 95°C for 15 s, 60°C for 30s, and 72°C for 30s. Consequently, at the end of the PCR cycles, the real-time PCR products were immediately analyzed using a ramping rate of 0.03°C/s from 60 to 95°C to calculate the dissociation curve to confirm that single PCR product was detected by SYBR Green I dye. No-template control reactions for every primer pair were also included on each reaction plate to check for external DNA contamination. Sequence-specific standard curves were generated using 10-fold serial dilutions of plasmid DNA, and then the value for the initial concentration of unknown samples was calculated by the software (version 1.7) provided with the ABI 7700 system. Samples were normalized using the housekeeping gene β-actin. Each measurement of sample was conducted in duplicate.
Mouse Gene-Specific Primer Sequences for CYP1A1, AhR, ARNT and β-Actin used in Quantitative Real-Time PCR.
Gene | Orientation | Sequence (5′-3′) | Positions | Product size (bp) | Accession no.a |
|---|---|---|---|---|---|
| CYP1A1 | sense | TCTCGTGGAGCCTCATGTACCT | 1170–1191 | 91 | NM 009992 |
| antisense | TGCCGATCTCTGCCAATCA | 1242–1260 | |||
| AhR | sense | CGTCCCTGCATCCCACTACTT | 2461–2481 | 131 | NM 013464 |
| antisense | GGACATGGCCCCAGCATAG | 2573–2591 | |||
| ARNT | sense | TGCGACGGAACAAGATGACA | 239–258 | 137 | NM 009709 |
| antisense | AGTTCCCCTCAAGGACTTCATGT | 353–375 | |||
| β-actin | sense | ATGGTGGGAATGGGTCAGAA | 210–229 | 118 | NM 007393 |
| antisense | CCATGTCGTCCCAGTTGGTAA | 307–327 |
Gene | Orientation | Sequence (5′-3′) | Positions | Product size (bp) | Accession no.a |
|---|---|---|---|---|---|
| CYP1A1 | sense | TCTCGTGGAGCCTCATGTACCT | 1170–1191 | 91 | NM 009992 |
| antisense | TGCCGATCTCTGCCAATCA | 1242–1260 | |||
| AhR | sense | CGTCCCTGCATCCCACTACTT | 2461–2481 | 131 | NM 013464 |
| antisense | GGACATGGCCCCAGCATAG | 2573–2591 | |||
| ARNT | sense | TGCGACGGAACAAGATGACA | 239–258 | 137 | NM 009709 |
| antisense | AGTTCCCCTCAAGGACTTCATGT | 353–375 | |||
| β-actin | sense | ATGGTGGGAATGGGTCAGAA | 210–229 | 118 | NM 007393 |
| antisense | CCATGTCGTCCCAGTTGGTAA | 307–327 |
Genbank accession number of corresponding gene, available at http://www.ncbi.nlm.nih.gov/.
Mouse Gene-Specific Primer Sequences for CYP1A1, AhR, ARNT and β-Actin used in Quantitative Real-Time PCR.
Gene | Orientation | Sequence (5′-3′) | Positions | Product size (bp) | Accession no.a |
|---|---|---|---|---|---|
| CYP1A1 | sense | TCTCGTGGAGCCTCATGTACCT | 1170–1191 | 91 | NM 009992 |
| antisense | TGCCGATCTCTGCCAATCA | 1242–1260 | |||
| AhR | sense | CGTCCCTGCATCCCACTACTT | 2461–2481 | 131 | NM 013464 |
| antisense | GGACATGGCCCCAGCATAG | 2573–2591 | |||
| ARNT | sense | TGCGACGGAACAAGATGACA | 239–258 | 137 | NM 009709 |
| antisense | AGTTCCCCTCAAGGACTTCATGT | 353–375 | |||
| β-actin | sense | ATGGTGGGAATGGGTCAGAA | 210–229 | 118 | NM 007393 |
| antisense | CCATGTCGTCCCAGTTGGTAA | 307–327 |
Gene | Orientation | Sequence (5′-3′) | Positions | Product size (bp) | Accession no.a |
|---|---|---|---|---|---|
| CYP1A1 | sense | TCTCGTGGAGCCTCATGTACCT | 1170–1191 | 91 | NM 009992 |
| antisense | TGCCGATCTCTGCCAATCA | 1242–1260 | |||
| AhR | sense | CGTCCCTGCATCCCACTACTT | 2461–2481 | 131 | NM 013464 |
| antisense | GGACATGGCCCCAGCATAG | 2573–2591 | |||
| ARNT | sense | TGCGACGGAACAAGATGACA | 239–258 | 137 | NM 009709 |
| antisense | AGTTCCCCTCAAGGACTTCATGT | 353–375 | |||
| β-actin | sense | ATGGTGGGAATGGGTCAGAA | 210–229 | 118 | NM 007393 |
| antisense | CCATGTCGTCCCAGTTGGTAA | 307–327 |
Genbank accession number of corresponding gene, available at http://www.ncbi.nlm.nih.gov/.
Western blotting.
Microsomal proteins (15 μg/lane) were resolved using 10% sodium dodecy1 sulfate gel electrophoresis (SDS–PAGE) and transferred to a nitrocellulose membrane (Hybond P, Amersham, UK). The primary antibody was the rabbit polyclonal anti-CYP1A1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA). The secondary antibody was alkaline phosphatase-linked goat anti-rabbit IgG (Promega, Madison, WI). The positive bands representing CYP1A1 protein were developed using the Western Blue Stabilized Substrate for Alkaline Phosphatase (Promega).
Statistical analysis.
All values were expressed as arithmetic means ± standard deviation. Group means for relative liver weights, EROD activity, and the mRNA expression of CYP1A1, AhR, and ARNT were evaluated using one-way analysis of variance (ANOVA). If the data were homogenous and normally distributed, multiple comparisons were made by Newman-Keuls test. If tests for normality or variance failed, the Kruskal-Wallis one-way ANOVA on ranks was carried out and followed with the Nemenyi test for multiple comparisons; p < 0.05 was considered as statistically significant.
RESULT
General Toxicity
No mice died during the study period. There was no significant difference in animal body weight between the four groups at each testing point in both experiments (data not shown).
Hepatic Toxicity
In Experiment I (Table 2), the relative liver weights in TCDD-treated (40 μg/kg) mice began to increase as early as 1 day, peaked after 3 days, and remained high till 28 days after TCDD exposure. Supplement of Vitamin A (2500 IU/kg) showed neither significant effect on the relative liver weights during the experiment, nor significant influence on the increase of relative liver weights induced by single TCDD dose.
Comparison of Hepatic Toxicity in Male C57BL/6 Mice with Different Treatments in Experiment I
Days after administration of TCDD | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 3 | 7 | 14 | 28 | |||||
| Relative liver weights (g/kg) | |||||||||
| Control group (A) | 56.1 ± 3.2 | 54.7 ± 2.9 | 53.2 ± 0.9 | 51.6 ± 1.7 | 46.2 ± 3.9 | ||||
| Vitamin A group (B) | 54.0 ± 2.3 | 52.1 ± 3.0 | 52.4 ± 0.6 | 52.8 ± 1.3 | 42.6 ± 1.3 | ||||
| TCDD group (C) | 65.0 ± 4.8ab | 78.9 ± 3.4ab | 68.2 ± 1.4ab | 61.1 ± 3.2ab | 54.5 ± 4.2ab | ||||
| Vitamin A + TCDD group (D) | 69.2 ± 5.8ab | 67.9 ± 5.1ab | 66.6 ± 2.8ab | 61.0 ± 1.4ab | 52.0 ± 5.3b | ||||
| Estimate of relative liver toxityc | |||||||||
| Control group (A) | − | − | − | − | − | ||||
| Vitamin A group (B) | − | − | − | − | − | ||||
| TCDD group (C) | + | ++ | +++ | +++ | ++ | ||||
| Vitamin A + TCDD group (D) | + | + | ++ | ++ | + | ||||
Days after administration of TCDD | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 3 | 7 | 14 | 28 | |||||
| Relative liver weights (g/kg) | |||||||||
| Control group (A) | 56.1 ± 3.2 | 54.7 ± 2.9 | 53.2 ± 0.9 | 51.6 ± 1.7 | 46.2 ± 3.9 | ||||
| Vitamin A group (B) | 54.0 ± 2.3 | 52.1 ± 3.0 | 52.4 ± 0.6 | 52.8 ± 1.3 | 42.6 ± 1.3 | ||||
| TCDD group (C) | 65.0 ± 4.8ab | 78.9 ± 3.4ab | 68.2 ± 1.4ab | 61.1 ± 3.2ab | 54.5 ± 4.2ab | ||||
| Vitamin A + TCDD group (D) | 69.2 ± 5.8ab | 67.9 ± 5.1ab | 66.6 ± 2.8ab | 61.0 ± 1.4ab | 52.0 ± 5.3b | ||||
| Estimate of relative liver toxityc | |||||||||
| Control group (A) | − | − | − | − | − | ||||
| Vitamin A group (B) | − | − | − | − | − | ||||
| TCDD group (C) | + | ++ | +++ | +++ | ++ | ||||
| Vitamin A + TCDD group (D) | + | + | ++ | ++ | + | ||||
Note. Data are expressed as arithmetic means ± SD (n = 5).
p < 0.05, significant difference compared with the control group at the same time point.
p < 0.05, significant difference compared with the vitamin A group at the same time point.
