





Abstract
In a striking contrast, matK is one of the most rapidly evolving plastid genes and also one of the few plastid genes to be retained in all nonphotosynthetic plants examined to date. DNA sequences of this region were obtained from photosynthetic and nonphotosynthetic plants of Orobanchaceae and their relatives. The resulting plastid DNA phylogeny was congruent with that recently obtained from analyses of rps2 and provided much better resolution. This phylogeny was then used to examine the relative degrees of evolutionary constraint of both the matK gene and the non–protein-coding regions that flank it inside the trnK intron. The method of subtree contrasts was introduced to compare levels of constraint. matK has evolved with a low but significant level of constraint on its amino acid sequence in both photosynthetic and nonphotosynthetic plants. Constraint is greater in photosynthetic than in nonphotosynthetic plants of this group. Domain X, thought to contain the active site of the protein, is not significantly more constrained than the rest of the protein. The portions of the flanking regions that are thought to form paired stem structures also show constraint, but in this case, there is no significant difference in degree of constraint between photosynthetic and nonphotosynthetic plants.
Introduction
If a gene is important to the fitness of an organism, deleterious mutations will be eliminated from populations by purifying selection (Li and Graur 1991 ). In protein-coding genes, purifying selection results in relatively fewer interspecific differences at nonsynonymous sites than at synonymous sites. This is an indication that the protein's amino acid sequence is evolving under conditions of functional constraint.
Parasitic plants provide evolutionary genetic mutants for investigation of gene function. The cessation of photosynthetic activity that has occurred in some parasites provides us with a valuable natural experiment—an opportunity to examine the genetic consequences of the shutdown of a major (perhaps the major) physiological process of most green plants (dePamphilis 1995 ; dePamphilis, Young, and Wolfe 1997 ). Genes not known to have an essential function outside of photosynthesis can be examined to see if they are still maintained in nonphotosynthetic plants (Wolfe, Morden, and Palmer 1992 ; dePamphilis 1995 ). Indeed, some of the plastid (chloroplast) genes of these plants have been lost or have become pseudogenes (dePamphilis 1995 ; Nickrent et al. 1998 ). If the genes have remained intact, their sequences and gene products can be examined for insight into whether they still have value to the plant.
In these nonphotosynthetic plants, genes on the plastid (chloroplast) genome may be much less constrained than are the same genes in photosynthetic plants. Nearly all of the plastid genome is composed of genes either directly involved in photosynthesis or involved in transcription and translation of plastid genes. Many plastid genes, including all those encoding photosynthetic proteins, are known to be completely expendable in at least one nonphotosynthetic plant, for they have been lost or have become pseudogenes in Epifagus virginiana (dePamphilis and Palmer 1990 ; Wolfe, Morden, and Palmer 1992 ). The monospecific Epifagus is holoparasitic, meaning that it has lost photosynthetic ability and derives its nutritional needs from other plants. It is a member of the family Orobanchaceae in the order Lamiales, and it has lost over half of the plastid DNA relative to Nicotiana, a photosynthetic member of the closely related order Solanales. The phylogeny of Epifagus and its relatives (dePamphilis, Young, and Wolfe 1997 ; Young, Steiner, and dePamphilis 1999 ) reveals several separate losses of photosynthesis in Orobanchaceae, which has recently been defined to include both photosynthetic and holoparasitic plants (Young, Steiner, and dePamphilis 1999 ). However, some genes are present as intact open reading frames (ORFs) in all plants of this group, even Epifagus. Although present in the holoparasites, these genes might experience less translational demand than in photosynthetic plants, due to the diminished role of the plastid in the holoparasites (Morden et al. 1991 ). Thus, purifying selection may be relaxed in these genes.
One of these persistent genes, matK, is found in every species we have examined. Despite this, its sequence is one of the least conserved of plastid genes (Olmstead and Palmer 1994 ; Soltis and Soltis 1998 ). matK is a popular choice for plant systematics studies, from the interspecific level (Johnson et al. 1996 ) to the ordinal level (Johnson and Soltis 1995 ; Hilu and Liang 1997 ). The coding region of matK (∼1,500 bp) and its flanking regions of a few hundred base pairs of DNA on each side together constitute a group II (lariat-forming) intron residing in the trnK gene (fig. 1).
In Epifagus, the matK gene is a full-length ORF. However, it is no longer inside an intron, because the trnK exons have been deleted, leaving matK a freestanding gene (Wolfe, Morden, and Palmer 1992 ). The plastid genome of Epifagus has experienced many other deletions as well. Only 42 plastid genes remain, compared with 112 genes in a typical photosynthetic plant such as Nicotiana tabacum (Wolfe, Morden, and Palmer 1992 ). In addition to the photosynthetic genes, a number of genes involved in gene expression have also been lost, including about half of the tRNAs, trnK among them. matK, however, appears to have been retained to aid in the splicing of other group II introns of the plastid genome (Ems et al. 1995 ). The persistence of matK, even in Epifagus, whose plastid genome has undergone such drastic change, raises the possibility that its function may be very important, even in holoparasites. If so, we might expect that relaxation of purifying selection has not occurred. Natural selection may continue to impose constraints on the evolution of this molecule, and these constraints may be similar to those found in related photosynthetic plants.
In photosynthetic plants, and in most holoparasites as well (except for Epifagus), the DNA of the trnK intron codes for two functions. It codes for a splicing enzyme, maturase K (MatK), and because the intron is a “self-splicing” ribozyme, the regions flanking the matK gene have a folded RNA structure thought to be required for splicing (Mohr, Perlman, and Lambowitz 1993 ). Although some group II introns have been observed to self-splice in vitro, they probably all require a chaperoning maturase to splice in vivo (Michel, Umesono, and Ozeki 1989 ; Mohr, Perlman, and Lambowitz 1993 ). MatK is thought to be distantly related to other maturases contained in other group II introns, but although many of these have reverse transcriptase (RT) domains and a domain X, the MatK protein has only the domain X and a small remnant of the RT domain. Thus, although the ancestor of MatK probably had reverse transcriptase ability, MatK has lost it. The domain X occupies a location similar to that of the “thumb” and “connection” domains of the HIV-1 RT, indicating a probable role in the binding of the intron RNA for splicing. In 34 published group II intron sequences from all kingdoms of organisms, both domain X and the remnant (regions V, VI, and VII) of the RT domain were more conserved than the rest of MatK (Mohr, Perlman, and Lambowitz 1993 ).
If purifying selection is conserving the function of the MatK protein, we expect the nonsynonymous substitution rate to be lower than the synonymous substitution rate in the protein-coding region. Likewise, if substitution rates in paired-stem regions of the RNA structure are lower than substitution rates in loops, it would indicate constraint due to RNA function.
Several methods exist for comparing amounts of purifying selection. Some are based on two-taxon comparisons (Zhang, Kumar, and Nei 1997 ). A more complete use of phylogenetic data is made in tree-based tests (Takezaki, Rzhetsky, and Nei 1995 ; Yang 1998 ; Suzuki and Gojobori 1999 ). Some of these tests (Yang 1998 ; Suzuki and Gojobori 1999 ) are designed for protein-coding regions; the other is a distance tree-based test for sister groups (Takezaki, Rzhetsky, and Nei 1995 ). Only part of the region we wish to analyze is protein-coding. We introduce a parsimony tree-based method, “subtree contrasts,” which can be used for coding and noncoding regions and is not limited to sister group comparisons.
Materials and Methods
Sampling
The holoparasitic and hemiparasitic (plants that parasitize other plants but also conduct photosynthesis) relatives of Epifagus together form a monophyletic group (dePamphilis, Young, and Wolfe 1997 ). The sister group to this clade contains the nonparasitic genus Lindenbergia. Together, the parasitic clade and Lindenbergia comprise the Orobanchaceae, as recently defined (Young, Steiner, and dePamphilis 1999 ). We will use the common name “broomrapes” to refer to the narrower Orobanchaceae sensuCronquist (1981) . Phylogenetic analyses (Young, Steiner, and dePamphilis 1999 ) have revealed seven Orobanchaceae clades and four close outgroup clades, all of which were sampled, as well as three more distant outgroups (fig. 2 ), for a total of 28 species. For all but three species, voucher locations are given in Young, Steiner, and dePamphilis (1999). For Harveya bolusii Kuntze, Hyobanche atropurpurea Bolus, and Hyobanche sanguinea L. (all from the Republic of South Africa), vouchers are on deposit at the Pennsylvania State University.
