




Abstract
The KEGG RPAIR database is a collection of biochemical structure transformation patterns, called RDM patterns, and chemical structure alignments of substrate-product pairs (reactant pairs) in all known enzyme-catalyzed reactions taken from the Enzyme Nomenclature and the KEGG PATHWAY database. Here, we present PathPred (http://www.genome.jp/tools/pathpred/), a web-based server to predict plausible pathways of muti-step reactions starting from a query compound, based on the local RDM pattern match and the global chemical structure alignment against the reactant pair library. In this server, we focus on predicting pathways for microbial biodegradation of environmental compounds and biosynthesis of plant secondary metabolites, which correspond to characteristic RDM patterns in 947 and 1397 reactant pairs, respectively. The server provides transformed compounds and reference transformation patterns in each predicted reaction, and displays all predicted multi-step reaction pathways in a tree-shaped graph.
INTRODUCTION
The complete genome sequence provides an insight into the metabolic capability of a particular organism through the process of metabolic reconstruction, which is based on known metabolic pathways and enzymes involved. We have been offering a web service, KEGG automatic annotation server (KAAS) (1), as a practical implementation of metabolic reconstruction. However, there are still many pathways that are not well characterized, such as biodegradation pathways of environmental compounds and biosynthesis pathways of secondary metabolites. One way to approach this problem is to utilize chemical logic of enzymatic reactions; namely, chemical structure transformation patterns of small molecules.
Such chemical logic is being organized in KEGG (2), especially in the KEGG REACTION and KEGG RPAIR databases (http://www.genome.jp/kegg/reaction/). KEGG REACTION is a collection of all known enzymatic reactions taken from the IUBMB Enzyme Nomenclature (3) and additional reactions taken from the KEGG metabolic pathways. KEGG RPAIR is a derived database containing biochemical structure transformation patterns for substrate–product pairs (reactant pairs) in KEGG REACTION. The biochemical transformation patterns are described by, what we call, the RDM pattern representing KEGG atom type changes at the reaction center atom (R), and its neighboring atoms on the different region (D) and the matched region (M) (4,5). All RDM patterns are manually curated after computationally generating structure alignments of reactant pairs (6).
The data stored in KEGG REACTION have been utilized in PathComp, a program to generate possible reaction paths between two given compounds, by repeatedly applying binary relationships of substrate–product pairs (7). Thus, PathComp is useful only when matching compounds are found in the KEGG REACTION database. In this article, we report a new program, PathPred, which is applicable even when no matching compounds are found in the database. This is because PathPred utilizes local RDM pattern matches reflecting generalized reactions shared among structurally related compounds. Current PathPred implementation is a multi-step reaction prediction server for biodegradation pathways of xenobiotic compounds and biosynthesis pathways of secondary metabolites as a first step towards a more comprehensive service for metabolic reconstruction.
There are other pathway prediction systems available, but they usually require user intervention. For example, the University of Minnesota Biocatalysis/Biodegradation Database (UM-BBD) (8) has a biodegradation pathway prediction system, UM-PPS, based on the structure transformation rules derived from their collection of microbial xenobiotics biodegradation pathways (9). UM-PPS predicts one or two reaction steps and the user needs to select the next starting compound from a divergent set of predicted reaction paths. In contrast, PathPred is a fully automatic server repeating prediction cycles until the pre-specified compound is reached, whether it is given by the user or it is a compound present in the KEGG metabolic pathway maps. Another unique feature of PathPred is its potential to link the prediction result to genomic information. The PathPred server reports new and alternative reaction steps irrespective of whether enzymes for these steps are found or not. If the enzyme is not known, the E-zyme tool (4,10) may be used to assign a possible EC number (up to the EC sub-subclass), which may then be used to search possible genes in the genome by sequence similarity of known genes with the same EC sub-subclass.
METHOD
Database
As of December 2009, the KEGG COMPOUND database contains 16 110 chemical compound entries, and the KEGG RPAIR database contains 12 032 reactant pair entries. Reactant pairs are binary relationships of chemical compounds manually defined from each KEGG REACTION entry, which generally consists of multiple substrates and multiple products, according to the EC number class. Thus, they are categorized into the following five types: (i) main pairs, describing changes of main compounds such as shown on the KEGG pathway map; (ii) cofac pairs, describing changes of cofactors for oxidoreductases; (iii) trans pairs, focused on transferred groups for transferases; (iv) ligase pairs, describing the consumption of nucleoside triphosphates for ligases; and (v) leave pairs, describing the separation or addition of inorganic compounds for such enzymes as lyases and hydrolases.
RDM pattern library
Each reactant pair entry contains the RDM pattern, which is manually extracted from the chemical structure alignment of two reactants. There is a tendency that the reactions in a specific category of KEGG pathways are characterized by a specific subset of RDM patterns (4). Thus, we identified 947 reactant pairs consisting of 739 compounds for xenobiotics biodegradation and metabolism, and 1397 reactant pairs consisting of 1015 compounds for biosynthesis of secondary metabolites. In this server, we use the RDM patterns of only the main pairs, namely, 853 main pairs consisting of 724 compounds for biodegradation of xenobiotics and 1126 main pairs consisting of 993 compounds for biosynthesis of secondary metabolites.
Algorithm
Figure 1 shows a flow chart of PathPred algorithm for the one-directional prediction. The first step of our method is a global similarity search of a query compound Q against the KEGG COMPOUND database by the SIMCOMP program (6, 11), a maximal common subgraph search program. The second step is a local similarity search against the RDM patterns of matched compounds Mi, to select the matched patterns Mi → Mi,j that are applicable to the query compound. The third step is transformation of the query compound Q to generate compounds Qi,j by following the matched patterns. The second and third steps are repeated (cycle A) as long as the generated compounds Qi,j and Mi,j can be used in the same way as the query compound Q and the matched compounds Mi. If not, the transformed compounds Qi,j are used as next queries for the global chemical structure search against the COMPOUND database (cycle B). This prediction cycle B may be repeated for a given number of times. The PathPred accepts bi-directional prediction when the user inputs the starting and final compounds. Then the prediction cycle is also performed from the final compound, which would limit the possible search space, and is repeated until the predicted pathway is connected to that from the initial query compound or the prediction cycle is repeated for a given number of times.
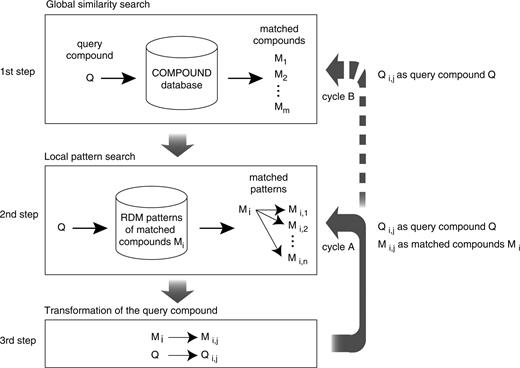
The protocol of PathPred.
Scoring
We define two scoring schemes in order to assign a plausibility value to each predicted pathway. The reaction score is designed to indicate the plausibility of each predicted reaction by using the Jaccard coefficient between the query compound atoms and the matched compound atoms. The score is weighted 3-fold at the RDM atoms in comparison to the other atoms, because they are considered especially important for the reaction. The pathway score is the average of individual reaction scores. The compounds in the pathway with high pathway scores are preferentially used as queries for the successive prediction cycles, in order to limit the search space for possible transformation patterns. In the case of bi-directional prediction, the similarity between the generated compounds and those in the other predicted pathways are also considered. Thus, the compounds are ranked by the sum of the pathway score and the maximum similarity score among the compounds in the other direction.
USAGE
Reference pathway selection
PathPred provides the RDM pattern libraries for two reference pathways; biodegradation pathways of xenobiotics in bacteria and biosynthesis pathways of secondary metabolites in plants. The user is requested to choose the reference pathway for either of them, which determines the subset of the RDM patterns to be utilized.
Query format
The user can input a query compound in the MDL mol file format, in the SMILES representation, or by the KEGG compound/drug identifier (C/D number). This compound, termed initial compound, corresponds to the compound to be degraded or the compound to be synthesized. The user may also input the end compound in biodegradation or the start compound in biosynthesis, which are called final compounds.
Options
When the size of query compound or the number of prediction cycle is large, the prediction may take a while. The user is requested to input the Email address, to which the URL to access the results will be notified once the calculation is completed. The parameter ‘Simcomp Threshold’ specifies the threshold of the chemical structure similarity score in the SIMCOMP computation. The parameter ‘Prediction cycle’ specifies the number of times cycle B in Figure 1 is used.
Output
The PathPred shows the prediction result as a tree-shaped graph (Figure 2A). The predicted pathway tree consists of compounds (nodes) and reactions (edges). The blue-colored C numbers are the compounds in known KEGG pathways and the black-colored CX numbers are the compounds that are not stored in KEGG. The light color node means that the same compound exists elsewhere in the tree. When the pathway reaches the final compound in the bi-directional prediction, it is highlighted in red. Consecutive edges in the same color indicate that the reactions are predicted by the consecutive reference reactions in the KEGG pathway. The thickness of the edge reflects the plausibility of the predicted reaction based on the reaction score. When the predicted pathway does not connect from the initial compound to the final compound in the bi-directional prediction, the result is shown in two separate trees. The graphical chemical transformations of the query are shown in each predicted pathway (Figure 2B). The scores in the white boxes show the pathway scores from the query to each compound and the scores in the blue boxes show the reaction score. The reference reactant pair images with coloring of RDM patterns are shown for each predicted reaction (Figure 3). Related KEGG REACTION and KEGG Orthology (orthologous gene group) entries are linked from the edges.
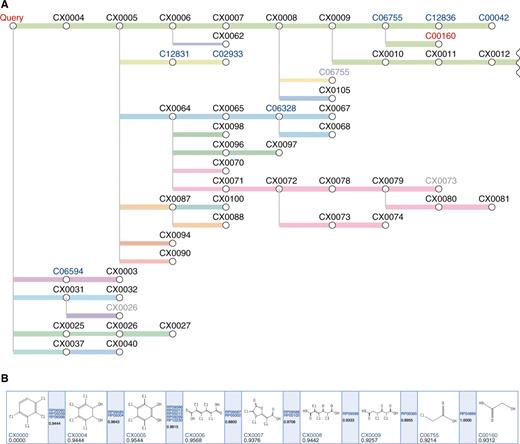
The example of the predicted pathway tree of tetrachlorobenzene biodegradation (A) and the detail of the top green pathway from the query compound (Query) to the final compound (C00160) (B). Structure images popup when the mouse is moved over nodes and edges in the tree if JavaScript is enabled in the web browser.
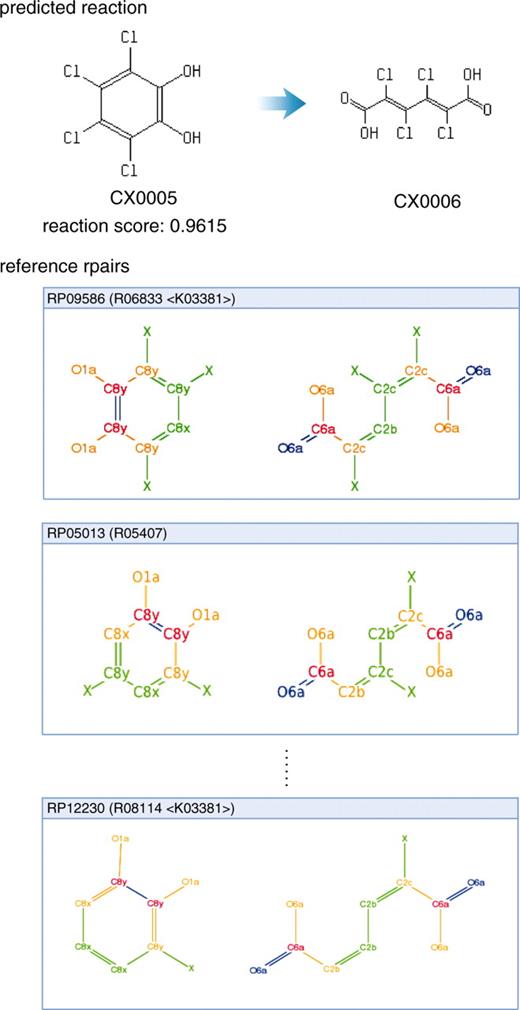
An example of the predicted reactions.
RESULT AND DISCUSSION
Figure 2 is an example of the biodegradation prediction, from 1,2,3,4-tetrachlorobenzene to glycolate (C00160). According to the UM-BBD, the tetrachlorobenzene is degraded along the pathway shown in Figure 2B from the query compound to 2,4-dichloro-3-oxoadipate (CX0009), which is shown as top green lines in Figure 2A. Thus, PathPred successfully predicted the biodegradation pathway with high plausibility. Furthermore, the tree shows other possible pathways, including biodegradations through known compounds such as 3,4,6-trichlorocatechol (C12831), 6-chlorobenzene-1,2,4-triol (C06328) and 1,2,4-trichlorobenzene (C06594). The degradation pathways of these compounds can be seen in the KEGG PATHWAY database from hyperlinks. Figure 3 shows the predicted reaction from tetrachlorocatechol (CX0005) to tetrachloromuconate (CX0006). It is suggested from the reference reactions that this reaction is catalyzed possibly by catechol 1,2-dioxygenase with the KEGG Orthology (KO) identifier of K03381. All possible enzyme genes in the KEGG GENES database that catalyze this reaction are accessible through this KO entry.
Figure 4 is an example of the biosynthesis prediction, from delphinidin (C05908) to gentiodelphin (C08641). This biosynthesis proceeds by addition of three glucoses (blue circles in Figure 4B) and two caffeoyl-CoAs (red circles) to delphinidin. In addition to known pathways in the KEGG, the prediction tree indicates that there are possible sequences of additional reactions. However, the reactant pair of the additional caffeoyl-CoA reaction corresponds to trans pairs, which was excluded from the reference reactant pair data set; therefore, PathPred could not predict accurately and the predicted reactions show additions of glucose and caffeoyl-CoA concurrently in one step. This type of problem may be improved by a more effective selection of reactant pairs in future releases.
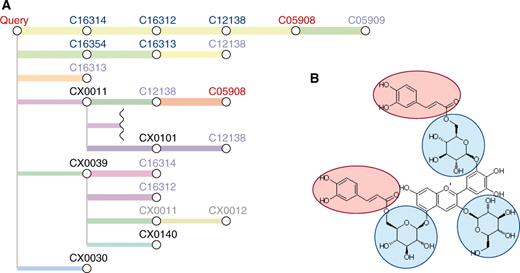
The example of the predicted pathway tree of gentiodelpin biosynthesis (A) and the structure of gentiodelpin (B).
The computation time depends on the size of the query compound, the number of prediction cycles and the size of the reference database. In the case of biodegradation from tetrachlorobenzene consisting of ten atoms (excluding hydrogen atoms) to glycolate consisting of five atoms, the computation allowing one prediction cycle B takes a few minutes. In contrast, in the case of biosynthesis from delphinidin consisting of 22 atoms to gentiodelphin consisting of 79 atoms, the computation allowing one prediction cycle B takes an hour. In a future release, plant biosynthesis pathways will be categorized into subclasses, such as phenylpropanoids, polyketides, terpenoids and alkaloids, which will reduce the size of the RDM pattern library and the computation time.
PathPred is a knowledge-based prediction system. The knowledge base, the KEGG RPAIR database, is continuously updated and expanded as more pathways are included in KEGG PATHWAY and more reactions are included in KEGG REACTION. This is especially true for the biosynthesis of plant secondary metabolites. We intend to increase the number of customized RDM data sets, for example, for drug metabolism and toxic compound metabolism. PathPred will be useful for detection of new and alternative reaction pathways and enzymes.
FUNDING
Grants from the Ministry of Education, Culture, Sports, Science and Technology and the Japan Science and Technology Agency. Computational resources were provided by the Bioinformatics Center, Institute for Chemical Research, Kyoto University. Funding for open access charge: Grant from Japan Science and Technology Agency.
Conflict of interest statement. None declared.
REFERENCES
Author notes
Present address: Daichi Shigemizu, Program in Bioinformatics and Systems Biology, Boston University, 44 Cummington Street, Boston, MA 02215, USA.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments