





Abstract
Chimpanzees (Pan troglodytes) have contributed to diverse fields of biomedical research due to their close genetic relationship to humans and in many instances due to the lack of any other animal model. This review focuses on the contributions of the chimpanzee model to research on hepatitis viruses where chimpanzees represented the only animal model (hepatitis B and C) or the most appropriate animal model (hepatitis A). Research with chimpanzees led to the development of vaccines for HAV and HBV that are used worldwide to protect hundreds of millions from these diseases and, where fully implemented, have provided immunity for entire generations. More recently, chimpanzee research was instrumental in the development of curative therapies for hepatitis C virus infections. Over a span of 40 years, this research would identify the causative agent of NonA,NonB hepatitis, validate the molecular tools for drug discovery, and provide safety and efficacy data on the therapies that now provide a rapid and complete cure of HCV chronic infections. Several cocktails of antivirals are FDA approved that eliminate the virus following 12 weeks of once-per-day oral therapy. This represents the first cure of a chronic viral disease and, once broadly implemented, will dramatically reduce the occurrence of cirrhosis and liver cancer. The recent contributions of chimpanzees to our current understanding of T cell immunity for HCV, development of novel therapeutics for HBV, and the biology of HAV are reviewed. Finally, a perspective is provided on the events leading to the cessation of the use of chimpanzees in research and the future of the chimpanzees previously used to bring about these amazing breakthroughs in human healthcare.
Introduction to Viral Hepatitis
Many viral infections can involve some inflammation of the liver or hepatitis. Those that have a specific tropism for the liver are designated as hepatitis A-E and share no virological relationship other than their tropism for the liver. The alphabetical designation of these viruses stems from the development of diagnostic assays capable of discrimination of the various infections that clinically manifest as hepatitis. The members of this group most devastating to humans are simply known as hepatitis A, B, and C. Chimpanzee (Pan troglodytes) models have been used extensively for research on vaccines and therapies for these viruses (Table 1). Hepatitis B (HBV) and HCV have a predilection for causing persistent infections that, over decades, induce serious liver diseases including cirrhosis and liver cancer. HAV most frequently causes an acute resolving infection that is most severe in those exposed after childhood and can progress to life-threatening fulminant hepatitis. A brief description of the viruses reviewed in this chapter is provided below with references for more extensive reviews.
Highlights of Chimpanzee Research
|
|
Highlights of Chimpanzee Research
|
|
The prevalence of HCV infection is 3% worldwide with an estimated 170 million people persistently infected. The U.S. adult population (~40–60 years of age) has a prevalence of approximately 4%. The disease is typically asymptomatic or mild for several decades; however, up to 20% of infected individuals eventually develop cirrhosis, and these individuals are at risk for progression to hepatocellular carcinoma (Alter and Seeff 2000; Thomas and Seeff 2005). End-stage liver disease due to HCV infection is the leading cause of liver transplantation in the United States, and HCV-associated liver cancer is now the most rapidly increasing cause of death due to cancer in the United States with an estimated 750,000 deaths annually. HCV liver disease is also a leading cause of morbidity and mortality in individuals co-infected with HIV whose HIV infection is under control with antiviral therapy, but who will experience more rapid progression of liver disease. HCV is a member of the Flaviviridae family within the Hepacivirus genus. HCV is a single-stranded, positive-sense, RNA virus with seven genotypes that display an exceptionally high genetic diversity (Simmonds et al. 2017; Smith et al. 2016). The genome contains a single open reading frame that encodes a polyprotein that is processed into 10 viral proteins by host and viral proteases (reviewed in Bartenschlager et al. 2013; Bukh 2016; Lindenbach and Rice 2005; Scheel and Rice 2013; Tellinghuisen et al. 2007). The proteins of greatest importance to antiviral therapy are NS3, NS5A, and NS5B (Bukh 2016; Gotte and Feld 2016). NS3 is the serine protease, which cleaves the nonstructural proteins into functional units and also contains the RNA helicase domain. NS5A is a unique, multi-domain, zinc-finger protein that has RNA binding activity and plays an essential role in viral assembly and replication (Nakamoto et al. 2014; Ross-Thriepland and Harris 2015). NS5B is the viral RNA-dependent RNA polymerase. In addition, the 5′ noncoding region contains an internal ribosome entry site that initiates translation and two binding sites for the liver-specific microRNA-122 that are essential for replication (Jangra et al. 2010; Jopling et al. 2008; Masaki et al. 2015) and have been targeted for antiviral therapy (Lanford et al. 2010). Currently, no vaccine is available for HCV, but a great deal of our understanding of the immune response to HCV comes from vaccine studies in chimpanzees, as is discussed later in this review.
Chronic HBV infection impacts over 250 million individuals and progresses to cirrhosis and liver cancer over decades (Chisari et al. 2010; Liang 2009). Annual deaths due to HBV-associated hepatocellular carcinoma are estimated at nearly 786,000 per year. Two vaccines for HBV were developed through research in chimpanzees. The initial vaccine was purified from the blood of infected individuals and inactivated. The efficacy as well as concerns over residual infectivity of this vaccine could only be addressed in the chimpanzee. The current vaccine for HBV is produced in yeast and contains the small surface protein designated HBV surface antigen (HBsAg). This was among the first recombinant DNA products used in man and was used for immunization of adults and infants. Recombinant DNA products were new at the time, and again concerns over efficacy and safety could only be addressed in the chimpanzee. This vaccine provides lifelong immunity and has provided immunity for entire generations where fully implemented. Although antivirals are available to suppress replication and disease progression, they must be taken for life. Curative therapies are not available, but novel new approaches will be discussed in this review. HBV is a member of Hepadnaviridae. The virion is partially double-stranded DNA that is the product of reverse transcription of a greater-than-genome-length pregenomic RNA (reviewed in Seeger and Mason 2015). This RNA is encapsidated along with the reverse transcriptase by a single protein, the core protein. Following reverse transcription, the capsid particle buds through the endoplasmic reticulum membrane and acquires a lipid envelop that is comprised of three HBV surface proteins that overlap due to initiation at three different start codons. The vaccine is comprised of the smallest of these proteins, HBsAg.
HAV is primarily transmitted by the fecal-oral route and is commonly acquired in early childhood in countries with poor sanitation. An estimated 1.4 million infections occur annually. Delay of exposure due to moderate or high sanitation in the absence of universal vaccination causes focal and occasionally large epidemics with a high disease burden. Infection in adults leads to jaundice in up to 70% of those infected with over 100,000 deaths annually. A protective vaccine is available but has not been widely implemented in most of the world. This vaccine is based on inactivated virus produced in tissue culture and was developed using multiple nonhuman primate (NHP) species, with the chimpanzee studies being crucial for approval. Efforts to derive a recombinant or peptide-based vaccine using the chimpanzee model were not successful. HAV is a member of the Hepatovirus family within the Picornaviridae genus (Ehrenfeld et al. 2010; Martin and Lemon 2006). Similar to HCV, the structure of the genome is positive-sense with a single polyprotein that is processed into 11 proteins. The capsid is comprised of four proteins, VP 1–4. Although conventionally classified as a nonenveloped virus due to the characteristics of excreted virus, recent discoveries have revealed secretion of particles in the blood that are cloaked in host membranes (Feng et al. 2013; Walker et al. 2015) that provide protection from neutralizing antibodies and may facilitate spread in the liver.
Discovery and Characterization of Transfusion-Associated NonA,NonB Hepatitis
The development of assays capable of distinguishing infections with HBV (previously serum hepatitis) and HAV (previously infectious hepatitis) led to observations of the transmission of a new type of hepatitis in hospitals involving blood or blood products. The agent was simply designated transfusion-associated NonA,NonB hepatitis (NANBH) (Feinstone et al. 1975), or essentially a virus with no name (Table 2). The difficulty of working on this agent cannot be overemphasized. There was no method to grow the virus in the laboratory using cell cultures, no technique to detect the virus, and no small-animal model that could be infected with the virus. The first breakthrough would occur when Harvey Alter, a scientist at NIH, collaborated with the Southwest Foundation for Biomedical Research (now Texas Biomedical Research Institute) to transmit the infection to a chimpanzee using human donor blood known to transmit the infection (Alter et al. 1978; Hollinger et al. 1978; Tabor et al. 1978). Since the virus could not be detected, the proof of transmission was based on the development of mild symptoms of hepatitis in the weeks following inoculation. In addition, infections were monitored by histological changes consistent with mild inflammatory disease in the liver and the detection by electron microscopy of membranous tubules in the cytoplasm of hepatocytes (Shimizu et al. 1979). Remarkably, the properties of the virus were defined using the chimpanzee model and conventional virological techniques, including the size of 50 to 80 nM based on filtration studies (Bradley 1985; He et al. 1987) and the presence of a lipid envelope based on loss infectivity after exposure to chloroform (Bradley et al. 1983; Feinstone et al. 1983b). The chimpanzee model was used to demonstrate that blood products were often contaminated with NANBH due to the use of large pools of plasma in manufacturing. Methods to inactivate the agent in blood products and render them safe for use in humans were based on infectivity studies in the chimpanzee, including UV light in the presence of beta-propiolactone (Prince et al. 1985) and solvent-detergent treatment (Horowitz et al. 1993; Prince et al. 1984, 1987). Long-term follow-up of chimpanzees and patients revealed that NANBH often induced chronic or persistent infections (Bradley et al. 1981), highlighting the need to develop methods to detect the virus and reduced transmission.