Sequencing
Most of the sequences were generated using an ABI 377 autosequencer (P.E. Biosystems) according to the manufacturer's instructions. However, some sequence data were generated manually with the Sequenase (U.S. Biochemicals) double-stranded method (dePamphilis, Young, and Wolfe 1997 ). Both strands were sequenced and translated to verify that the protein-coding regions contained no internal stop codons. Sequencing primers and PCR conditions were the same as in Young, Steiner, and dePamphilis (1999), with the exception that three additional primers (matK-79R, 5′-CATAGTGCGATANAGYCAAAACAAG-3′; matK319R, 5′-CCACAATAAAAGCAAAYCCCTCTG-3′; matK1449F, 5′-ATTGGAAGGATTTYTTATGTCGG-3′) were routinely used, and two additional primers were used for the broomrapes (matK810F-OR, 5′-TYTTGTGAATGTTTTTGTAAAGG-3′; matK1356R-OR, 5′-GAGTATTTTTGTGTTTCCGAGCC-3′). Sequences of nearly the entire trnK intron were generated for most taxa; three photosynthetic taxa (Tozzia, Verbascum, and Veronica) have so far only been sequenced for the matK coding region. The matK gene of Epifagus is homologous to that of photosynthetic plants, but the flanking regions are not (see Ems et al. [1995] and Results). Boschniakia strobilacea did not yield an amplification product using the usual PCR primers located in the two trnK exons, so a smaller region was amplified and sequenced using internal primers. All sequences have been deposited in GenBank (AFO51977–AFO52003, CHNTXX, EPFCPCG). The aligned data set is also available (http://depcla4.bio.psu.edu/trnKintron).
Alignment
In several previous studies (e.g., Soltis et al. 1996 ; Plunkett, Soltis, and Soltis 1997 ; Xiang, Soltis, and Soltis 1998 ), indels within the matK coding region were easy to align by eye, but in one (Gadek, Wilson, and Quinn 1996 ), indel-containing regions were removed from the phylogenetic analysis.
In this study, the data set for the trnK intron (which includes the matK gene) includes at least 96 indels, making sequence alignment a complex task. Because coding and flanking regions are expected to have different evolutionary dynamics, the matK gene was aligned separately from the flanking regions. Many alignments were generated for each region using CLUSTAL W (Thompson, Higgins, and Gibson 1994 ). The one best alignment was chosen for each region using a phylogenetic optimality criterion (Wheeler 1995 ). Specifically, we chose the alignment that produced parsimony trees with the least homoplasy, as judged by the rescaled consistency index (RC; Farris 1989 ). For the coding-region alignment, we explored gap weights (G) of 5–17 and gap extension weights (E) of 1–7, with and without transition/transversion (ti/tv) weighting. In the flanking regions, because the lack of a reading frame was assumed to permit more frequent indel events, we explored G = 2–10 and E = 1–5, with and without ti/tv weighting. In these analyses, gaps were coded as indel characters (see Phylogenetic Analysis, below). Alignments were also refined manually and compared against the CLUSTAL alignments.
Phylogenetic Analysis
Maximum-parsimony analyses of nearly the entire intron were conducted, beginning 19 bp after trnK exon 1 and ending 26 bp before trnK exon 2 (fig. 1 ). This region was approximately 2,420 bp long. Separate analyses of the coding region and of the combined flanking regions were conducted. Additional indel characters were included (23 characters for the coding region and 73 for the flanking regions). Simple gaps (those that did not overlap with other gaps) were each coded as single characters, regardless of length. Complex regions of overlapping gaps were found in both coding and flanking regions. Each of these was coded as a single multistate, unordered character (Baum, Sytsma, and Hoch 1994 ). The full data set was also analyzed without these indels to determine their effect. PAUP* was used for both analyses, utilizing 1,000 random-addition replicates and tree bisection-reconnection (TBR) branch swapping. Bootstrap support (Felsenstein 1985 ) was calculated for each node on the phylogeny of the entire data set.
Constraint Analysis
In order to compare levels of constraint, we developed a parsimony-based method called “subtree contrasts.” The branch segments of the phylogeny (fig. 2 ) were subdivided into subtrees (fig. 3 ). In this case, we wanted to compare evolutionary changes that occurred in photosynthetic plants with those changes that occurred in holoparasitic plants, so this became the basis of our subtree choice. The photosynthetic subtree consisted of branches connecting all taxa except the holoparasites (those shown in bold in fig. 2 ). Because slightly different taxa were included in the coding-region and flanking-regions analyses, the subtrees differed slightly (fig. 3A and D ). Two holoparasitic subtrees were used in each analysis. The broomrape subtree consisted of the branches connecting species of Epifagus, Orobanche, and Boschniakia (fig. 3B and E ). The Harveya-Hyobanche subtree consisted of the branches connecting those taxa (fig. 3C and F ). In practice, the subtrees were generated by deleting taxa in PAUP*.
The actual changes observed on photosynthetic versus holoparasitic subtrees were considered samples of photosynthetic and holoparasitic evolution, respectively, and statistical comparisons were made. In doing so, the characters were divided into contrasting subsets, such as third positions versus first and second positions combined, or stem-paired region characters versus loop characters. The number of changes (number of steps) per character was then generated using the DIAG option of the DESCRIBE command in PAUP*. Each character's number of steps was used as an observation of the average number of steps per character over a chosen portion of the phylogeny (subtree) and subset of characters. Because every character contributes an independent observation, this method has great power. The output from the DIAG option was imported into JMP (SAS Institute 1994 ) to generate means and perform statistical tests.
Analysis of the Protein-Coding Region
To examine constraint on protein evolution, we used the coding region only and compared changes at third codon positions, which were mostly (∼70%) synonymous, with changes at first and second positions combined, which were mostly (∼98%) nonsynonymous. Assuming that third codon positions were evolving in an unconstrained manner, tests of constraint were conducted by testing whether the mean number of steps at third codon positions differed from the mean at first and second positions combined. Because the number of steps per character was not normally distributed, but followed a Poisson distribution, t-tests and linear regressions could not be used. A more suitable test, Wilcoxon's nonparametric test, was used. The data take the form of a list of DNA sequence positions, each with two variables. The first tells whether the position belonged to codon position type “first and second codon positions” or “third codon positions.” The second contains the number of steps. The test, in effect, asks whether there are two distributions for base changes relative to the two codon position types. The following questions were addressed: Are the bases at third codon positions changing more frequently than those at first and second positions for the photosynthetic taxa? For holoparasites? For the broomrapes alone? A “yes” to any of these questions was taken as evidence of constraint in the group examined.
We also tested whether molecular evolution in the holoparasites was less constrained than that in the photosynthetic taxa. Subtree contrasts were made using the ordinal logistic regression model (Sokal and Rohlf 1995 , pp. 767–778; Sall and Lehman 1996 , pp. 224–234). The data set was constructed with a list of positions. One variable was called GH and had a value of G for the data from the photosynthetic subtree and H for the data from the holoparasitic subtrees. The third variable (POS) told whether the position belonged to the class “first and second codon positions” or “third codon positions.” The dependent variable was ordinal and contained the number of steps. Because so few characters had more than three steps, the “number of steps” variable was divided into four categories: 0, 1, 2, and >2. It is the significance of the interaction effect that determines whether the degrees of constraint differ, for example, among photosynthetic and holoparasitic plants of this data set. This method allows comparison of different subtrees that may have different ages, numbers of species, or amounts of molecular evolution.
The distribution of amino acid changes along the gene was generated for photosynthetic and holoparasitic subtrees. Each amino acid was coded as variant (1) or invariant (0), and a correlation was sought between the distributions based on the two subtrees.
Analysis of Flanking Regions
The same subtree contrast method was used to look for constraint on the RNA secondary structure. The flanking region was divided into bases involved in paired stem structures (stem-pair bases) and others (nonstem bases). For all taxa in this study, stem-pairing was assumed to occur as in Nicotiana (Michel, Umesono, and Ozeki 1989 ). If RNA secondary structures are not conserved, the statistical tests will be weakened but may still be revealing. We also assumed (for the purposes of the test) that nonstem bases were not constrained in their evolution. If this assumption is false, and they are constrained, then the chance of detecting significant levels of constraint in the stem-pair bases is weakened but may still show whether levels of constraint differ in the two regions. Once again, the phylogeny was based on the entire intron, but the five taxa that were not sequenced for the flanking regions were removed prior to identifying subtrees. The same set of constraint tests was performed, this time on stem-pair bases versus nonstem bases.
Coding Versus Flanking Regions
To compare substitution rates in the coding region with those of the flanking regions, we chose the subtree of photosynthetic taxa for which all regions were sequenced (fig. 3D ). We also eliminated regions containing indels, because including these might underestimate rates. Again, we used the subtree contrasts method to test whether coding regions and flanking regions differed in their average substitution rates.
Results
PCR Amplification, Alignment, and Phylogeny
Of the 58 taxa of Lamiales tested so far, only four (E. virginiana, Conopholis americana, B. strobilacea, and Cistanche phelypaea) consistently fail to allow the amplification of the trnK region. This could be an indication that these taxa (all Orobanchaceae holoparasites) lack the trnK exons, as we know to be the case for Epifagus (Morden et al. 1991 ; Wolfe, Morden, and Palmer 1992 ). It could also be that the primer-binding sites have diverged too much for successful amplification. Using internal primers, we obtained sequences of the matK ORF from all of these except Conopholis.
When the entire Epifagus plastid sequence was published, matK was reported to be 200 bp shorter at the 5′ end than in other angiosperms (Wolfe, Morden, and Palmer 1992 ). However, Mohr, Perlman, and Lambowitz (1993) noted that a sequencing error resulting in a frameshift may have occurred. We have sequenced matK from an individual from the same Michigan population of Epifagus (dePamphilis and Palmer 1990 ) from which the published sequence derived, as well as an individual from a population from Indiana, and have found both to contain full-length ORFs. With the exception of one nucleotide, our sequence from the Michigan individual agrees with the published sequence, so we use the corrected sequence in our analysis.
The best alignment parameters for the matK gene (under the stated optimality criterion of maximizing RC) were gap weight (G) = 15 and gap extension weight (E) = 5, with a ti/tv weight of ½. For the ∼700 -bp 5′-flanking region and the ∼230-bp 3′-flanking region, which were aligned in a combined analysis, the best parameters were G = 3 and E = 2, with a ti/tv weight of 1/1. Additional efforts to improve the alignment by eye resulted in trees with equal or lower RCs. This alignment shows that length differences are many times more common in the flanking regions than in the coding region (table 1 ).
After alignment, the approximately 2,420 bases provided 2,804 aligned bases. All regions, along with their 96 coded indel characters, were then combined into one data set. The number of parsimony-informative characters was 792 (table 1 ). The flanking regions contained nearly the same density of parsimony-informative characters (0.312/bp) as the coding region (0.336/bp; table 1 ). A single, completely resolved, most-parsimonious tree was found (fig. 2 ) that agreed with previous analyses of plastid rps2 and the matK coding region (Young, Steiner, and dePamphilis 1999 ) and improved on them by resolving the position of Schwalbea and by increased bootstrap support values. The indel characters contain less homoplasy (consistency index [CI] = 0.745) than the substitution characters (CI = 0.573), and their inclusion is required for maximal phylogenetic resolution. Excluding the 96 indel characters yielded two most-parsimonious trees. One was the same as that in figure 2 ; the other differed in the placement of Schwalbea americana, which was placed on the branch between Lindenbergia philippinensis and the other 19 parasites. Excluding all sequence bases in indel-containing regions (1,330 bp) and analyzing the remaining 1,475 bp left only 620 informative characters, and the resulting analysis had more homoplasy (RC = 0.406; CI = 0.564) than the analysis in which indel characters were included (RC = 0.432; CI = 0.585). It also yielded a single most-parsimonious tree, but the topology was inconsistent with those obtained in the other analyses and the rps2 analysis. Analysis of the coding region alone (indels included) gave the same topology as analysis with all regions included. Analysis of the flanking regions alone (indel characters included) gave a different topology from the analysis of all regions. This topology placed Boschniakia hookeri as sister to a clade consisting of Orobanche, Alectra, Striga, Harveya, and Hyobanche, but the two topologies were otherwise the same. For these reasons, the single most-parsimonious tree from the analysis of all regions plus indel characters will thus be referred to as the best phylogeny (fig. 2 ).
Protein-Coding Region
The phylogeny (fig. 2 ) was then used to test for significant differences in substitution rates among codon positions as an indication of the level of constraint on the matK gene. Wilcoxon tests of evolutionary changes on photosynthetic and holoparasitic subtrees (fig. 3 ) were used to examine constraint on the gene, first among photosynthetic plants and then among holoparasites. The photosynthetic subtree had a greater rate of change at third codon positions than at the other positions (χ2 = 50.1, df = 1, P < 0.0001; table 2 ), suggesting that the gene is evolving under constraint. Third-position change averaged 1.7 (±0.4) times the (per-base-pair) change at first and second positions. We then analyzed the holoparasites. Because the number of steps on the Harveya-Hyobanche subtree was too small to analyze separately, the steps on the two holoparasitic subtrees were added together and used to represent holoparasitic evolution. In the holoparasitic subtrees, constraint on matK was also observed (χ2 = 15.2, df = 1, P < 0.0001). In the broomrapes alone, constraint was evident as well (χ2 = 13.2, df = 1, P = 0.0003). Using the interaction effect tests of the ordinal logistic regression model, matK in the holoparasites was significantly less constrained than in photosynthetic plants (χ2 = 4.96, df = 1, P = 0.0259). Likewise, matK in the broomrapes alone was less constrained than in photosynthetic plants (χ2 = 5.85, df = 1, P = 0.0156).
To test for differences in constraint among protein domains, we used the photosynthetic plant subtree, because constraint is more intense in photosynthetic plants. The protein domain X did not differ significantly from the rest of the gene in degree of constraint (χ2 = 2.68, df = 1, P = 0.102). Likewise, the region containing domains V, VI, VII, and X did not differ significantly from the rest of the gene in degree of constraint (χ2 = 1.63, df = 1, P = 0.201).
Although the protein domains tested did not show differences in constraint, when amino acid sites were examined individually, there was an excellent correlation (r2 = 0.205, P < 0.0001) between sites that were invariant on the photosynthetic subtree (fig. 3A ) and those that were invariant over the holoparasitic subtrees (fig. 3B and C ).
Intron RNA Secondary Structure
We then looked for evidence of constraint on the ribozyme structure, comparing the stem-pair regions with nonstem regions. Paired stem regions show less change than nonstem regions in photosynthetic plants (χ2 = 54.9, df = 1, P < 0.0001; table 2 ), holoparasites combined (χ2 = 53.4, df = 1, P < 0.0001), and the broomrapes alone (χ2 = 51.0, df = 1, P < 0.0001). The combined holoparasites do not differ significantly in degree of constraint when compared with photosynthetic plants (χ2 = 0.121, df = 1, P = 0.738). Likewise, the subtree of broomrapes alone does not differ from that of photosynthetic plants in degree of constraint (χ2 = 0.228, df = 1, P = 0.633). For photosynthetic plants, the nonstem regions show 2.3 times the rate of change of the stem-pair regions.
Coding Versus Flanking Regions
We also estimated the relative substitution rates for the various regions using the subtree of photosynthetic plants for which both regions were sequenced (fig. 3D ). For these comparisons, we excluded indel-containing regions. In terms of steps per character over the photosynthetic subtree, which is a relative indication of evolutionary rate, the flanking regions averaged 0.365 and the coding region averaged 0.486. These two values were significantly different (P = 0.0005). Within the flanking regions, the nonstem regions are expected to be relatively unconstrained, as are the third codon positions of the coding region. Nonstem regions averaged 0.474 steps per character, and third codon positions averaged 0.673. These two values were significantly different (P = 0.0003).
Discussion
In a remarkable contrast, the matK gene is one of the most rapidly evolving plastid genes and one of the few protein-coding genes to be retained in Epifagus and the other holoparasites we sequenced. In fact, the only known case in which matK is inactivated is in three related orchid genera, where it appears to be a pseudogene (Jarrell and Clegg 1995 ). Most of the other protein-coding genes retained in Epifagus are involved directly in transcription or translation and may be necessary as long as any plastid genes are expressed. MatK's splicing function may also be required. However, in Epifagus, MatK is not needed to splice trnK, which has been deleted. In these nonphotosynthetic plants, MatK's most vital role may be splicing other group II introns, such as those in rps12, rpl2, and clpP (Ems et al. 1995 ). How Epifagus translates plastid genes without trnK and half of the tRNA genes is still a mystery. However, trnK is retained in many holoparasites, where it may still be an important part of the translational machinery. If so, the need to splice the two trnK exons would also require retention of the intron's ribozyme function.
Parsimony-Based Constraint Analyses
Although we refer to taxa as “photosynthetic” and “holoparasitic” for the sake of convenience, we compared rates of change only in Orobanchaceae and their close relatives. All hypotheses were tested based on a phylogeny generated from the intron sequences themselves. The accuracy of this plastid DNA phylogeny does not depend on these sequences alone, but is confirmed by analyses of the rps2 gene (Young, Steiner, and dePamphilis 1999 ). Trees derived from rps2 are less resolved than those based on matK, but they are congruent.
Protein Constraint
These analyses allowed us to see that both the matK gene and its flanking regions continue to evolve constrained by purifying selection, even after loss of photosynthesis. Our tests show that there is significant constraint on MatK evolution in the photosynthetic plants we examined, but the level of constraint is low, as has been reported for most angiosperms examined (table 3 ). The ratio of third-position changes to the average of the other positions is only 1.7 (±0.4). This is similar to the ratio of 1.4 for matK in the Saxifragaceae (Johnson and Soltis 1995 ) and is a much lower level of constraint than is found in rbcL, which has a ratio of 5.5 in the Saxifragaceae (Johnson and Soltis 1995 ). The level of constraint is not significantly reduced in domain X or domains V, VI, VII, and X. However, similar sites are invariant in the photosynthetic plants and in the holoparasites, suggesting a similarity in the constraint they have experienced.
Consistent with the generally low level of constraint on matK, amino acid variation in general is not concentrated in particular regions of the gene. However, the two spots of lowest variation (28% and 88% of the way from the start codon to the stop codon) are also the two least variable regions detected in a much wider survey of GenBank sequences (Hilu and Liang 1997 ). The second of these spots is within domain X.
Ribozyme Constraint
Both the photosynthetic plants and the holoparasites in this study show constraint on the evolution of the putative paired stem regions. The nonstem regions change 2.3 times as fast as the paired stem regions. The paired stem regions are constrained in the holoparasites at nearly the same level as in their photosynthetic relatives. This suggests that despite the loss of photosynthesis and the deletions that have occurred in the plastid of the holoparasites, the self-splicing ribozyme function of the trnK intron is still important, perhaps because the trnK gene is still necessary. Both matK and the rest of the trnK intron remain selectively constrained in the holoparasites that have them. This fact deepens the mystery behind the loss of trnK in Epifagus (Wolfe, Morden, and Palmer 1992 ).
The comparison of coding and flanking regions shows an intriguing result. Among photosynthetic plants, the substitution rate in the flanking regions is only 54% of that at third codon positions in the coding region. Even when the stem-pair regions are excluded, the remaining nonstem regions have only 70% of the substitution rate of third positions, suggesting that parts of the nonstem regions are also under constraint of some kind. However, indels are much more frequent in the flanking regions (at least 61 per 1,000 bp, as compared with at least 14 per 1,000 bp for the coding region). Selection is constraining substitutions in the flanking region, while still allowing a large number of indels.
The subtree contrasts method presented here can be used to test for differences and two-way interactions whenever a phylogeny can be divided into subtrees. The subtrees can be monophyletic or paraphyletic. For example, warm-blooded and cold-blooded vertebrate subtrees could be tested for differing substitution rates and for differing degrees of constraint on protein evolution. The method could be adapted for the use of synonymous and nonsynonymous sites, rather than the first, second, and third positions used here. It also could be adapted for the use of likelihood-based estimates of branch lengths. When only one subtree in each category is analyzed, the conclusions hold only for those subtrees examined, but if multiple subtrees are available for each category, the capacity for generalization will increase (dePamphilis 1995 ).
Pamela Soltis, Reviewing Editor
Keywords: group II intron purifying selection ribozyme matKEpifagus virginiana, parasitic plant subtree contrasts
Address for correspondence and reprints: Nelson D. Young, Department of Biology, Trinity University, San Antonio, Texas 78212. E-mail: nyoung@trinity.edu.
Table 1 Phylogenetically Informative Characters for matK Coding and Flanking Regions
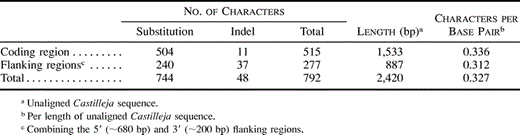
Table 1 Phylogenetically Informative Characters for matK Coding and Flanking Regions
Table 2 Differences Among Regions Provide Evidence of Constraint on Sequence Evolution of matK Coding and Flanking Regions
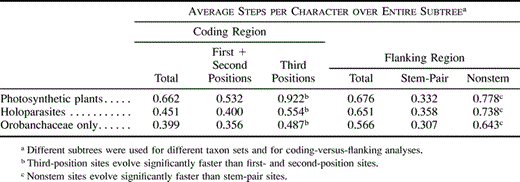
Table 2 Differences Among Regions Provide Evidence of Constraint on Sequence Evolution of matK Coding and Flanking Regions
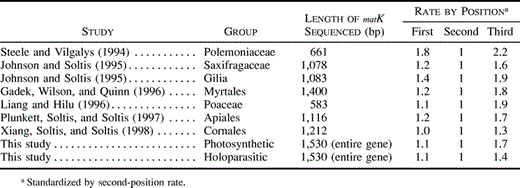
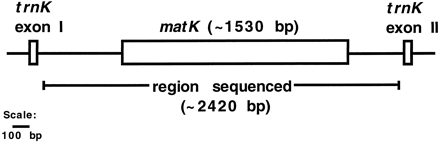
Fig. 1.—Sketch of the plastid gene trnK and its matK-containing intron
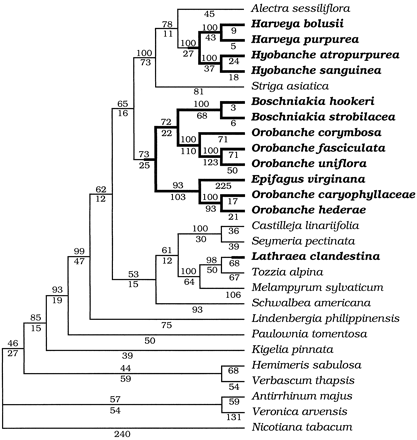
Fig. 2.—Most-parsimonious tree of coding and noncoding regions combined, based on a heuristic search involving 1,000 random-addition replicates and using tree bisection-reconnection (TBR) branch swapping. Bootstrap values (based on 1,000 replicates) are shown above each branch; the branch length (number of changes) is show below. The tree length is 2,926, the rescaled consistency index is 0.432, and the consistency index (excluding uninformative characters) is 0.585. Holoparasites are shown in bold
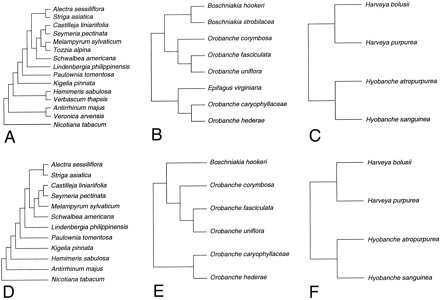
Fig. 3.—Subtrees used to measure amounts of evolutionary change for photosynthetic plants and holoparasites. All of the subtrees were taken from parsimony trees whose topology was based on the analysis of the whole trnK intron. Subtrees A, B, and C were used in the coding-region analysis. Subtree A represented photosynthetic plants, and subtrees B and C represented holoparasites. Likewise, subtrees D, E, and F (same as C) were used in the flanking-regions analysis. In that analysis, subtree D represented photosynthetic plants, and subtrees E and F represented holoparasites
We thank J. Alison, the Missouri Botanical Garden, K. Steiner, L. Musselmann, M. Wetherwax, R. Olmstead, K. Kirkman, R. Wyatt, and J. Armstrong for help in obtaining some of the plants or DNAs used in this study; T. Barkman, J. Leebens-Mack, J. Lyons-Weiler, D. McCauley, O. Pellmyr, S. Kumar, and J. Groman for discussions of the text; and D. Swofford for making PAUP*, version 4.0, available for analyses. Financial support for this research was provided by NSF grant (DEB-9120258) and NSF equipment grant (DBI-9604814) to C.W.D.
literature cited
Baum, D. A., K. J. Sytsma, and P. C. Hoch.
Cronquist, A.
dePamphilis, C. W.
dePamphilis, C. W., and J. D. Palmer.
dePamphilis, C. W., N. D. Young, and A. D. Wolfe.
Ems, S., C. W. Morden, C. Dixon, K. H. Wolfe, C. W. dePamphilis, and J. D. Palmer.
Felsenstein, J.
Gadek, P. A., P. G. Wilson, and C. J. Quinn.
Hilu, K. W., and H. Liang.
Jarrell, D. C., and M. T. Clegg.
Johnson, L. A., J. L. Schultz, D. E. Soltis, and P. S. Soltis.
Johnson, L. A., and D. E. Soltis.
Liang, H., and K. W. Hilu.
Michel, F., K. Umesono, and H. Ozeki.
Mohr, G., P. S. Perlman, and A. M. Lambowitz.
Morden, C. W., K. H. Wolfe, C. W. dePamphilis, and J. D. Palmer.
Nickrent, D. L., C. W. dePamphilis, A. D. Wolfe, A. E. Colwell, N. D. Young, and R. J. Duff.
Olmstead, R. G., and J. D. Palmer.
Plunkett, G. M., D. E. Soltis, and P. S. Soltis.
Soltis, D. E., R. K. Kuzoff, E. Conti, R. Gornall, and K. Ferguson.
Soltis, D. E., and P. S. Soltis.
Steele, K. P., and R. Vilgalys.
Suzuki, Y., and T. Gojobori.
Takezaki, N., A. Rzhetsky, and M. Nei.
Thompson, J. D., D. G. Higgins, and T. J. Gibson.
Wheeler, W. C.
Wolfe, K. H., C. W. Morden, and J. D. Palmer.
Xiang, Q.-Y., D. E. Soltis, and P. S. Soltis.
Yang, Z.
Young, N. D., K. E. Steiner, and C. W. dePamphilis.

{kind=link}
{kind=link}
{kind=link}