Toxicity as assessed in the report by Smith et al. (1998).
Comparison of Hepatic Toxicity in Male C57BL/6 Mice with Different Treatments in Experiment I
Days after administration of TCDD | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 3 | 7 | 14 | 28 | |||||
| Relative liver weights (g/kg) | |||||||||
| Control group (A) | 56.1 ± 3.2 | 54.7 ± 2.9 | 53.2 ± 0.9 | 51.6 ± 1.7 | 46.2 ± 3.9 | ||||
| Vitamin A group (B) | 54.0 ± 2.3 | 52.1 ± 3.0 | 52.4 ± 0.6 | 52.8 ± 1.3 | 42.6 ± 1.3 | ||||
| TCDD group (C) | 65.0 ± 4.8ab | 78.9 ± 3.4ab | 68.2 ± 1.4ab | 61.1 ± 3.2ab | 54.5 ± 4.2ab | ||||
| Vitamin A + TCDD group (D) | 69.2 ± 5.8ab | 67.9 ± 5.1ab | 66.6 ± 2.8ab | 61.0 ± 1.4ab | 52.0 ± 5.3b | ||||
| Estimate of relative liver toxityc | |||||||||
| Control group (A) | − | − | − | − | − | ||||
| Vitamin A group (B) | − | − | − | − | − | ||||
| TCDD group (C) | + | ++ | +++ | +++ | ++ | ||||
| Vitamin A + TCDD group (D) | + | + | ++ | ++ | + | ||||
Days after administration of TCDD | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 3 | 7 | 14 | 28 | |||||
| Relative liver weights (g/kg) | |||||||||
| Control group (A) | 56.1 ± 3.2 | 54.7 ± 2.9 | 53.2 ± 0.9 | 51.6 ± 1.7 | 46.2 ± 3.9 | ||||
| Vitamin A group (B) | 54.0 ± 2.3 | 52.1 ± 3.0 | 52.4 ± 0.6 | 52.8 ± 1.3 | 42.6 ± 1.3 | ||||
| TCDD group (C) | 65.0 ± 4.8ab | 78.9 ± 3.4ab | 68.2 ± 1.4ab | 61.1 ± 3.2ab | 54.5 ± 4.2ab | ||||
| Vitamin A + TCDD group (D) | 69.2 ± 5.8ab | 67.9 ± 5.1ab | 66.6 ± 2.8ab | 61.0 ± 1.4ab | 52.0 ± 5.3b | ||||
| Estimate of relative liver toxityc | |||||||||
| Control group (A) | − | − | − | − | − | ||||
| Vitamin A group (B) | − | − | − | − | − | ||||
| TCDD group (C) | + | ++ | +++ | +++ | ++ | ||||
| Vitamin A + TCDD group (D) | + | + | ++ | ++ | + | ||||
Note. Data are expressed as arithmetic means ± SD (n = 5).
p < 0.05, significant difference compared with the control group at the same time point.
p < 0.05, significant difference compared with the vitamin A group at the same time point.
Toxicity as assessed in the report by Smith et al. (1998).
In Experiment II (Table 3), daily exposures to low dose of TCDD (0.1 μg /kg body weight) caused increases in relative liver weights. After 28 or 42 days of TCDD dosing, supplement of vitamin A (2000 IU/kg) significantly attenuated the increases of relative liver weights induced by TCDD.
Comparison of Hepatic Toxicity in Male C57BL/6 Mice with Different Treatments in Experiment II
Days after administration of TCDD | |||||
|---|---|---|---|---|---|
| 14 | 28 | 42 | |||
| Relative liver weights (g/kg) | |||||
| Control group (A) | 52.7 ± 3.5 | 44.6 ± 3.5 | 39.7 ± 1.5 | ||
| Vitamin A group (B) | 51.6 ± 2.9 | 43.3 ± 1.7 | 38.5 ± 1.3 | ||
| TCDD group (C) | 62.3 ± 3.8ab | 54.1 ± 2.3ab | 53.1 ± 2.2ab | ||
| Vitamin A + TCDD group (D) | 59.4 ± 3.5ab | 44.7 ± 1.9c | 47.7 ± 0.8abc | ||
| Estimate of relative liver toxityd | |||||
| Control group (A) | − | − | − | ||
| Vitamin A group (B) | − | − | − | ||
| TCDD group (C) | + | ++ | +++ | ||
| Vitamin A + TCDD group (D) | + | + | + | ||
Days after administration of TCDD | |||||
|---|---|---|---|---|---|
| 14 | 28 | 42 | |||
| Relative liver weights (g/kg) | |||||
| Control group (A) | 52.7 ± 3.5 | 44.6 ± 3.5 | 39.7 ± 1.5 | ||
| Vitamin A group (B) | 51.6 ± 2.9 | 43.3 ± 1.7 | 38.5 ± 1.3 | ||
| TCDD group (C) | 62.3 ± 3.8ab | 54.1 ± 2.3ab | 53.1 ± 2.2ab | ||
| Vitamin A + TCDD group (D) | 59.4 ± 3.5ab | 44.7 ± 1.9c | 47.7 ± 0.8abc | ||
| Estimate of relative liver toxityd | |||||
| Control group (A) | − | − | − | ||
| Vitamin A group (B) | − | − | − | ||
| TCDD group (C) | + | ++ | +++ | ||
| Vitamin A + TCDD group (D) | + | + | + | ||
Note. Data are expressed as arithmetic means ± SD (n = 5).
p < 0.05, significant difference compared with the control group at the same time point.
p < 0.05, significant difference compared with the vitamin A group at the same time point.
p < 0.05, significant difference compared the TCDD group at the same time point.
Toxicity as assessed in the report by Smith et al. (1998).
Comparison of Hepatic Toxicity in Male C57BL/6 Mice with Different Treatments in Experiment II
Days after administration of TCDD | |||||
|---|---|---|---|---|---|
| 14 | 28 | 42 | |||
| Relative liver weights (g/kg) | |||||
| Control group (A) | 52.7 ± 3.5 | 44.6 ± 3.5 | 39.7 ± 1.5 | ||
| Vitamin A group (B) | 51.6 ± 2.9 | 43.3 ± 1.7 | 38.5 ± 1.3 | ||
| TCDD group (C) | 62.3 ± 3.8ab | 54.1 ± 2.3ab | 53.1 ± 2.2ab | ||
| Vitamin A + TCDD group (D) | 59.4 ± 3.5ab | 44.7 ± 1.9c | 47.7 ± 0.8abc | ||
| Estimate of relative liver toxityd | |||||
| Control group (A) | − | − | − | ||
| Vitamin A group (B) | − | − | − | ||
| TCDD group (C) | + | ++ | +++ | ||
| Vitamin A + TCDD group (D) | + | + | + | ||
Days after administration of TCDD | |||||
|---|---|---|---|---|---|
| 14 | 28 | 42 | |||
| Relative liver weights (g/kg) | |||||
| Control group (A) | 52.7 ± 3.5 | 44.6 ± 3.5 | 39.7 ± 1.5 | ||
| Vitamin A group (B) | 51.6 ± 2.9 | 43.3 ± 1.7 | 38.5 ± 1.3 | ||
| TCDD group (C) | 62.3 ± 3.8ab | 54.1 ± 2.3ab | 53.1 ± 2.2ab | ||
| Vitamin A + TCDD group (D) | 59.4 ± 3.5ab | 44.7 ± 1.9c | 47.7 ± 0.8abc | ||
| Estimate of relative liver toxityd | |||||
| Control group (A) | − | − | − | ||
| Vitamin A group (B) | − | − | − | ||
| TCDD group (C) | + | ++ | +++ | ||
| Vitamin A + TCDD group (D) | + | + | + | ||
Note. Data are expressed as arithmetic means ± SD (n = 5).
p < 0.05, significant difference compared with the control group at the same time point.
p < 0.05, significant difference compared with the vitamin A group at the same time point.
p < 0.05, significant difference compared the TCDD group at the same time point.
Toxicity as assessed in the report by Smith et al. (1998).
Either single or repeated exposures to TCDD caused liver damage with hepatocyte hydropic changes, necrosis, and inflammatory cell infiltration (Fig. 1). In single-exposed mice, the liver damage aggravated with time, reached the most severe after 14 days, and began to ameliorate after 28 days; in repeated exposed mice, the liver damage showed an aggravating tendency, and reached its peak after 42 days. In mice treated with TCDD + vitamin A, the liver damage was less severe than that in TCDD-treated mice in both experiments (Tables 2 and 3).
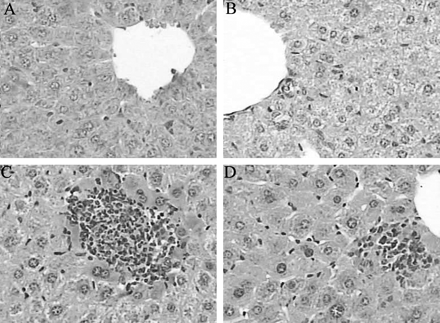
Histology of liver of the mice killed after 7 days: control (A), vitamin A (B), a single oral dose of 40 μg /kg TCDD (C), and vitamin A + single oral dose of 40 μg /kg TCDD (D). Control and vitamin A-treated livers demonstrate normal histology with sinusoids clearly visible between plates of uniform hepatocytes. TCDD-treated liver shows focal necrosis with inflammatory cell infiltration. In vitamin A + TCDD–treated mice, the magnitude of liver injury is less severe than that in the TCDD-treated mice. Original magnification 300×.
Hepatic EROD Activity
In both experiments, TCDD induced high hepatic EROD activity, while supplement of vitamin A had no apparent effect. In Experiment I, the EROD activity began to increase as early as 1 day following single exposure to TCDD, peaked after 3 days, and remained high until 28 days. The EROD activity increased in TCDD-treated mice by 42-fold over the control mice at 7 days. In Experiment II, repeated TCDD treatment increased EROD activity with time and reached a 44-fold over the control mice at 42 days. In both experiments, the EROD activity in TCDD + vitamin A–treated mice was lower than that in the TCDD-treated mice. The EROD activities in TCDD + vitamin A–treated mice was 17% lower 7 days after single exposure to TCDD and 28% lower (p < 0.05) after 42 days repeated treatment of TCDD, compared with the TCDD-treated mice, respectively (Fig. 2).
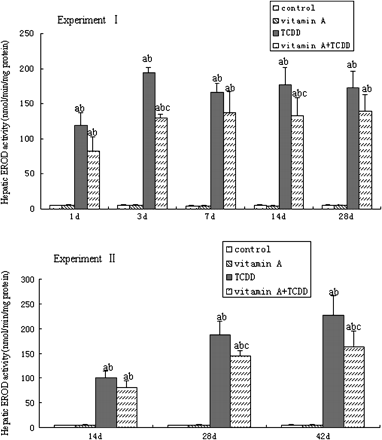
Time course of hepatic CYP1A1 activities in male C57BL/6 mice with different treatments. All bars are expressed as arithmetic means ± standard deviation (n = 4). ap < 0.05, significant difference compared with the control group. bp < 0.05, significant difference compared with the vitamin A group. cp < 0.05, significant difference compared with the TCDD group.
Hepatic CYP1A1 mRNA Expression
Mice liver CYP1A1 mRNA expression was quantified by real-time PCR (Fig 3). TCDD significantly increased the expression of liver CYP1A1 mRNA in both experiments, however, supplement of vitamin A showed no obvious effects on it. In Experiment I, CYP1A1 mRNA expression began to increase as early as 1 day following single exposure to TCDD, peaked after 7 days and remained high until 28 days. CYP1A1 mRNA expression increased by 97-fold in TCDD-treated mice over the control mice at 7 days. In Experiment II, repeated TCDD treatment increased CYP1A1 mRNA expression with time and approached to 47-fold over the control mice at 42 days. In both experiments, the CYP1A1 mRNA expression in TCDD + vitamin A–treated mice was lower than that in the TCDD-treated mice. The magnitude of CYP1A1 mRNA expression in TCDD + vitamin A–treated mice was 25% lower 3 days after single exposure to TCDD and 68% lower (p < 0.05) after 42 days of repeated treatment with TCDD, compared with the TCDD-treated mice, respectively.
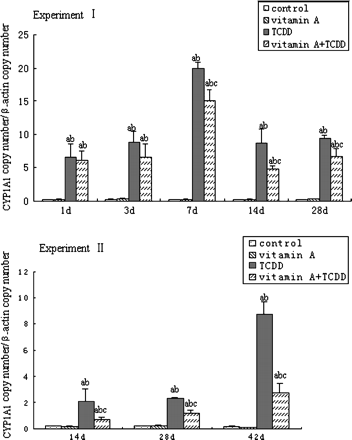
Time course of CYP1A1 mRNA expression in mouse liver. All bars are expressed as arithmetic means ± standard deviation (n = 4). ap < 0.05, significant difference compared with the control group. bp < 0.05, significant difference compared with the vitamin A group. cp < 0.05, significant difference compared with the TCDD group.
Hepatic CYP1A1 Protein Expression
Hepatic CYP1A1 protein expression was detected by Western blotting (Fig. 4). CYP1A1 protein was not detectable in the livers treated with vehicle (control) or vitamin A at each checking time. The magnitude of positive bands in TCDD-treated mice was significantly higher than that in the mice treated with TCDD + vitamin A. CYP1A1 protein levels increased and peaked at the time of 7 or 42 days after TCDD dosing in two experiments, respectively, while supplementation with vitamin A reduced such increase.
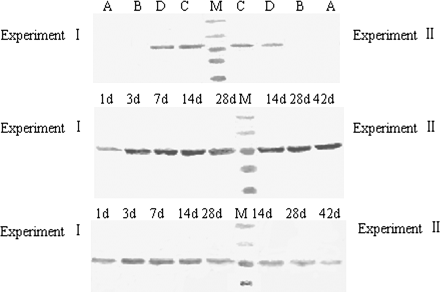
CYP1A1 protein expression in mouse liver detected by western blotting. 15 μg of total microsomal protein was loaded in each lane and resolved on a 10% SDS–PAGE gel, transferred to nitrocellulose membrane, and incubated with rabbit polyclonal antibody for the CYP1A1 protein (Santa Cruz). The positive bands representing CYP1A1 protein were developed using the Western Blue Stabilized Substrate for Alkaline Phosphatase (Promega). Top: Immunoblots of CYP1A1 in representative mouse administered vehicle (control, line A), vitamin A (line B), TCDD (line C), or vitamin A + TCDD (line D) and killed after 14 days with a single oral dose of 40 μg/kg TCDD (left) and with repeated exposure to 0.1 μg/kg TCDD (right). Middle: Time course of CYP1A1 protein expression in TCDD-treated mice. Bottom: Time course of CYP1A1 protein expression in vitamin A + TCDD–treated mice.
Hepatic AhR mRNA Expression
AhR mRNA expression in mice livers was quantified by real-time PCR (Fig. 5). TCDD significantly increased the expression of AhR mRNA in both experiments; however, supplements of vitamin A did not affect it at each checking time. In Experiment I, hepatic AhR mRNA expression began to increase as early as 1 day following single exposure to TCDD, peaked after 7 days, and remained high until 28 days. The magnitude of AhR mRNA expression was increased by 4.5-fold in TCDD-treated mice over the control mice after 7 days. In Experiment II, TCDD increased AhR mRNA expression with time and approached 3.3-fold over the control mice after 42 days. In both experiments, the AhR mRNA expression in TCDD + vitamin A–treated mice was lower than that in the TCDD-treated mice. The magnitude of AhR mRNA expression in TCDD + vitamin A–treated mice was 52.6% lower (p < 0.05) 3 days after single exposure to TCDD and 43.8% lower (p < 0.05) after 28 days repeated treatment of TCDD, compared with the TCDD-treated mice, respectively.
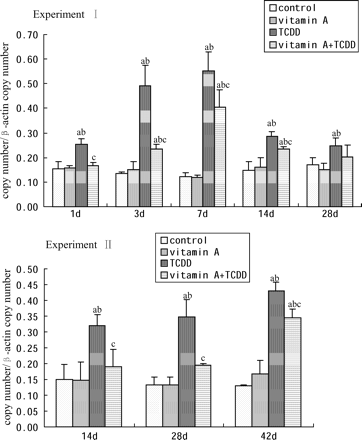
Time course of AhR mRNA expression in mouse liver. All bars are expressed as arithmetic means ± standard deviation (n = 4). ap < 0.05, significant difference compared with the control group. bp < 0.05, significant difference compared with the vitamin A group. cp < 0.05, significant difference compared with the TCDD group.
Hepatic ARNT mRNA Expression
Liver ARNT mRNA expression in mice was quantified by real-time PCR (Fig. 6). Though TCDD increased the expression of ARNT mRNA, the differences were not significant compared with the control mice. Vitamin A did not affect the expression of ARNT mRNA during the test.
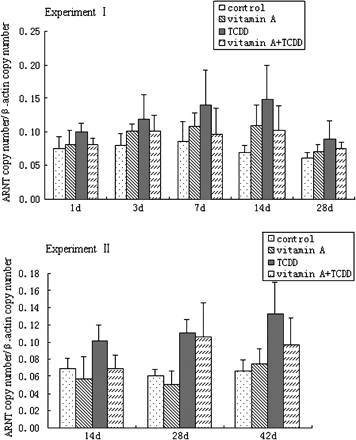
Time course of ARNT mRNA expression in mouse liver. All bars are expressed as arithmetic means ± standard deviation (n = 4). There was no significant difference in the expression of ARNT mRNA compared with the control group at the same time point (p > 0.05).
DISCUSSION
Exposure to TCDD results in various adverse biological effects in experimental animals, such as hepatic damage, thymic atrophy and immune dysfunction, and several types of cancer (Pohjanvirta and Tuomisto, 1994). Changed vitamin A homeostasis may be related to some TCDD-induced toxicity, such as the wasting syndrome (Fletcher et al., 2001; Zile, 1992). Stohs et al. (1984) found that vitamin A abolished the hepatic lipid peroxidation caused by TCDD, but exhibited little influence on the lethality induced by TCDD. In this study, we found that both a single oral dose of 40 μg/kg or repeated dose of 0.1 μg/kg TCDD significantly increased the relative liver weight and caused liver damage, and that the liver damage in TCDD + vitamin A–treated mice was less severe than that in the TCDD-treated mice, which demonstrates that supplementation of vitamin A attenuates the liver damage caused by TCDD.
TCDD induces CYP1A1 activity and gene expression in various tissues (Charles and Shiverick, 1997; Kress and Greenlee, 1997). Increased CYP1A1 expression may contribute toward some toxic effects of TCDD (Nebert, 1991). Both CYP1A1 mRNA and protein are virtually undetectable in untreated animals or cells, but following exposure to TCDD, can be among the most abundant in the cell (Nebert, 1989). Experiments showed that antisense blockade of CYP1A synthesis in developing fish embryos attenuated TCDD-induced developmental toxicity, supporting the idea that CYP1A family member's gene expression contributes to TCDD-induced toxicity (Teraoka et al., 2003). Uno et al. (2004) observed that CYP1A1 (−/−) mice, which lack a functional CYP1A1 gene and have no overt phenotype (Dalton et al., 2000), are partially resistant to high-dose TCDD-induced toxicity, including hepatocyte hypertrophy. The present study demonstrated that TCDD (both a single dose of 40 μg/kg and repeated doses of 0.1 μg/kg TCDD) strongly induced the hepatic microsomal CYP1A1 enzyme activity and expression in mice, and the liver damage was preceded by induction of CYP1A1. Therefore, the TCDD-induced CYP1A1 might contribute to liver damage. Modulation of CYP1A1 activity and expression influences the biological effects of environmental pollutants. Hence, the decrease in the activation of carcinogens through modulation of CYP1A1 activity has been hypothesized as a possible preventive mechanism. The inhibitory ability of retinoids (all-trans-retinol, all-trans-retinal, all-trans-retinoic acid, and retinol-palmitate) on CYP1A1-dependent 7-ethoxycoumarin deethylation was demonstrated (Inouye et al., 1999). Huang et al. (1999) also reported that the preventive effect of retinoids on B(a)P-induced carcinogenesis might be partially due to their specific inhibition of B(a)P activation catalyzed by CYP1A1. In this study, we found that supplementation of vitamin A not only attenuated the increased hepatic EROD activity by TCDD, but also reduced the CYP1A1 mRNA and protein expression induced by TCDD. There was no increase in relative liver weight 28 days after treatment with TCDD and vitamin A compared to control groups. Yet there was marked enzyme induction at this time point. Based on our findings, vitamin A may reduce the liver damage caused by TCDD by complex mechanisms, including attenuation of TCDD-induced CYP1A1 expression.
Transcriptional activation of CYP1A1 induced by TCDD is mediated via AhR, a cytosolic protein that belongs to the basic helix-loop-helix protein family. Upon ligand binding, AhR translocates to the nucleus, where it binds to a nuclear transcription factor, ARNT. The heterodimer of AhR and ARNT binds to consensus sequences in the regulatory domains of the CYP1A1 gene and stimulates its transcription (Hankinson, 1995; Schmidt and Bradfield, 1996). Huang et al. (2003) reported that single oral dose of 50 μg/kg TCDD increased AhR mRNA expression significantly, 7 days after dosing, but did not alter the ARNT mRNA expression in mouse liver. In this study, we also found that both a single oral dose of 40 μg/kg or repeated exposure of 0.1 μg/kg TCDD, increased the expression of AhR mRNA, but did not affect the expression of ARNT mRNA in mouse liver. Moreover, the time course of CYP1A1 mRNA and AhR mRNA were similar. Wanner et al. (1996) observed that all-trans-RA, as the biologically active form of vitamin A, could decrease the expression of the AhR and ARNT in proliferative keratinocytes. In our study, we also found that supplementation of vitamin A significantly attenuated the increase in AhR mRNA induced by TCDD. Therefore, vitamin A may decrease the TCDD-increased CYP1A1 expression through the reduction in AhR mRNA. AhR, as a ligand-activated transcription factor, may induce the expression of many different genes. It is possible that the reduction in AhR expression might have an ameliorative effect on liver damage caused by TCDD in a CYP1A1-dependent and independent manner.