Milestones in HCV Research
| 1975 | New hepatitis agent recognized, NANBH |
| 1978 | NANBH transmitted to chimpanzees |
| 1989 | HCV cloned from chimpanzee serum |
| 1990 | HCV antibody assay developed to screen donor blood |
| Realization of chronic infections in 3% of adults | |
| 1997 | Validation of infectious clones in chimpanzees |
| 1999 | Development of HCV replicon model |
| 2005 | In vitro replication of HCV in tissue culture |
| 2006 | Transmission from culture to chimp and back to culture |
| 2000–2014 | Era of drug development |
| Preclinical testing for HCV antivirals in chimpanzees | |
| 2014 | FDA approval of first curative cocktail for HCV |
| 1975 | New hepatitis agent recognized, NANBH |
| 1978 | NANBH transmitted to chimpanzees |
| 1989 | HCV cloned from chimpanzee serum |
| 1990 | HCV antibody assay developed to screen donor blood |
| Realization of chronic infections in 3% of adults | |
| 1997 | Validation of infectious clones in chimpanzees |
| 1999 | Development of HCV replicon model |
| 2005 | In vitro replication of HCV in tissue culture |
| 2006 | Transmission from culture to chimp and back to culture |
| 2000–2014 | Era of drug development |
| Preclinical testing for HCV antivirals in chimpanzees | |
| 2014 | FDA approval of first curative cocktail for HCV |
Milestones in HCV Research
| 1975 | New hepatitis agent recognized, NANBH |
| 1978 | NANBH transmitted to chimpanzees |
| 1989 | HCV cloned from chimpanzee serum |
| 1990 | HCV antibody assay developed to screen donor blood |
| Realization of chronic infections in 3% of adults | |
| 1997 | Validation of infectious clones in chimpanzees |
| 1999 | Development of HCV replicon model |
| 2005 | In vitro replication of HCV in tissue culture |
| 2006 | Transmission from culture to chimp and back to culture |
| 2000–2014 | Era of drug development |
| Preclinical testing for HCV antivirals in chimpanzees | |
| 2014 | FDA approval of first curative cocktail for HCV |
| 1975 | New hepatitis agent recognized, NANBH |
| 1978 | NANBH transmitted to chimpanzees |
| 1989 | HCV cloned from chimpanzee serum |
| 1990 | HCV antibody assay developed to screen donor blood |
| Realization of chronic infections in 3% of adults | |
| 1997 | Validation of infectious clones in chimpanzees |
| 1999 | Development of HCV replicon model |
| 2005 | In vitro replication of HCV in tissue culture |
| 2006 | Transmission from culture to chimp and back to culture |
| 2000–2014 | Era of drug development |
| Preclinical testing for HCV antivirals in chimpanzees | |
| 2014 | FDA approval of first curative cocktail for HCV |
Cloning of HCV and Validation of Molecular Tools for HCV Drug Discovery
An intensive international effort was devoted to the isolation of this virus with no name, which resulted in more than a few false reports. To aid in the validation of claims of discovery, Harvey Alter developed a coded panel of human serum with known patient histories. The isolation of HCV finally occurred when a team of scientists at Chiron Laboratories led by Michael Houghton cloned a small fragment of the viral genome (Alter and Houghton 2000; Alter et al. 1989; Choo et al. 1989). This discovery quickly provided the basis of an assay to screen for antibodies to HCV in human blood, making the blood supply safe, 15 years after the original description of a new hepatitis in hospitals. The initial clone was isolated from a lambda gt11 phage expression library produced using infected chimpanzee serum and then screened with the serum from a NANBH patient as the source of antibody. A single clone of approximately 150 nucleotides (clone 5-1-1) was detected. The dependence of this feat on the skill and perseverance on the part of the scientists involved is without doubt, but the essential role of the chimpanzee must also be appreciated. The chimpanzee serum was provided by Dan Bradley at the CDC and had been documented to have exceptionally high levels of virus by inoculating chimpanzees with highly diluted samples in a titration study. The fact that a single clone was detected out of millions of gt11 plaques suggests that the clone would have been missed if the viral titer of the starting material had not been of such high level. In fact, production of additional libraries from the same source failed to produce a second clone of HCV (Houghton 2009). The use of the 5-1-1 clone permitted selection of overlapping clones until the entire open reading frame of the polyprotein had been captured. This would begin the struggles to develop an infectious clone and culture system suitable for drug discovery.
The production of a full-length infectious clone would present a new hurdle to be overcome only after great difficulty (Table 2). One of greatest hurdles was identification of the true 3′ terminus of the genome. The cloning of the 3′ terminus of HCV was hampered due to the low levels of starting RNA for amplification and the lack of a polyA tail from which to prime cDNA synthesis (Blight and Rice 1997; Kolykhalov et al. 1996; Tanaka et al. 1995, 1996). Even after the entire sequence was available, the validation of infectious clones was not straightforward. In the absence of a culture system, the assay to validate infectivity was the direct inoculation of in vitro transcribed synthetic RNA into the liver of a chimpanzee. The difficulty of this task is emphasized by the efforts of Kolykhalov and co-workers, who inoculated 34 full-length clones into chimpanzees without identification of an infectious clone (Kolykhalov et al. 1997). The first infectious clones were produced by making a consensus sequence based on multiple full-length sequences from the same starting material (Kolykhalov et al. 1997; Yanagi et al. 1997). Subsequently, multiple infectious clones were constructed and validated in the chimpanzee by correcting nonconsensus residues from various strains (Beard et al. 1999; Hong et al. 1999; Lanford et al. 2001; Yanagi et al. 1998, 1999a); this included clones representing genotypes 3 and 4 (Gottwein et al. 2010). Importantly, representatives of all six major genotypes have been shown to induce infections in chimpanzees (Bukh et al. 2010), providing characterized material for future studies.
Despite the development of infectious clones, no tissue culture system was available for analysis of viral replication or drug screening. Critical studies continued in the chimpanzee to examine the infectivity of modified clones. The essential nature of the conserved 98 nucleotide stem loop structure at the 3′ NCR (Kolykhalov et al. 2000; Yanagi et al. 1999b), the requirement of p7 for infectivity (Sakai et al. 2003), and the nonessential nature of the E2 HVR1 (Forns et al. 2000) were all demonstrated in the chimpanzee in this manner.
Perhaps the most significant development with regard to drug discovery was the creation of replicons that lack the ability to produce infectious virus but nonetheless allow analysis of all of the functions required for RNA replication in tissue culture (Lohmann et al. 1999). Even this breakthrough would be limited to specific viral isolates and replication in a single human liver cancer cell line, Huh7. Substantial adaptation occurred in the replicon in achieving high levels of replication (Blight et al. 2000; Lohmann et al. 2001), leading to concerns that specific targets may have subtle but significant differences between the in vitro model and virus in the liver. Nonetheless, most drugs were either developed or perfected by iterative modification prior to preclinical trials in chimpanzees and eventually human trials. The NS5A region displayed the most evident differences between viral sequences supporting replication in tissue culture and virus in the liver. Adaptive mutations occurring in NS5A in the replicon resulted in clearly attenuated replication in chimpanzees and reverted to the wild-type sequence (Bukh et al. 2002). These findings and the lack of a molecular assay for NS5A function caused some concern in approaching NS5A as a drug target, yet high throughput assays with drug libraries repeatedly selected hits that involved NS5A. The isolation of the JFH1 strain (Kato et al. 2003; Wakita et al. 2005) that permitted direct infection of cells in culture for the first time also pointed to differences in replication in vitro and in the liver. A derivative of the Huh7 cell line, Huh7.5 (Blight et al. 2002), was required to attain high levels of replication, and virus produced in culture was highly attenuated in chimpanzees with low level replication and a limited duration of viremia (Kato et al. 2008; Lindenbach et al. 2006; Prentoe et al. 2011, 2016; Wakita et al. 2005). An infectious clone representing the genotype 1a, H77-S, was optimized by incorporation of a series of mutations to increase production of infectious virus following transfection of permissive cells (Yi et al. 2006). This clone was the first to induce a chronic infection in the chimpanzee (Yi et al. 2014). Despite the advances in molecular biology and in vitro cultivation methods, over 40 years after description of NANBH in the clinic, replication of isolates from patients in tissue culture is still not possible.
The Innate Immune Response to HCV
Viral RNA is sensed by a number of host proteins, which in turn stimulate the production of type I interferon (IFNα and IFNβ) and type III IFN (IFNλ 1–4) through complex signaling pathways. In infected hepatocytes, Toll-Like Receptor 3 (TLR3) and DExD-box helicase receptors (RIG-I and MDA5) recognize viral RNAs via “pathogen-associated molecular patterns,” resulting in the activation of transcription factors including interferon regulatory factor 3 (IRF-3) and NF-κB. In addition, PKR binds dsRNA, becomes activated by phosphorylation, and contributes to the activation of NF-κB. IRF-3 and NF-κB in conjunction with other transcription factors induce transcription of IFNβ. A distinct subset of “interferon stimulated genes” (ISGs) is also directly induced by IRF-3, while NF-κB promotes the expression of proinflammatory cytokines. In addition to the production of IFNs by infected cells, plasmacytoid dendritic cells (pDCs) produce IFNα following recognition by TLR7 of viral RNAs secreted from infected cells (Garaigorta and Chisari 2009; Takahashi et al. 2010). Type I and III IFNs interact with their receptors to initiate the JAK-STAT signaling pathway and the production of hundreds of ISGs, bringing about a potent antiviral response. HCV subverts the innate immune responses by several mechanisms. The NS3/4A protease blocks both TLR3 and RIG-I pathways and prevents the activation of IRF-3 (Foy et al. 2003, 2005; Li et al. 2005a) and NF-κB. The TLR3 pathway is blocked by cleavage of the adaptor protein TRIF (Li et al. 2005a; Wang et al. 2009), while the RIG-I pathway is blocked by NS3/4A cleavage of the adapter protein MAVS (aka IPS-1, CARDIF, or VISA) (Li et al. 2005b; Meylan et al. 2005). NS5A may play multiple roles in blocking the response to IFNα through interactions with PKR.
Early studies with NANBH explored therapeutics with no direct mechanism to measure efficacy except for the improvement of symptoms and decrease in serum levels of the liver enzyme ALT. In 1986, interferon-α was found to reduce ALT in 50% of NANBH patients, but most relapsed following cessation of therapy. By 1995, ribavirin was combined with IFNα, providing a cure for up to 35% of patients, but most patients could not tolerate the 1 year of therapy required to obtain a cure. The mechanism by which ribavirin enhanced the activity of IFNα was not understood, since it showed no significant activity as a monotherapy. In 2001, the introduction of PEGylated-IFNα with ribavirin would increase the cure rate to as high as 50% following 1 year of therapy, but the improved therapy was still plagued with multiple adverse reactions.
The complexity of the mechanism of IFNα efficacy for HCV and the variable response rate would be explored for more than a decade. The introduction of total genome microarrays provided an opportunity to directly explore the molecular events in the liver during chronic HCV infection and IFNα therapy. Studies of hepatic gene expression during acute HCV infection in chimpanzees revealed a rapid innate immune response occurring days after infection involving the induction of hundreds of ISG transcripts, some of which were elevated by 100-fold or more (Bigger et al. 2001; Su et al. 2002). The ISG response parallels the rise and decline of viremia during acute-resolving infection, leaving no doubt that the changes are in response to viral RNA. Chronic HCV infection also evokes a strong ISG response within the liver in chimpanzees (Figure 1) and humans (Bigger et al. 2001, 2004; Lanford et al. 2006, 2007; Sarasin-Filipowicz et al. 2008; Su et al. 2002), yet this response does not impact viral persistence. Indeed, treatment of chimpanzees chronically infected with HCV with exogenous IFNα failed to reduce viral RNA even when introduced at exceptionally high levels (Lanford et al. 2007). The ISG response in the liver of infected chimpanzees appeared to be maximally induced, and addition of exogenous IFNα provided no increase in ISG expression, while ISG expression in PBMC of the same chimpanzees was highly induced. This would be designated the Null IFN response in both chimpanzees and humans. Chimpanzees uniformly have high ISG levels during chronic and acute infection (Figure 1), and almost every cell expresses high levels of ISG15 (Lanford et al. 2011). In contrast, humans appear to be divided into two groups, high versus low ISG levels, prior to treatment. An association between high ISG levels and lack of response to therapy agrees with data from the chimpanzee, and in humans this correlates with polymorphisms in IL28B region (Ge et al. 2009; Suppiah et al. 2009; Tanaka et al. 2009).
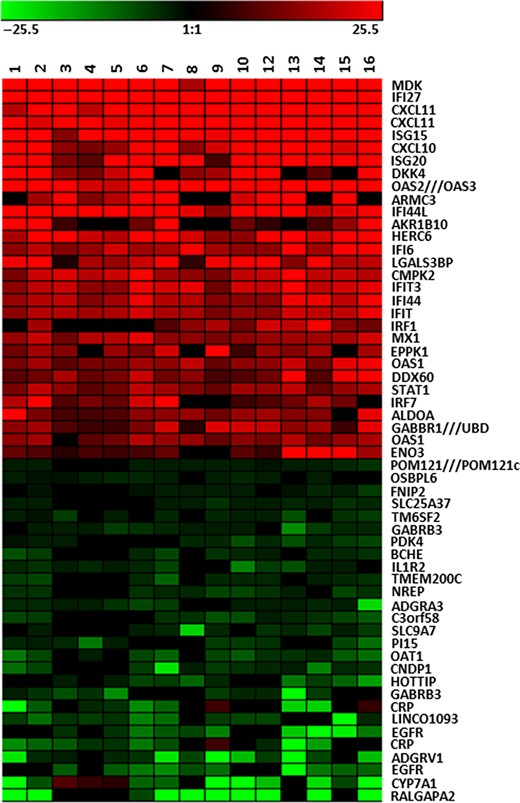
Heat Map of Hepatic Gene Expression in HCV Chronic Chimpanzees. The 30 most highly induced genes in the liver of HCV chronically infected chimpanzees are shown in red. The analyses involve total genome microarray analysis from 16 liver samples from HCV-infected chimpanzees in comparison to 6 uninfected chimpanzees. The 30 genes with the greatest decrease in expression in infected liver are shown in green. The genes increased in expression are primarily ISGs as determined by comparison to uninfected animals treated with IFNα. These genes are uniformly expressed at high levels during HCV infection. The genes decreased in expression may reflect some genes specifically regulated by IFN, but the response is variable in different chimpanzees.
Extrapolation of the levels of viral RNA in the liver and assumptions of RNA copy number per cell suggested that <10% of hepatocytes are infected (Bigger et al. 2004; Lanford et al. 2006, 2007). These estimates are in agreement with detection of infected cells using multiphoton microscopy and in situ hybridization (Liang et al. 2009; Wieland et al. 2014). One possible conclusion from these studies is that host responses limit the spread of the virus in the liver by inducing an ISG response in uninfected cells, but viral proteins subvert the antiviral activity in infected cells, allowing persistence of the infection. The stimulus for hepatic ISG expression in most cells is not likely viral replication and may be the production of IFNs by nearby pDCs (Dreux et al. 2012; Lau et al. 2008; Takahashi et al. 2010).
Experimental Vaccines for HCV Infection
Spontaneous resolution of acute HCV infection in some humans and chimpanzees confers long-lived immunity that can sharply reduce the risk of persistent infection upon reexposure to the virus. This observation has provided a strong foundation for identification of protective immune responses and development of protective HCV vaccines. The first test of whether chimpanzees with naturally acquired immunity are protected against reinfection was undertaken approximately 25 years ago, shortly after discovery of the virus (Farci et al. 1992; Prince et al. 1992). Rechallenge of immune animals resulted in viremia and histologic changes in the liver consistent with infection. Because reinfection was even observed after sequential challenge with the same HCV strain virus (Farci et al. 1992; Prince et al. 1992), it was concluded that immunity was absent or weak and raised concerns for vaccine development. This early experience with HCV clearly differed from HAV and HBV, where natural resolution of infection or vaccination provided robust sterilizing immunity in chimpanzees.
A series of insightful studies about a decade later altered this view. Reinfection after challenge of immune chimpanzees with HCV was confirmed (Bassett et al. 2001; Major et al. 2002). However, peak viremia was sharply attenuated and cleared rapidly when compared with the prolonged course of primary infection (Bassett et al. 2001; Dahari et al. 2010; Major et al. 2002). Protection was long-lived; some animals were challenged several years after resolution of the first infection (Bassett et al. 2001; Major et al. 2002). Protection also extended to HCV genotypes different from the one that established the first resolving infection (Lanford et al. 2004; Prince et al. 2005). For instance, in one study, recovery from a genotype 1 HCV infection protected animals against persistent infection with a mixed inoculum of genotype 1, 2, 3, and 4 viruses that were up to 30% different at the amino acid level (Lanford et al. 2004). It is important to emphasize that naturally acquired protection sometimes failed (Bukh et al. 2008; Prince et al. 2005). In one remarkable example, a chimpanzee was partially or completely protected following five homologous rechallenges with a genotype 1a virus and six heterologous rechallenges with genotype 1b and 2a viruses (Bukh et al. 2008). A final challenge with the genotype 1a virus that established the initial resolving infection resulted in persistence (Bukh et al. 2008).
Collectively, these animal studies provided a compelling rationale for development of a pan-genotypic HCV vaccine and suggested that protection does not require sterilizing immunity. Prevention of persistence is considered a more realistic objective for vaccination given the largely asymptomatic nature of acute hepatitis C, an apparent absence of latent HCV genomes that could cause a relapsing infection, and the capacity to cure any breakthrough infections with direct acting antivirals (DAA).
Mechanisms of Protective Immunity
Precisely how natural resolution of HCV infection provides protection from persistence is still not fully understood. Chimpanzee studies have provided evidence that T cells and antibodies contribute to control of HCV replication (Dahari et al. 2010).
T Cells and Protective Immunity
Early chimpanzee studies demonstrated that successful control of virus replication occurs 2 to 3 months after HCV challenge. The sharp reduction in viremia was kinetically associated with upregulation of genes associated with an adaptive immune response like IFN-γ gene expression in liver (Bigger et al. 2004; Su et al. 2002) and expansion of HCV-specific T cells in blood and liver (Cooper et al. 1999; Thimme et al. 2002). Importantly, reexposure of animals to HCV resulted in accelerated IFN-γ and T cell responses that were associated with rapid clearance of infection (Bassett et al. 2001; Grakoui et al. 2003; Major et al. 2002; Nascimbeni et al. 2003; Shoukry et al. 2003). These findings are similar to those from humans with acute hepatitis C (Klenerman and Thimme 2012). Depletion of CD4+ or CD8+ T cells from chimpanzees with naturally acquired immunity immediately before rechallenge with HCV provided additional direct evidence of their contribution to infection control (Grakoui et al. 2003; Shoukry et al. 2003). Antibody-mediated depletion of CD4+ helper T cells resulted in persistent HCV infection instead of rapid resolution (Grakoui et al. 2003). Memory CD8+ T cells in these animals selected for escape mutations in class I epitopes and lost antiviral effector functions. Depletion of CD8+ cytotoxic T cells in a second set of animals resulted in prolonged virus replication (Shoukry et al. 2003). Together, the temporal kinetic associations between virus control and T cell immunity and the altered outcome of reinfection after antibody-mediated T cell depletion provide strong evidence that adaptive cellular immunity protects against virus persistence (Dahari et al. 2010).
Antibodies and Protective Immunity
Cell culture assays to measure antibody neutralization of HCV were not available until approximately 15 years after the discovery of HCV. The chimpanzee model was therefore important in early analyses of humoral immune responses. Co-incubation of HCV with serum from a chronically infected patient prior to transfer to a chimpanzee prevented infection, providing the first evidence of antibody-mediated HCV neutralization (Farci et al. 1994). Passive immunization of a chimpanzee with a neutralizing monoclonal antibody before HCV challenge was also shown to prevent apparent infection (Morin et al. 2012). Sterilizing immunity mediated by neutralizing antibodies may offer limited protection, however. In one recent study, a chimpanzee was passively immunized with broadly neutralizing serum antibodies from a patient with a persistent genotype 1a HCV infection (Bukh et al. 2015). Challenge of the chimpanzee with a mixture of genotype 1a, 4a, 5a, and 6a HCV isolates that were readily neutralized in cell culture assays resulted in infection. There was no apparent control of the heterologous HCV genotypes (Bukh et al. 2015), highlighting the challenge of inducing broadly protective sterilizing immunity by vaccination. Prolonged suppression of the homologous genotype 1a virus was observed (Bukh et al. 2015), suggesting that antibodies may contribute to control of an established infection. This result confirmed early observations that transfer of polyclonal (Krawczynski et al. 1996) or monoclonal (Morin et al. 2012) anti-HCV antibodies after challenge delayed but did not prevent the onset of virus replication in some chimpanzees. The observations also provided direct support for observations from human studies, where rapid development of neutralizing antibodies was associated with reduced virus replication and resolution of acute infection (Dowd et al. 2009; Lavillette et al. 2005; Osburn et al. 2014; Pestka et al. 2007).
Preventing HCV Infection by Vaccination
Many different experimental HCV vaccines elicited humoral and/or cellular immunity when assessed for immunogenicity in rodents (Liang 2013). Very few of the vaccines were assessed for protection of chimpanzees from infection (Liang 2013). The vaccines differed in design, reflecting uncertainty about the relative contribution of antibodies versus T cells to protection from persistence. For instance, vaccines to elicit antibodies against the HCV envelope proteins E1 and E2 (Houghton 2011; Puig et al. 2004) and T cells against nonstructural proteins (Folgori et al. 2006) have been assessed in chimpanzees, as have multi-component vaccines designed to elicit humoral and cellular responses (Elmowalid et al. 2007; Rollier et al. 2004). All these vaccines protected at least some chimpanzees from HCV persistence. Two vaccines designed to elicit neutralizing antibody or T cell responses have advanced to human clinical trials.
Vaccination to Prime Neutralizing Antibodies
An antibody vaccine developed by Michael Houghton and his colleagues is comprised of recombinant envelope glycoproteins E1 and E2 in a microfluidized oil:water emulsion adjuvant (Houghton 2011). It elicited pan-genotypic neutralizing antibodies and provided sterilizing immunity in a subset of vaccinated animals (Meunier et al. 2011). Breakthrough infections were observed in other animals, but they were more likely to resolve than infections in mock vaccinated controls (Houghton 2011). It remains uncertain if protection from persistence was mediated by antibodies or the strong CD4+ T cell response that this vaccine is known to induce. Based on these results in the chimpanzee model, the E1/E2 vaccine was assessed for immunogenicity in humans at low risk for exposure to HCV. Vaccination generated neutralizing antibodies (Law et al. 2013) and strong HCV-specific CD4+ T cell responses (Frey et al. 2010) similar to those detected in immunized chimpanzees. Efforts to refine this vaccine approach are underway by Houghton and colleagues. Future human trials are likely and could advance the concept that vaccines eliciting antibodies can alter the course of HCV infection.
Vaccination to Prime T Cells
The novel hypothesis that vaccination to prime T cell immunity might be sufficient to prevent HCV persistence has been advanced by Folgori, Nicosia, and colleagues at Okairos Corporation (now Glaxo Smith Kline). Chimpanzees immunized with adenovirus and plasmid DNA vectors encoding nonstructural proteins NS3 to NS5b developed robust memory CD4+ and CD8+ T cell immunity (Folgori et al. 2006). Upon challenge with HCV, the vaccine-primed T cell response was rapidly recalled and was associated with profound suppression of acute phase viremia (Folgori et al. 2006). Rechallenge studies in chimpanzees supported the concept that the T cell response alone could be protective. Chimpanzees with a T cell response but no detectable neutralizing antibody following viral clearance from the initial infection were protected against rechallenge (Bukh et al. 2008). Human subjects primed and boosted with a similar vaccine comprised of two serologically distinct adenovirus vectors elicited similar T cell responses against nonstructural HCV proteins (Barnes et al. 2012). Further refinement by boosting with a modified vaccinia virus Ankara vector elicited stronger, multifunctional T cells that targeted epitopes conserved in many HCV genotypes (Swadling et al. 2014). The ability of this vaccine to protect against HCV persistence is being assessed in subjects at risk for infection because of injection drug use (Clinical Trials.gov NCT01436357).
The chimpanzee model of HCV infection has provided critical support for the concept that a protective HCV vaccine is feasible and that the goal of vaccination should be prevention of persistence and not sterilizing immunity. In the absence of clear surrogate markers of immune protection that existed for other hepatotropic viruses, the model also provided proof that candidate HCV vaccines reduced and did not increase the risk of persistent infection before human clinical trials were undertaken. The study of antiviral immunity and vaccine-mediated protection have established an important legacy for chimpanzee research as efforts to reduce the global burden of HCV infection continue.
The Path to a Cure for HCV and ramifications for HBV Co-infections
The Challenge of Resistance to HCV Antivirals
With the development of the replicon model (Lohmann et al. 1999), the tools were available for high throughput drug screening, and many pharmaceutical and biotech companies were placing major resources into this effort. The period between 2000 and 2014 was an era of intense drug discovery, preclinical testing in chimpanzees, and clinical trials in humans. During this same period, scientists were using the same tools to provide a greater understanding of the functions for HCV proteins (reviewed in Liang 2009; Lindenbach and Rice 2005; Scheel and Rice 2013; Tellinghuisen et al. 2007). Despite the rapid pace of scientific discovery, the development of a curative therapy was a daunting challenge. Infected individuals produce approximately one trillion virions per day with a substantial genomic diversity due to error-prone replication. Mutations preexist that could provide resistance to any single or double antiviral therapy. Eventually, four antiviral targets would be selected as the finalists for further drug development, although others continued to be pursued. The most actively pursued targets would be the NS3 protease, NS5A, non-nucleoside inhibitors of the NS5B RNA polymerase, and nucleos(t)ide analogues that inhibited RNA replication (Gotte and Feld 2016). The proof of concept for each class of antiviral was demonstrated in the chimpanzee, and examples of these antivirals will be discussed here. The best of the antiviral drugs could reduce the level of virus by 10,000-fold in just 3 days. However, resistance to these drugs developed immediately, literally in days. The virus levels in the blood declined rapidly just to rebound due to expansion of virus with genetic mutations for resistance. There were initial concerns that therapies would create populations of patients resistant to all of the drugs. This led to research on alternative targets that promised lower probability of resistance and among these was the miR122 inhibitor, Miravirsen (Lanford et al. 2010). However, with the use of cocktails of selected drugs, it soon became clear that infection in chimpanzees and humans could be cured by cocktails of two to four direct-acting antivirals.
Antiviral Efficacy Studies in Chimpanzees Lead the Path to a Cure
Early studies in chimpanzees demonstrated the high potency of direct-acting antivirals, while at the same time providing the first glimpse of how rapidly resistance could occur in vivo. A benzothiadiazine nonnucleoside inhibitor of the NS5B RNA polymerase (A-83,7093) reduced viremia by 2.5 logs within 2 days of oral dosing, but resistant virus was detected almost immediately while the animals were still on therapy (Chen et al. 2007). Resistance mutations could be detected on day 2 with 67% of the virus containing at least one resistance mutation, which increased to 87% by day 5. More troubling was the persistence of mutations after cessation of therapy. The rapid emergence of resistant viruses during viral decline suggests that these mutations not only preexist in the quasispecies, but exist as replication complexes in hepatocytes and are the primary source of viral production early after antiviral treatment when the decline of WT from the circulation correlates with the half-life of viral clearance. The potency of NS3 protease inhibitors was examined in chimpanzees with numerous compounds. One particular example highlighted both the rapid development of resistance and the generation of unfit mutants that did not persist in the absence of selective pressure from the drug. EA-058 was orally dosed for only 2.5 days and reduced the viral load by 4-log10 (Pilot-Matias et al. 2009). Sequencing of virus on day 4, the nadir for viral load, revealed that 80% of viral genomes contained resistance mutations. However, the mutations clearly reduced the fitness for replication, since only WT sequence could be detected after a few days in the absence of drug. Low fitness mutants probably do not represent a hurdle to retreatment of people that fail therapy, but prolonged dosing may provide an opportunity for compensatory mutations that restore fitness to the resistant virus.
One of the early studies of critical importance in this context evaluated a highly potent nucleoside analogue in chimpanzees (Carroll et al. 2009). MK-608 resulted in suppression of HCV below the limits of detection during 37 days of therapy, and virus remained undetectable for 12 days after cessation of therapy. One conclusion from this study is that resistant mutants of nucleoside analogues were highly unfit and persisted below the limit of detection. The first evidence of the potential for a cure came from experiments conducted with a combination of MK-608 (nucleoside analogue) and MK-7009 (NS3 protease inhibitor) (Olsen et al. 2011). Double therapy was conducted for 37 days with extension of dosing the protease inhibitor for a total of 84 days. One of three chimpanzees developed resistance during monotherapy 28 days after stopping the nucleoside analogue. The second chimpanzee rebounded 21 days after discontinuation of both drugs. One chimpanzee exhibited a sustained viral response at 6 months after therapy with no detectable virus and by definition was cured of HCV. Although the group size was small and only one animal was cured, this provided proof that an HCV cure could be attained with as few as two potent antivirals.
The concerns over resistance led to evaluation of other targets for therapy including host factors required by HCV for replication. One particularly attractive target was miR-122, a highly abundant, liver-specific microRNA. HCV has two binding sites for miR-122 in the 5′ noncoding region of the RNA genome, and both are essential for maintaining RNA abundance (Jangra et al. 2010; Jopling et al. 2008; Masaki et al. 2015). The drug SPC3649 or Miravirsen is an antisense oligonucleotide that targets miR122. This drug is the first example of a DNA-based therapy that is highly efficacious when administered systemically, and the success of the drug relies on the use of the Locked Nucleic Acid technology that provides enhanced stability and higher binding affinity for the oligonucleotide. In contrast to direct-acting antivirals, Miravirsen resulted in a slow decline of virus over weeks rather than days, but no resistance developed with extended dosing. Miravirsen was administered once per week for 12 weeks and induced a 2.6-log drop in viral RNA levels that persisted for months after the last dose of drug (Lanford et al. 2010). The slow decline of virus is due to the need to load the liver with the antisense and completely sequester miR-122, while the stability of the antisense in the liver maintained antiviral activity for extended periods. The drug provided an extremely high genetic barrier to resistance. No adaptive or antiviral resistant mutations were detected in the 5′ NCR. Although Miravirsen progressed to phase II human trials, the need for antivirals that targeted host factors was reduced by the rapid development and FDA approval of curative cocktails of direct-acting antivirals. The antivirals such as Miravirsen may still play essential roles in treatment of some individuals if resistance to DAA cocktails proves significant in the future.
In 2014, the FDA announced the approval of the first interferon-sparing all-DAA combination regimen for HCV. Harvoni is a combination of a nucleoside analogue and an NS5A inhibitor. By 2016, three additional cocktails were approved: VIEKIRA XR, Zepatier, and Epclusa. All these cocktails provide a >95% cure rate with a once-per-day dosing over 12 weeks and minimal side effects. This represents the first cure of a chronic viral disease and represents the cumulative efforts of thousands of scientists over 40 years, since the description of NANBH. Most importantly, these therapies would not be available today without the important contribution of the chimpanzee model, from the discovery of HCV to the development of the cure.
Hepatitis B Virus Reactivation after HCV Cure
Unexpectedly, in October of 2016, the FDA released a drug-safety communication warning of the risk of hepatitis B reactivation in some patients treated with HCV direct-acting antivirals. This communication included an FDA Boxed Warning, the highest of the FDA warnings, for all of the direct-acting antivirals for HCV, including the approved cocktails. At the time of the warning, the FDA had received reports of 24 cases, including one that required a transplant and two that resulted in death. The mechanism of HBV reactivation is not understood. Presumably, HCV chronic infection suppresses HBV, and the cure of the infection can lead to reactivation of HBV replication and disease. The most likely mechanisms would involve the immune response to HCV. The high level of innate immune response to HCV in the liver, as discussed above, may have an impact on HBV suppression. Reactivation of HBV during therapeutic immune suppression and chemotherapy are well documented and can lead to lethal outcomes. The American Association for the Study of Liver Disease conducted a survey that provided information on 188 patients with HBV reactivation during chemotherapy; 57% required hospitalization, 37% required intensive care, and 23% died. These patients were reactivations during chemotherapy, not following HCV cure. Sufficient data are not available to determine the outcome of HBV reactivation following HCV cure. Clearly, HBV reactivation will continue to be a problem in many areas of medical care, but the inclusion of HCV cure as one of them was unexpected.
Immune Status Following HCV Cure from Antiviral Therapy
It is uncertain if immune responses recover in cured individuals or provide protection against reinfection. This concept has been tested in one chimpanzee cured of chronic infection with DAA (Callendret et al. 2014). CD8+ T cells visualized in liver during chronic infection were still present in liver 2 years after termination of virus replication by DAA therapy. Rechallenge resulted in rapid expansion of some HCV-specific T cells (Callendret et al. 2014). However, they provided only transient control of HCV replication, and a second persistent infection was established when the virus rapidly acquired escape mutations in class I epitopes (Callendret et al. 2014). This important study serves as a starting point for detailed studies of immune reconstitution and protection in humans at risk of HCV exposure after successful DAA treatment.
Novel Therapeutics for HBV
The HBV vaccine developed in chimpanzees in the 1980s is used worldwide and has resulted in a generation of immune individuals where fully implemented. However, this is of little benefit to the 350 million people chronically infected with HBV and at high risk of end-stage liver disease and liver cancer. Nucleos(t)ide analogue therapies are highly effective at suppressing viral replication and reducing diseas but must be taken for life. Cure of HBV infection is not yet possible, but novel therapeutic approaches are being developed. The challenge with HBV is the persistence of the highly stable covalently closed circular DNA that is the transcriptional template for all HBV proteins and the pregenomic RNA (Seeger and Mason 2015). This DNA is nonreplicating and is present in the nucleus of nondividing hepatocytes, providing remarkable persistence even in individuals that have apparently resolved infection, as discussed above under reactivation of HBV infection. The chimpanzee model of HBV infection has been essential to our understanding of this disease. An adequate review of this literature is not possible in this chapter, but several comprehensive reviews are available (Chisari et al. 2010; Guidotti et al. 2015; Wieland 2015).
This section will describe two novel approaches that were among the last tested in chimpanzees prior to the ban of research and both subsequently moved to human trials. The first is an immune modulator that is an agonist for Toll-Like Receptor 7 (TLR7) and the second is a novel small interfering RNA (RNAi) that targets HBsAg as an immune modulation approach. TLR7 is a receptor present on various immune cells including plasmacytoid dendritic cells and B cells that when stimulated activates the innate immune response and aides in orchestration of the adaptive immune response. GS-9620 is an orally available TLR7 agonist that was tested in HBV chronically infected chimpanzees (Lanford et al. 2013). Two rounds of 4 weeks of therapy provided potent activation of innate immune response followed by a T cell response. The viral load declined by 100-fold, and remarkably the reduction in virus persisted for many months after cessation of therapy, suggesting that the immune status of the animals had been altered (Figure 2). Evaluation of the liver for cells expressing HBV core antigen revealed a dramatic drop in the number of positive cells during therapy. This same drug has shown promise in rhesus macaques (Macaca mulatta) infected with SIV. In some animals treated with HIV antiviral therapy and then given GS-9620, virus did not rebound after stopping therapy. GS-9620 is in clinical trials as a candidate for reducing HIV-latent viral reservoirs.
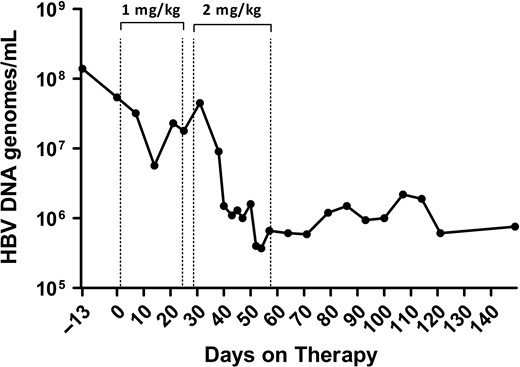
TLR7 agonist GS-9620 induces a decrease in HBV DNA in the serum of a chimpanzee. A chimpanzee chronically infected with HBV was treated with oral GS-9620 therapy at 1 mg/kg or 2 mg/kg, with three times per week dosing for four weeks at each level. The line graph illustrates the decline in viremia as determined by quantitative PCR (genomes/ml). A maximum of 2.2 log decrease in viremia was observed, and suppression of HBV persisted for months after discontinuing therapy, suggesting an alteration in the immune response of the chimpanzee to HBV. Figure modified from Gastroenterology 2013;144(7):1508–1517.
ARC-520 is an RNAi antiviral that targets HBV transcripts and includes technology to target the liver and release the small interfering RNA from the endosomal compartment without degradation (Wooddell et al. 2013). The RNAi triggers the degradation of all HBV transcripts including surface antigen (HBsAg). One of the concepts behind this therapy is the prevailing hypothesis that HBsAg suppresses the immune response and thus supports persistence of the virus. A study in HBV chronically infected chimpanzees was designed for ARC-520 in combination with nucleoside therapy to reflect patients that would likely be on nucleoside therapy prior to use of ARC-520. As expected, nucleoside monotherapy as a lead-in had little effect on HBsAg, but reduced viremia by multiple logs. In contrast, initiation of ARC-520 provided profound reduction in HBsAg, with a maximum reduction of more than 2 logs of HBsAg (Figure 3) (Wooddell et al. 2017). ARC-520 provided greater reduction of HBsAg in HBeAg positive chimpanzees compared to HBeAg negative chimpanzees. During the course of these studies, it was determined that substantial HBsAg is derived from integrated HBV DNA, rather than cccDNA. This observation accounted for the lower percent reduction of HBsAg in HBeAg negative animals. As predicted, repositioning of the small interfering RNA target sequences to effectively eliminate transcripts from integrated DNA dramatically increased the percent reduction of HBsAg in HBeAg negative chimpanzees (Wooddell et al. 2017). This highly promising technology progressed to human clinical trials.
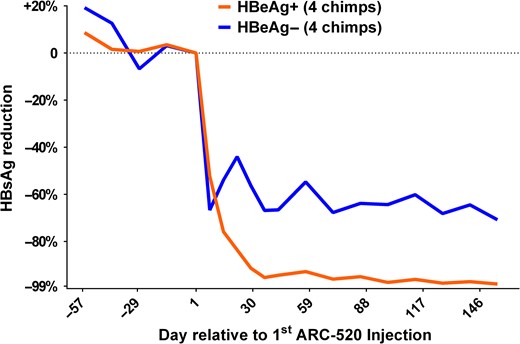
Decline in serum HBsAg levels in chimpanzees chronically infected with HBV during therapy with ARC-520. ARC-520 is an RNAi antiviral that targets HBV transcripts. The RNAi triggers the degradation of all HBV transcripts including surface antigen (HBsAg). Four chimpanzees positive (HBeAg+) or negative (HBeAg−) for serum HBV e-antigen were treated with a lead-in period with monotherapy of standard of care nucleoside analogues (days −57 to 0). This therapy had minimal impact on HBsAg but reduced serum HBV DNA levels by multiple logs (data not shown). Initiation of ARC-520 therapy resulted in rapid decline in HBsAg levels, with up to a 2-log decline. ARC-520 provided greater reduction of HBsAg in HBeAg positive chimpanzees compared to HBeAg negative chimpanzees. Figure derived from data in Wooddell et al. RNAi-based treatment of chronically infected patients and chimpanzees implicates integrated hepatitis B virus DNA as a source of HBsAg. Sci Transl Med, In press.
NHP Studies of Hepatitis A
Hepatitis A is an acute inflammatory disease of the liver. It is an ancient disease with worldwide distribution. The causative agent, HAV, is an atypical member of the Picornaviridae, a large and diverse family of positive-strand RNA viruses (Ehrenfeld et al. 2010). NHPs have figured prominently in the history of HAV research, contributing in three distinct areas of research: characterization of virologic and immunologic features of infection, development of efficacious HAV vaccines, and (more recently) demonstration of the unusual dual lifestyle of HAV as both a naked, nonenveloped virus and a membrane-cloaked, quasi-enveloped virion (Feng et al. 2014).
Human HAV represents the type species of the genus Hepatovirus, a genus that has recently been shown to include multiple viruses infecting a variety of small mammals, including bats, rodents, shrews, and hedgehogs (Drexler et al. 2015). It differs substantially in sequence and structure from other mammalian picornaviruses, with its capsid structure sharing features common to primitive, picornavirus-like insect viruses (Wang et al. 2015). Human HAV is capable of infecting a variety of Old World and New World NHPs, including chimpanzees (Dienstag et al. 1975; Maynard et al. 1975a), rhesus and cynomolgus macaques (Macaca fascicularis) (Amado et al. 2010; Shevtsova et al. 1988), green monkeys (Cercopithecus aethiops) (Shevtsova et al. 1988), marmosets and tamarins (Saguinus mystax and S. labiatus) (Maynard et al. 1975b; Provost et al. 1978), and owl monkeys (Aotus trivirgatus) (Lemon et al. 1982). Viruses have been recovered from naturally infected NHPs held in captivity, and two-way transmission of virus between captive NHPs and their handlers was common prior to the availability of effective vaccines (Dienstag et al. 1976a). An HAV strain recovered from an African green monkey may represent a true simian strain, close to human HAV in nucleotide sequence but with minor differences in antigenic structure and perhaps species-specific pathogenicity (Arankalle and Ramakrishnan 2009; Emerson et al. 1996). Almost half a century has lapsed since the discovery that human HAV can be transmitted experimentally to NHPs. Only recently has it been recognized that infection is also possible in mice with genetic deficits in innate immunity (Hirai-Yuki et al. 2016a).
HAV is spread primarily by fecal-oral transmission, and it was first identified by electron microscopy in the feces of experimentally infected human prisoner volunteers (Feinstone et al. 1973). Prior to its identification, however, the virus was recognized to infect and cause disease in tamarin/marmosets (Holmes et al. 1969). A series of studies in experimentally infected chimpanzees, tamarin/marmosets, and owl monkeys subsequently established the hepatotropic nature of HAV (Dienstag et al. 1976b; Keenan et al. 1984; Lanford et al. 2011; Maynard et al. 1975a; Schaffner et al. 1977; Schulman et al. 1976). Acute inflammatory liver injury was found to occur coincident with the appearance of antibodies to the virus (anti-HAV), typically after a relatively lengthy 2- to 3-week subclinical phase (Figure 4). Fecal shedding of virus and a low magnitude viremia persist during much of this subclinical period (Cohen et al. 1989; Taylor et al. 1993). NHPs have been infected by oral as well as intravenous inoculation of virus, with the required infectious dose of virus 10,000-fold greater when given orally versus intravenously to chimpanzees or tamarin/marmosets (Purcell et al. 2002).
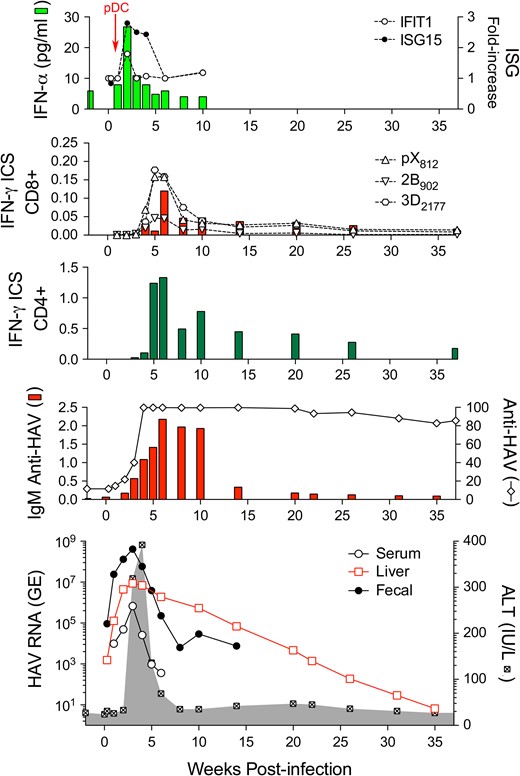
Virologic and immunologic events during acute HAV infection in a chimpanzee inoculated intravenously with wild-type HAV. The bottom panel shows the presence of viral RNA (GE, genome equivalents) in serum (GE/ml), feces (GE/gm), and liver tissue (GE/μg total RNA) in relationship to serum alanine aminotransferase (ALT) activity shown in the shaded zone. The prolonged persistence of intrahepatic HAV RNA is surprising. The panel immediately above shows total anti-HAV antibody (% blocking in a competitive ELISA assay) and IgM anti-HAV (ELISA O.D.) The next two panels show frequencies of HAV-specific CD4+ and CD8+ T cells among peripheral blood mononuclear cells, as determined in an IFN-γ intracellular staining (ICS) assay. CD8+ cells were also quantified on the basis of staining with tetramers targeting epitopes in pX, 2B, and 3Dpol. Note the difference in scale between CD4+ and CD8+ T cell frequencies. The top panel shows type I IFN responses to HAV infection as reflected in minimal and only early serum IFN-α levels detectable by cytokine ELISA, and minimal increases in intrahepatic expression of IFN-stimulated genes IFIT1 and ISG15. pDCs were detected in liver tissue only at 1 week after viral challenge (arrow). Figure reproduced from Curr Opin Virol 2015 Apr; 11:7–13.
Studies in chimpanzees demonstrated large amounts of virus in bile (Schulman et al. 1976), suggesting that most virus shed in feces is derived from the liver. Other studies in tamarin/marmosets and chimpanzees revealed the presence of viral antigen within hepatocytes and hepatic Kupffer cells; lesser amounts were present within germinal centers of the spleen and lymph nodes and along the glomerular basement membrane in the kidney (Mathiesen et al. 1978; Shimizu et al. 1978). Extensive efforts to document an enteric site of virus replication in orally and intravenously infected tamarin/marmosets and owl monkeys provided generally negative results (Asher et al. 1995; Mathiesen et al. 1978, 1980). The only evidence for replication within the gut comes from studies of orally inoculated owl monkeys, in which viral antigen was putatively identified by immunofluorescence in isolated epithelial cells in crypts within the small intestine (Asher et al. 1995). Small amounts of virus have been found within saliva from chimpanzees (Cohen et al. 1989), the source of which is uncertain. This large body of work effectively defined the virologic parameters of acute HAV infection. The results of experimental infections in humans done prior to and following World War II, as well as subsequent observational clinical studies, suggest that HAV pathogenicity is very similar in humans (Boggs et al. 1970; Feinstone et al. 1973; Havens 1946; Krugman et al. 1959). Fulminant hepatitis is a rare complication of infection in humans and has been observed in a single infected chimpanzee (Theamboonlers et al. 2012). Importantly, unlike other viruses that cause acute hepatitis in humans, HAV is not capable of establishing long-term persistent infections in either humans or NHPs. However, fecal HAV shedding may persist for up to 5 months in infected infants (Rosenblum et al. 1991).
Studies done over 80 years ago demonstrated that symptomatic infectious hepatitis (presumably hepatitis A) can be prevented in humans by prophylactic administration of pooled human serum immune globulin (Gellis et al. 1945). This early work effectively established the existence of serum anti-HAV as a strong correlate of immunity. Vaccines in use today are Salk-style, formalin-inactivated, and alum-adjuvanted vaccines prepared from virus propagated in cell culture (Fiore et al. 2006). Studies done in tamarin/marmosets by Philip Provost and Maurice Hilleman at the Merck Laboratories provided early proof-of-principle for such vaccines by demonstrating the protective potential of formalin-inactivated virus recovered from the liver of infected animals (Provost and Hilleman 1978). Subsequent studies carried out by the US Army confirmed that a vaccine produced by formalin inactivation of cell culture-derived virus was capable of protecting owl monkeys against subsequent, wild-type virus challenge (Binn et al. 1986). An important step prior to the registration trials of commercially produced vaccines was the demonstration that serum antibodies from humans immunized by a candidate vaccine protected against virus challenge in chimpanzees (Purcell et al. 1992). Thus, studies in NHPs provided data that contributed directly to the development of vaccines that have since protected millions of persons from HAV infection.
Early studies in NHPs also explored the potential for attenuated hepatitis A vaccines based on the reduced virulence of virus passaged in cell culture. Patient-derived isolates of HAV replicate, but only poorly, in a variety of cultured mammalian cell lines (Binn et al. 1984). HAV was first isolated in cell culture at Merck following serial passage of virus in tamarin/marmosets (Provost and Hilleman 1979). With continued passage in cell culture, the virus adapts and replicates more rapidly and to higher titer, and in some cases with cytopathic effect (Daemer et al. 1981; Lemon et al. 1991). Cell culture adaptation is associated with key changes in the nucleotide sequences of the nonstructural HAV 2B protein and a translational control element, the internal ribosome entry site located within the 5′ nontranslated segment of the viral RNA genome (Funkhouser et al. 1994; Jansen et al. 1988). Passage of the virus in cell culture was shown to lead to a progressive loss of virulence in chimpanzees, raising early hopes that such virus might be used for development of an attenuated vaccine (Feinstone et al. 1983a; Karron et al. 1988; Provost et al. 1983). However, these studies also suggested that the reduced virulence of cell culture-passaged virus was due to a reduced capacity for replication in vivo. This was confirmed subsequently by poor immunogenicity of an attenuated vaccine candidate in humans (Midthun et al. 1991), resulting in the eventual abandonment of this vaccine strategy.
In recent years, studies in experimentally infected NHPs have provided fresh insight into the nature of the immune response to HAV and how the virus is cleared from the liver. Studies done recently in chimpanzees, prior to the NIH moratorium on such studies, revealed that acute infection engenders only paltry intrahepatic innate immune responses (Lanford et al. 2011). Much less transcriptional activation of interferon-stimulated genes was observed than in acute, resolving hepatitis C virus infection. This lack of a strong interferon response is likely due to the ability of HAV-encoded proteases to degrade human MAVS, TRIF, and NEMO, key host proteins involved in the induction of interferon responses to virus infection (Qu et al. 2011; Wang et al. 2014; Yang et al. 2007). A weak type-I interferon response was evident during only the first 2 weeks of infection in chimpanzees (Figure 4), prior to the onset of liver injury and peak fecal shedding of virus, and was associated with transient infiltration of the infected liver by plasmacytoid dendritic cells (Feng et al. 2015; Lanford et al. 2011).
HAV RNA was found to persist for up to 10 months within the liver following resolution of biochemical evidence of liver injury in these chimpanzees (Figure 4) (Lanford et al. 2011). Whether this RNA reflects the presence of infectious virus in the liver is uncertain, but it was not associated with persistent fecal shedding. Declines in intrahepatic viral genome copy numbers correlated well with the intrahepatic abundance of multi-functional, virus-specific CD4+ T cells (Zhou et al. 2012). Such studies cannot be done in human subjects due to the inability to access acutely infected liver tissue, and they are relevant to our understanding of the immune mechanisms mediating control of other, more pathogenic human hepatitis viruses. The results of these chimpanzee studies suggest a novel paradigm for cytokine-mediated CD4+ T cell control of HAV infection that is distinct from earlier concepts of a primary role for cytotoxic CD8+ T cell immunity (Walker et al. 2015). However, B cell responses, which dominate host transcriptional changes in the acutely HAV-infected liver (Lanford et al. 2011), and antibody are key to immunity against reinfection.
Finally, the viremia associated with acute HAV infection in chimpanzees has been shown to be comprised primarily, if not exclusively, of low-density, membrane-cloaked, quasi-enveloped virions (Feng et al. 2013). These studies confirmed the biological relevance of cell culture studies showing that HAV is released in a noncytolytic, exosome-like fashion from infected cells completely cloaked in host-derived membranes. This unusual mechanism for egress of a classically nonenveloped virus involves engagement of the host ESCRT-III (endosomal sorting complex required for transport) complex by fully assembled capsids (Feng et al. 2013). The quasi-enveloped virus is resistant to neutralizing antibodies but is neutralized following its entry into hepatocytes and transport to an endolysosomal compartment, where its membranes are degraded. Virus shed in feces of infected chimpanzees lack such membranes, presumably because they are removed by the detergent action of bile salts during passage of the virus through the biliary tract (Feng et al. 2013; Hirai-Yuki et al. 2016b). This dual lifestyle allows for stealthy spread of membrane-cloaked virus within the infected host and a high degree of physical stability of naked, nonenveloped virions promoting environmental transmission between hosts. These studies provide insight into how a proteinaceous virus capsid can gain egress across the hydrophobic plasma membrane without cell lysis. They have blurred the classic distinction between enveloped and nonenveloped viruses (Feng et al. 2013) and confirm the continuing importance of NHP models in dissecting pathogenic mechanisms of human hepatitis viruses.
Hepatocellular Carcinoma in Chimpanzees
Thirty-nine years have passed since the first experimental transmission of NANBH to chimpanzees (Alter et al. 1978; Hollinger et al. 1978). The long-term consequences of persistent infection with HCV and HBV appear to be minimal. With increasing follow-up and duration of infections, it is clear that few chimpanzees experience progressive liver disease due to chronic viral hepatitis. Historically, two chimpanzees with hepatocellular carcinoma (HCC) were reported at the LEMSIP facility, but only one of the tumors was subsequently confirmed as HCC (Muchmore et al. 1988; Tabor et al. 1994). The Southwest National Primate Research Center has maintained viral and pathological surveillance on both infected and uninfected chimpanzees for over two decades. Although some animals exhibit mildly elevated liver enzymes, inflammatory histological changes and fibrosis are minimal or absent. The lack of contributing cofactors such as alcohol consumption, obesity, and high-iron diet may play a role in the mild nature of the infections in chimpanzees. We previously reported two additional cases of HCC, one each with persistent HCV and HBV infections (Lanford et al. 2008). The HCV-infected animal exhibited mild portal fibrosis, but cirrhosis was not present. The HBV-infected animal had metastatic HCC and advanced cirrhosis. Cumulative data from three research facilities and two sanctuaries now include 10 cases of HCC, only six of which are associated with chronic hepatitis infection. Considering that fewer than 100 chimpanzees are persistently infected with HCV at the various facilities housing infected animals and most have been infected for less than three decades, it is actually surprising that this number of HCC cases have been detected. With the exception of the one HBV case, all of the tumors arose in the absence of cirrhosis. Although cirrhosis is often considered a prerequisite for development of HCV-associated HCC in humans, recent studies have revealed an increasing number of human cases of HCC in the absence of cirrhosis (Yeh et al. 2010). This raises obvious questions of whether HCV possesses direct oncogenic activity. Interestingly, the first transgenic mice expressing HCV proteins developed spontaneous HCC, one with the entire ORF and one with core protein (Lerat et al. 2002; Moriya et al. 1998). Nonetheless, HCC in the absence of hepatitis infection suggests that at least some chimpanzees may have genetic factors leading to the development of HCC.
Chimp Haven, the IOM Committee, and Closure
The first laboratories dedicated to research with chimpanzees were funded in 1930 as the Yale Laboratories for Primate Biology run by Dr. Robert Yerkes (Table 3). NIH began funding chimpanzee breeding in the United States in 1960 with the creation of the National Primate Research Centers. This would bring an end to importation of animals for research as the breeding colonies became established, and in 1975 the ban on importation was formalized with the Endangered Species Act. The use of chimpanzees for research increased with the discovery of AIDS and the causative virus HIV; however, the need for chimpanzees in HIV research rapidly declined with the development of the rhesus macaque model of SIV infection. NIH began to evaluate whether the current chimpanzee population would exceed the needs for biomedical research efforts on HCV and HBV. In 1995, NIH initiated a moratorium on the breeding of NIH-supported chimpanzees. At the same time, an effort was initiated to provide long-term care for a portion of the chimpanzee population in a federally funded sanctuary. Part of this effort was initiated at one of the research facilities using chimpanzees, the Southwest Foundation for Biomedical Research, today known at Texas Biomedical Research Institute and home of the Southwest National Primate Research Center. The concept was championed by Linda Brent, the head of the behavioral sciences group at Texas Biomed and the eventual founder and President of Chimp Haven. In 1995, Chimp Haven Inc. was founded as a nonprofit organization, and four years later, 200 acres in Louisiana was donated to the sanctuary. In 2002, Congress approved up to $30 million for construction and chimpanzee care under the Chimpanzee Health Improvement, Maintenance and Protection Act. The first 31 chimpanzees arrived in 2005, all animals from Texas Biomed. The Chimp Haven website (March 2017) reports that over 300 animals have been taken in and over 200 are still alive on campus. Funding from the Chimpanzee Act became exhausted by 2013, requiring that the federal government expand funding for this program. Following an Insitute of Medicine report on chimpanzees in 2011, the NIH announced an interim policy to stop funding research in chimpanzees but allowed for the possibility of a colony of 50 federally owned chimpanzees for future research needs. The plan for a colony of 50 animals was never implemented, and in 2016 NIH announced that it would not maintain a reserve colony and that no invasive research on chimpanzees would be supported. In 2015, the federal government further restricted research in chimpanzees when the U.S. Fish and Wildlife ruled that even chimpanzees born in captivity at research centers were endangered and special permits would be required for use in research.
History of NIH Chimpanzees
| 1930 | Yerkes starts Yale Laboratories for Primate Biology |
| 1960 | NIH NPRCs founded and include chimpanzee breeding |
| 1975 | End of importation and implementation of CITES |
| 1986 | Increased breeding for HIV vaccine development |
| 1995 | NIH moratorium on breeding |
| 1995 | Chimp Haven Inc. created as non-profit org |
| 2002 | Congress funds CHIMP Act—Federal Sanctuary |
| 2011 | Institute of Medicine review of need for chimpanzees |
| 2011 | NIH initial halt on research in chimpanzees |
| 2015 | Fish & Wildlife designates captive chimpanzees as endangered |
| 2016 | NIH projects that all chimpanzees will be in federal sanctuary by 2026 |
| 1930 | Yerkes starts Yale Laboratories for Primate Biology |
| 1960 | NIH NPRCs founded and include chimpanzee breeding |
| 1975 | End of importation and implementation of CITES |
| 1986 | Increased breeding for HIV vaccine development |
| 1995 | NIH moratorium on breeding |
| 1995 | Chimp Haven Inc. created as non-profit org |
| 2002 | Congress funds CHIMP Act—Federal Sanctuary |
| 2011 | Institute of Medicine review of need for chimpanzees |
| 2011 | NIH initial halt on research in chimpanzees |
| 2015 | Fish & Wildlife designates captive chimpanzees as endangered |
| 2016 | NIH projects that all chimpanzees will be in federal sanctuary by 2026 |
History of NIH Chimpanzees
| 1930 | Yerkes starts Yale Laboratories for Primate Biology |
| 1960 | NIH NPRCs founded and include chimpanzee breeding |
| 1975 | End of importation and implementation of CITES |
| 1986 | Increased breeding for HIV vaccine development |
| 1995 | NIH moratorium on breeding |
| 1995 | Chimp Haven Inc. created as non-profit org |
| 2002 | Congress funds CHIMP Act—Federal Sanctuary |
| 2011 | Institute of Medicine review of need for chimpanzees |
| 2011 | NIH initial halt on research in chimpanzees |
| 2015 | Fish & Wildlife designates captive chimpanzees as endangered |
| 2016 | NIH projects that all chimpanzees will be in federal sanctuary by 2026 |
| 1930 | Yerkes starts Yale Laboratories for Primate Biology |
| 1960 | NIH NPRCs founded and include chimpanzee breeding |
| 1975 | End of importation and implementation of CITES |
| 1986 | Increased breeding for HIV vaccine development |
| 1995 | NIH moratorium on breeding |
| 1995 | Chimp Haven Inc. created as non-profit org |
| 2002 | Congress funds CHIMP Act—Federal Sanctuary |
| 2011 | Institute of Medicine review of need for chimpanzees |
| 2011 | NIH initial halt on research in chimpanzees |
| 2015 | Fish & Wildlife designates captive chimpanzees as endangered |
| 2016 | NIH projects that all chimpanzees will be in federal sanctuary by 2026 |
The final chapter on chimpanzees is still in progress. Several research facilities maintain a population of chimpanzees that are retired. NIH estimates that it will be 2026 before all of the animals can be moved to Chimp Haven based on available space and the expected attrition from old age. This remains controversial with animal rights advocates requesting expansion of construction of new facilities at Chimp Haven to accelerate the time frame, while those currently caring for chimpanzees at biomedical research facilities cite that the movement of geriatric animals from the environment and caregivers that they have known since birth is detrimental to the animals. The existing facilities at research centers are equivalent to those at Chimp Haven with indoor/outdoor housing, grass-covered playgrounds, and elevated structures for climbing. Due to the nature of the research centers, they have more extensive medical facilities and larger behavioral and veterinarian teams to provide care for the animals, especially aging animals, while Chimp Haven has a few large wooded enclosures that the chimpanzees get to experience on a rotation basis. Both types of facilities provide an exceptional environment for the animals with extensive efforts for enrichment of their lives. An unbiased observer would conclude that the animals are enjoying their retirement.
General Summary
The advances in research on hepatitis viruses have been heavily dependent on the chimpanzee model. Today, entire generations are immune to HAV and HBV due to vaccines developed in the chimpanzee that are in widespread use globally. Eradication of these viruses may be possible in the distant future as the pool of chronically infected individuals continues to diminish as each new generation achieves higher rates of vaccination. However, HBV chronic infection currently affects over 250 million people, with liver cancer being a common outcome. The loss of the chimpanzee model has stymied development of curative therapies for this infection. Future efforts will depend on the development of improved mouse models and perhaps new nonhuman primate models. For HCV, due to decades of research in chimpanzees, antiviral therapies are available that require a short course of treatment with minimal side effects and eradicate a virus that has induced progressive liver disease and resisted immune elimination for decades. However, treatment of the 170 million chronically infected individuals is an unrealistic expectation with these therapies. Here too, the loss of the chimpanzee model has stymied the development of vaccines. Human vaccine trials will be lengthy, as numerous candidate vaccines will need evaluation, and will be difficult to coordinate within high risk populations. The use of other species of nonhuman primates can provide essential information on novel vaccine strategies, even though efficacy cannot be evaluated in species other than the chimpanzee. Future generations will certainly look favorably upon the contribution of the chimpanzee to human healthcare as the positive impact of the vaccines and therapies developed in the chimpanzee becomes increasingly apparent with time.
Acknowledgments
The authors would like to thank Helen Hawn for excellent editorial assistance with the manuscript. The authors have the following funding associated with this review: Southwest National Primate Research Center P51-OD011133 (R.E.L.), R01-AI103083 (S.M.L.), and U19-AI109965 (S.M.L.), R37AI47367 (C.M.W.), R01AI096882 (C.M.W.), and U01AI131313 (CMW).
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}