Most functions of vitamin A are mediated through the binding of its active metabolite, RA, to nuclear receptors (Chambon, 1996). Although TCDD could increase the all-trans-RA contents in rats (Schmidt, 2003), TCDD prevents some gene expression regulated by retinoic acid (Krig et al., 2002). Moreover, Inouye et al. (1999) pointed out that the inhibitory ability of all-trans retinoic acid on CYP1A1-dependent 7-ethoxycoumarin deethylation is not as strong as that of all-trans-retinol. In this paper, there was no difference in the all-trans-RA contents between the TCDD-treated mice and the mice treated by TCDD and vitamin A (data not shown), yet there was marked CYP1A1 and AhR mRNA induction. Therefore, it is still an open question how vitamin A inhibits the TCDD-induced CYP1A1 or AhR mRNA expression.
This study demonstrated that both single oral dose of 40 μg/kg and repeated doses 0.1 μg/kg TCDD could increase the AhR mRNA expression and the CYP1A1 expression and activity, which might in turn be involved in TCDD-induced liver damage. Supplementation of vitamin A could partially inhibit the expression of CYP1A1 and AhR and attenuate the liver damage caused by TCDD.
This study was supported by Natural Science Foundation Grant (Z02037) of Education Department of Guangdong Province and Natural Science Foundation Grant (021221) of Guangdong Province.
References
Bradford, M. M. (
Charles, G. D., and Shiverick, K. T. (
Chambon, P. (
Dalton, T. P., Dieter, M. Z., Matlib, R. S., Childs, N. L., Shertzer, H. G., Genter, M. B., and Nebert, D. W., (
Fletcher, N., Hanberg, A., and Håkansson, H. (
Flodstrom, S., Busk, L., Kronevi, T., and Ahlborg, U. G. (
Gonzalez, F. J., and Gelboin, H. V. (
Håkansson, H., Manzoor, E., and Ahlborg, U. G. (
Hankinson, O. (
Honkakoski, P., and Negishi, M. (
Huang, D. Y., Ohnishi, T., Jiang, H., Furukawa, A., and Ichikawa, Y. (
Huang, P., Ceccatelli, S., Hoegberg, P., Sten, Shi, T. J., Håkansson, H., and Rannug, A. (
Inouye, K., Mae, T., Kondo, S., and Ohkawa, H. (
Kress, S., and Greenlee, W. F. (
Krig, S. R., Chandraratna, R. A., Chang, M. M., Wu, R., and Rice, R. H. (
Maden, M., Gale, E., and Zile, M. (
Morris, D. L., Jeong, H. G., Jordan, S. D., Kaminski, N. E., and Holsapple, M. P. (
Nebert, D. W., (
Nebert, D. W. (
Nelson, D. R., Koymans, L., Kamataki, T., Stegeman, J. J., Feyereisen, R., Waxman, D. J., Waterman, M. R., Gotoh, O., Coon, M. J., Estabrook, R. W., et al. (
Okey, A. B., Riddick, D. S., and Harper, P. A. (
Pohjanvirta, R., and Tuomisto, J. (
Schmidt, J. V., and Bradfield, C. A. (
Schmidt, C. K., Hoegberg, P., Fletcher, N., Nilsson, C. B., Trossvik, C., Håkansson, H., and Nau, H. (
Smith, A. G., Clothier, B., Robinson, S., Scullion, M. J., Carthew, P., Edwards, R., Luo, J., Lim, C. K., and Toledano, M. (
Stohs, S. J., Hassan, M. Q., and Murray, W. J. (
Shou, M., Korzekwa, K. R., Crespi, C., Gonzalez, F. J., and Gelboin, H. V. (
Teraoka, H., Dong, W., Tsujimoto, Y., Iwasa, H., Endoh, D., Ueno, N., Stegeman, J. J., Peterson, R. E., and Hiraga, T. (
Uno, S., Dalton, T. P., Sinclair, P. R., Gorman, N., Wang, B., Smith, A. G., Miller, M. L., Shertzer, H. G., and Nebert, D. W. (
Wanner, R., Panteleyev, A., Henz, B. M., and Rosenbach, T. (
Wei, X., Loi, C. M., Jarvi, E. J., and Vestal, R. E. (
Whitlock, J. P. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments