





Synopsis
Identifying the factors that structure host-associated microbiota is critical to understand the role these microbes may play in host ecology and evolutionary history. To begin to address this question we investigate the diversity and persistence of the bacterial community of the giant Neotropical bullet ant, Paraponera clavata. We included samples from four widely dispersed locations to address the role geography plays in shaping these communities. To understand how the digestive tract can filter bacterial communities, we sampled mouth and gut communities. To investigate the stability of community members we sampled wild caught and individuals kept on a sterile diet. Only a single bacterial taxon in the Firmicutes is consistently present across individuals, indicating a remarkably simple “core” bacterial community for the giant Neotropical bullet ant. Geography did not explain host bacterial diversity, but we did find significant reductions in diversity between the mouth and the gut tract. Lastly, our diet manipulations highlight the importance of controlled experiments to tease apart persistent microbial communities from environmental transients.
Evolutionary biologists and ecologists have long been interested in understanding the extrinsic and intrinsic factors that structure biological diversity across space and time. But only recently has it been appreciated that many individual organisms are actually conglomerates of hosts and symbiotic microorganisms that have important effects on each other (Mindell 1992; Margulis 1993; Zilber-Rosenberg and Rosenberg 2008). Part of the reason these intimate associations were not fully appreciated in the past was due to the difficulty in documenting and describing this symbiotic diversity. Advances in sequencing technologies have provided an unprecedented opportunity to begin to understand these symbiotic relationships at a scale that was not previously possible. For example, high-throughput sequencing technologies are providing critical insights into the diversity and role of microbial communities for human health and disease (Hanski etal. 2012; Gilbert etal. 2016), frequency of horizontally acquired bacterial genes in eukaryotic genomes (Crisp etal. 2015), and the unintended impact antibiotics may have on human health (Manichanh etal. 2010; Macklaim etal. 2015).
These tools are providing insights into the diversity of associations that are present across the tree of life, including among the insects (Dillon and Dillon 2004; Jones etal. 2013). From these studies of insect hosts we are learning the role microbes play in nutrition (Douglas 1998; Moran etal. 2005; Engel etal. 2012) and mating preference (Sharon etal. 2010). Even among the ants (Hymenoptera: Formicidae) we now appreciate that microbes can play important roles in diet and nutrition (Feldhaar etal. 2007; Russell etal. 2009) and immune defense (de Souza etal. 2009).
Although we are now in an era when documenting the diversity of host-associated microbes is possible from across the tree of life, we still know little about their function and persistence. To address these two important aspects of host–symbiont interaction, careful manipulation and functional experiments are required, although in most cases this will not be an easy task. Understanding “core” (those that are present in all or the majority of individuals) bacterial communities from the digestive tract may be one of the most fundamental questions we can address through comparing samples from across a wide geographic range and/or comparing wild type/wild caught gut communities with those of individuals that have been reared on controlled diets. These comparisons will allow a better understanding of microbes that are transient members that are introduced to the digestive tract from the environment, including their diet, but may not become residents of the stable “core” gut community. In most cases, the core community is that which we aim to understand, as these are the bacteria that exert the strongest influences on their hosts.
The giant Neotropical bullet ant, Paraponera clavata, is notorious for the pain of the venom that is delivered through their well-developed sting (Schmidt etal. 1984). At several centimeters in length, these ants are conspicuous members of Neotropical forests from Nicaragua in the north to Bolivia and Brazil in the south (Murphy and Breed 2007). These social insects are solitary foragers that can group recruit and have generalized diets that range from active prey capture to feeding on plant-derived resources (McCluskey and Brown 1972; Hölldobler and Wilson 1990). Although not much work has been done to document the microbial community associated with this species, a previous study by Kautz etal. (2013) screened 23 individuals of P. clavata from nine colonies from Peru for three putative “parasitic” bacterial lineages, Wolbachia, Spiroplasma, and Asaia, and found no individuals infected with Wolbachia, two individuals from two colonies infected with Spiroplasma, and one individual from one colony infected with Asaia. In another study of a bacterium associated with the giant Neotropical bullet ant, Larson etal. (2014) screened 59 colonies of P. clavata for Bartonella-like Rhizobiales and found this bacterium is present in 14 colonies, although its functional role is not understood. These two studies together suggest that the diversity of bacteria from P. clavata is not well understood and for those bacteria that have been screened, it is not clear if they are persistent, stable members of the microbiota of this species.
In this study we investigate the diversity and persistence of the bacterial community of the digestive tract of the giant Neotropical bullet ant. As we are interested in the role that the local environment may play in structuring bacterial communities, we included samples from four distinct locations across four countries. To understand tissue specificity, we included samples from each end of the digestive tract (mouth cavity and gut). Lastly, to test whether the diversity and abundance of wild-caught host-associated bacteria of P. clavata are stable community members (persistent regardless of diet or environment), we conducted a controlled diet manipulation experiment.
Materials and methods
Sample preparation
For this study we included 74 samples of the giant Neotropical bullet ant, P. clavata. These samples included 72 adult workers and 1 pupa and 1 larva collected in Brazil, Costa Rica, French Guiana, and Peru (Supplementary Table S1). Samples were either stored in 95% ethanol following the protocol of Moreau etal. (2013) immediately upon collection (wild caught; N = 50) or kept alive for the diet manipulation experiment (diet manipulation; N = 24). Live ants were maintained in a field laboratory at ambient temperature in containers that were cleaned with 10% bleach and 95% ethanol prior to adding the ants to the containers directly in the field. These diet manipulation ants were provided unlimited sterile PCR water and sterile laboratory-grade 30% sucrose, which was homogenized by shaking the mixture in a sterile 25 mL tube, for 10 days before preservation in 95% ethanol. A complete list of specimens included in this study is provided in Supplementary Table S1.
Dissections were performed to isolate the (1) gut (crop, midgut, and hindgut; N = 46) and (2) mouth cavity (also called the infrabuccal pocket which is an oral cavity present in all ants; N = 26) to assess bacterial diversity associated with these tissue specific regions for all samples. Ants were dissected using a stereomicroscope (Leica M205 C) in a sterilized chemical fume hood. Before dissection each individual ant sample was removed from the collection vial using sterile forceps and placed in a Petri dish with 95% ethanol for 2 min, followed by a 1 min soak in 5% bleach (sodium hypochlorite), and a final rinse in sterilized water to surface sterilize the specimen. The digestive tract was dissected under sterile ddH2O by carefully removing the abdominal segments, ensuring that individual parts remained intact, and placed in an individual 1.5 mL collection tube. Next, the contents of each mouth infrabuccal pocket was dissected out and placed in an individual 1.5 mL collection tube. Between dissections, forceps and Petri dishes were washed with ddH2O, 10% bleach, 95% ethanol, and the forceps were flamed.
DNA extraction was completed using the Powersoil DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA, USA). We followed the “modified PowerSoil” protocol of Rubin etal. (2014), which includes a proteinase K digestion step to ensure that host tissue was sufficiently dissolved to allow isolation of endosymbiotic bacterial DNA.
454 pyrosequencing
Bacterial tag-encoded FLX titanium amplicon pyrosequencing (bTEFAP) was performed by the Research and Testing Laboratory (Lubbock, TX, USA) as described in Dowd etal. (2008). The 16S rRNA universal eubacterial primers 28F (5′-GAGTTTGATCITGGCTCAG) and 519R (5′-GWATTACCGCGGCKGCTG) were used to amplify ∼500 base pairs (bp) of the variable regions V1-V3 under the following conditions: 94 °C for 3 min followed by 32 cycles of 94 °C for 30 s, 60 °C for 40 s, and 72 °C for 1 min, with a final elongation step at 72 °C for 5 min. All 74 samples, plus two negative controls consisting of blank DNA extractions, were multiplexed on a single 454 run. Our 454 data is deposited in the GenBank Sequence Read Archive (submission number SUB2353702).
Quantitative polymerase chain reaction to determine bacterial copy numbers
Real-time quantitative polymerase chain reaction (qPCR) was performed to estimate the abundance of bacteria in each sample. We used the universal bacterial 16S rRNA primers 515F (5′-GTGCCAGCMGCCGCGGTAA) and 806R (5′-GGACTACHVGGGTWTCTAAT) (Caporaso etal. 2012). All qPCR reactions where performed in triplicate on a CFX Connect Real-Time System (Bio-Rad, Hercules, CA, USA) using SsoAdvanced 2X SYBR green supermix (Bio-Rad) and 2 µL of DNA extract. Standard curves were generated from Escherichia coli 16S rRNA. To determine 16S rRNA gene copy number per microgram of DNA, we measured the DNA concentration of each sample on a Qubit (Life Technologies, Grand Island, NY, USA) (Supplementary Table S1).
To test for differences in 16S rRNA copy number among sample types we used t-tests of the log-transformed mean SQ values standardized by total DNA concentration as determined by Qubit fluorometry.
Bacterial community analysis
All 16S rRNA pyrosequencing reads were first demultiplexed by the sequencing facility. Reads were denoised using Acacia v. 1.52 (Bragg etal. 2012), truncated at 250 bases and further processed using UPARSE (Edgar 2013). We clustered the resulting sequences at 97% similarity and filtered chimeras using the standard pipeline of UPARSE with reference-based chimera detection. Those samples with fewer than 100 reads were discarded. Representative sequences were aligned against the SILVA database (Quast etal. 2013) using PyNAST and a phylogeny reconstructed using fasttree as implemented in QIIME v1.9.1 (Caporaso etal. 2010a). Those operational taxonomic units (OTUs) that failed to align were excluded from all further analyses. Bacterial taxonomic classifications of our sequences were obtained using the web-based RDP classifier (Wang etal. 2007) with an 80% confidence level. Those classified as chloroplasts were also excluded. We summarized the proportions of identified taxa in each sample and calculated the amount of bacterial diversity shared between sample OTU counts with weighted UniFrac metric (Lozupone and Knight 2005; Lozupone etal. 2007) implemented in QIIME v1.9.1 (Caporaso etal. 2010b). Similarity of samples was visualized for the weighted UniFrac analysis using principal coordinates analysis plots. Significant differences between alpha diversities of different groups was determined by Wilcoxon rank sum tests. The variation within community type was determined by weighted UniFrac beta diversity comparisons within all samples of a particular type (e.g. “gut”) and the differences in this variation between groups assessed using Wilcoxon rank sum tests.
We conducted three distinct analyses on our dataset. First, we examined the role of geography on structuring host bacterial diversity in wild caught samples. Second, we examined the bacterial communities across host tissue in both wild caught and our diet manipulation experiment. Third, we compared whether the diversity and abundance of host-associated bacteria was affected by our diet experiment.
We estimated alpha diversity by averaging 1000 replicates of rarefactions. The number of observed OTUs, the Chao1 estimator, and the equitability index implemented in QIIME were used to estimate alpha diversity.
Results
Analysis of 454 pyrosequencing data
Of our 74 samples of P. clavata, 6 samples (SK1790a.2g, SK1790b.2p, SK1796b.3g, SK1797a.1g, RS0296b.2g, RS0297b.2g; Supplementary Table S1) along with the 2 negative controls, all yielded no sequences. We obtained a total of 288,680 16S rRNA sequencing reads from our 68 remaining samples. Average read length was 424 bp. After denoising, 233,107 sequences remained. Reads clustered into 732 OTUs, of which 29 were found to be chimeric and excluded from further analyses. Seven samples (SK1797a.2g, SK1797a.2p, SK1797a.3g, SK1797a.3p, RS0293b.1g, RS0293b.2g, RS0293b.3g) were excluded because after collection we realized they had fungus growing out of all of their joints leading to contamination concerns. After quality control and chimera detection, 210,564 reads remained representing 672 OTUs in 61 samples (Supplementary Table S1). The number of reads after quality control per sample ranged from 1 to 37,876 (mean ± SD: 3062 ± 5989). Nine samples had fewer than 100 reads (1–44) and were presumed failures so were discarded. Therefore, 52 samples remained with minimum sequence count of 203 and mean count of 3060 representing 663 OTUs with 159,117 total sequences. From these 49 OTUs failed to align to the SILVA database and 12 OTUs (2494 sequences) were identified as chloroplast sequences (16,809 sequences) by the RDP classifier and these were removed from all further analyses. This left 602 OTUs represented by 139,814 sequences in 52 samples with 62–37,870 reads per sample (Supplementary Tables S2 and S3).
We recovered 15 bacterial phyla associated with our 52 included samples (39 wild caught and 13 diet manipulation samples). The most common phyla across all samples were Proteobacteria (50.1% of reads), Firmicutes (30.8% of reads), and Actinobacteria (8.0%) (Fig. 1; Supplementary Tables S2 and S3). Two “core” OTUs are present in more than half of all wild caught gut samples regardless of locality and were classified as members of the Firmicutes in the genus Tumebacillus (Supplementary Table S4). The first, OTU_4, is present in 22/28 samples. The relative abundance of this taxon within samples varies widely between <1% and as high as 95% (RS0294b.3g). Those six samples that are missing OTU_4 are not limited to particular colonies or geographic regions. The second common OTU is OTU_601, which is also classified as Tumebacillus. This taxon is less abundant when present than OTU_4 and only reaches its highest abundance of 58% of reads in a single sample with very few reads (RS0297b.1g).
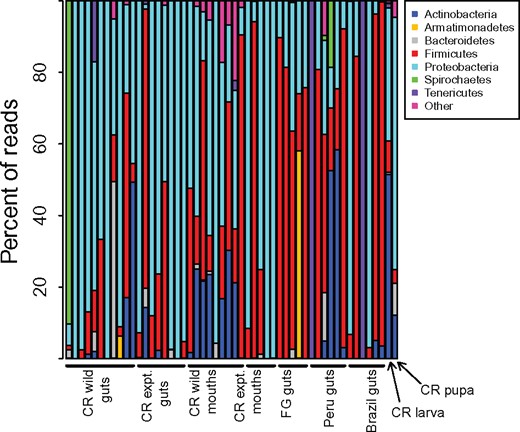
Percentage of communities composed of the most abundant bacterial phyla. Only phyla that make up at least 1% of total sequence data are shown. CR: Costa Rica, FG: French Guiana, expt.: experimental diet manipulation.
Two OTUs were present in more than half of all 11 wild caught gut samples from Costa Rica, regardless of collection locality. The first, OTU_4, is present in eight samples. The abundance of OTU_4 reached as high as 27% (215 reads) of all reads in a sample (SK1797b.2g) but was present at a maximum of 7.7% in all other samples. The second common OTU in Costa Rican wild caught gut samples was OTU_109 (Proteobacteria; Granulobacter), which was present in 6 of 11 samples. This OTU reached 59 reads in on sample (SK1793b.3g) at a relative abundance of 2.9% but never had more than 8 reads in any of the other samples. It made up 3.9% of reads in SK1793b.1g but this sample only had a total of 77 reads.
Five OTUs were present in >50% of the nine mouth pocket samples (Supplementary Table S5). OTU_4 was, again, the most common taxon, present in seven of nine samples at varying levels from <1% to 45%. OTU_601 was present in six samples at levels of 1.1% or lower. OTU_16 (Proteobacteria; Ralstonia), OTU_18 (Proteobacteria; Rhodoplanes), and OTU_92 (Firmicutes; Weissella) were all present in five of the mouth samples. In only two instances did these OTUs reach levels >10%: OTU_18 was at 36.0% in SK1793b.3p and OTU_92 was at 23.4% in SK1793b.2p.
In the eight diet treatment gut samples, there were nine OTUs present in more than half of the samples (Supplementary Table S6). OTU_4 is present in seven samples and OTU_109 and OTU_601 are both present in six of the samples. OTU_2 (Proteobacteria; Asaia) is present in five, OTU_7 (Proteobacteria; Frateuria) is present in four, OTU_156 (Proteobacteria; Asaia) is present in five, OTU_385 (Proteobacteria; Asaia) is present in five, and OTU_400 (Proteobacteria; Asaia) is present in five. The same set of OTU’s was present in more than half of diet-manipulated mouth samples except for OTU_109 (Supplementary Table S7). Three additional OTU’s were also present in more than half of these samples: OTU_298 (Proteobacteria; Asaia), OTU_465 (Proteobacteria; Gluconobacter), and OTU_545 (Proteobacteria; Asaia). OTU_4 (Firmicutes: Tumebacillus) is quite clearly one of the most commonly present bacterial taxa across P. clavata tissues and geographic range.
Five of eleven wild Costa Rican guts had Rhizobiales present at least at a low level and five of eight diet manipulated Costa Rican guts had Rhizobiales present.
Role of geography on structuring host gut bacterial diversity
The principal coordinates analysis (PCoA) of all wild caught P. clavata bacterial communities shows that there is no clustering based on geography or colony for either guts or mouths (Fig. 2). Indeed, gut beta diversities within countries were not significantly different than between countries (Wilcoxon signed rank test, p = 0.37).
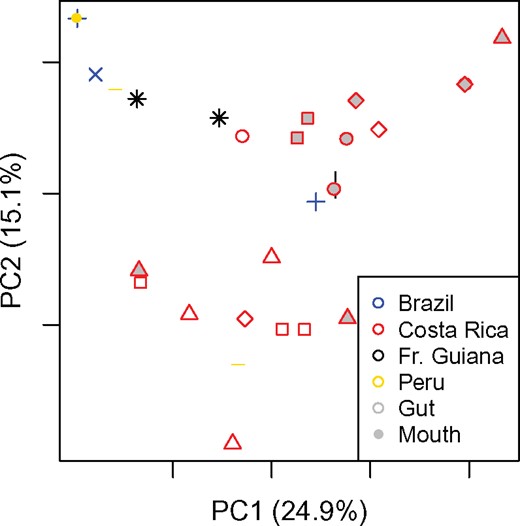
Weighted UniFrac principal coordinates analysis plot of all wild caught samples. Shapes indicate the colony from which samples were drawn. Colors indicate the country of collection as denoted in legend inset.
Mouths have greater diversity
Bacteria in the mouth may or may not be passed to the rest of the digestive tract. To examine the diversity of bacterial communities in the mouth infrabuccal pocket and gut (crop, midgut, and hindgut), we compared samples from these two tissues for wild caught individuals. Wild mouths and wild guts differ significantly in number of bacterial OTU’s (Wilcoxon rank sum test; P = 0.0004). There were 396 OTUs found in wild mouths from Costa Rica and 115 found in guts, and only 66 shared between the two organs, suggesting that there is likely some amount of filtering that occurs between the mouth and gut.
Role of diet in structuring bacterial diversity
To investigate the influence of diet on host bacterial community structure, we included samples from wild caught colonies of the giant Neotropical bullet ant and conspecifics from some of the same colonies after keeping on a diet of sucrose water in the laboratory.
The distribution of read counts for guts and mouths differed substantially. Generally, more reads were recovered from mouths than guts. We expect that this is the result of a larger proportion of DNA of bacterial origin versus ant-derived DNA in the mouths. When examining each type of sample independently, we, therefore, rarefied to different numbers of sequences. For mouths, we rarefied to 1200. Only a single mouth from the quality filtered dataset had fewer than this number of sequences (SK1790a-3p) and was, therefore, discarded from analyses. Guts, on the contrary, were rarefied to 600 reads and, even at this far lower level three samples were eliminated (SK1793b.1g: 77 reads, SK1793a.3g: 164 reads, SK1793a.2g: 220 reads). Sterile sucrose diets clearly influence both the structure and diversity of bacterial communities in P. clavata. Although the number of taxa (alpha diversity) in guts does not change significantly (Wilcoxon rank sum test; P = 0.22), the number of taxa drops significantly in mouths (p = 0.006) when kept in sterile conditions. The same pattern holds for Chao1 and equitability measures of alpha diversity (P = 0.12 for both Chao1 and equitability differences in guts and P = 0.01 and P = 0.02 for Chao1 and equitability for differences in mouths; Supplementary Table S8). Weighted UniFrac distances between communities in wild guts are significantly greater than between those guts with sterile diets (Wilcoxon rank sum test; P = 0.0001, Fig. 3). Mouths do not differ significantly (P = 0.87). Chao1 and equitability measures of alpha diversity of bacterial symbiont communities in guts become less variable when fed sterile diets (Supplementary Table S8).
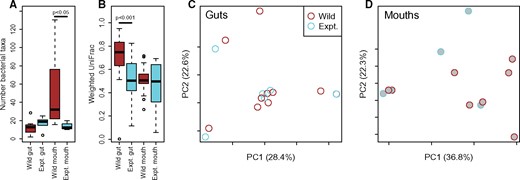
Comparisons of wild caught and experimentally manipulated P. clavata from Costa Rica. (A) Alpha diversity (observed taxa) from each treatment and body part. Significance is determined by Wilcoxon rank sum tests. (B) Weighted UniFrac distances between samples within each group. Significance is determined by Wilcoxon rank sum tests. Weighted UniFrac principal coordinates analyses of guts (C) and mouths (D) from Costa Rica.
Abundance of bacteria across samples
The vast majority of mouth infrabuccal pockets yielded DNA at concentrations below the minimum sensitivity of the Qubit. We, therefore, were unable to study the quantity of bacteria in the mouths of this species. Several guts were also below the minimum sensitivity that we could assess and were, therefore, discarded. The guts from wild caught Costa Rican specimens had higher quantities of bacteria than those fed sterile diets, but not significantly so (Supplementary Fig. S1; t-test: P = 0.69, t = 0.40).
DISCUSSION
Although no organism exists in isolation, we are beginning to appreciate that, for many multicellular eukaryotes including insects, their intimate associations with microbes may be more important to the host's ecology and evolutionary history than previously understood. This seems especially true for the digestive tract of animals where symbiotic microbiota may be critical to their host’s ability to digest food and process the resulting nutrients (Douglas 1998; Moran etal. 2008; Douglas 2009). Documenting the microbial diversity associated with the digestive tract is the first step in understanding the potential importance of these microbes. However, the added challenge of determining which members of the measured community persist over time and dietary fluctuation rather than transients introduced through food or the environment is essential for understanding the impact of these symbioses.
To address this question we studied the digestive tract of the giant Neotropical bullet ant, P. clavata. We sampled both the mouth cavity and gut (crop, midgut, and hindgut) to explore potential tissue specificity across four distinct geographic locations. We also sampled individuals from wild caught populations and did a controlled diet experiment on individuals from those same colonies to gain an understanding of what are persistent, and potentially “core,” members of the digestive tract in these ants.
Although we did find a limited “core” microbiota associated with our wild caught P. clavata samples, our diet manipulation experiment may provide a clearer picture of the persistent digestive tract bacterial community. From our wild caught gut samples from four countries, only one bacterial genus was shared across >50% of the samples (OTU_4: Firmicutes: Tumebacillus). This group of Gram-positive bacteria contains bacteria that are commonly found in soil and associated with a diversity of hosts.
In our diet manipulation experiment of the giant Neotropical bullet ant, we again found OTU_4 in seven of nine samples. In addition, we found eight other OTUs in >50% of our diet manipulation gut samples, the majority of which are members of the family Acetobacteraceae and one of which is a member of Xanthomonadales. These OTU’s were also present in wild caught samples, although not present in high frequency across these samples. It is interesting that we find a diversity of OTUs from the Acetobacteraceae, after feeding the ants on a sterile sugar-based diet, which contain acetic acid bacteria that are known to oxidize sugars or ethanol and produce acetic acid during fermentation. This suggests that these bacteria may be responding to a shift to predominantly sugar-based nutrition, a flexibility that may be advantageous in natural conditions as these ants are often observed feeding at extrafloral nectaries.
Unlike in the gut, the wild caught bacterial diversity for the mouth cavity for these solitary foragers may reflect what was eaten last. Once fed on a sterile diet the diversity and identity of the bacteria decreased in the mouth cavity (Fig. 3). We acknowledge that any experiment comparing wild caught and laboratory manipulated individuals introduces potential confounding factors, although we did attempt to limit these.
Regarding the bacteria that have been found in this species in previous studies, we found a number of Asaia OTU’s in our P. clavata samples, suggesting that this genus may be even more common than found by Kautz etal. (2013). That study also found two of 23 individuals infected with Spiroplasma, a taxon not represented in the communities examined here indicating that it exists at very low frequency in this species. In a previous study by Larson etal. (2014), P. clavata was screened for the presence of Bartonella-like Rhizobiales, hypothesizing that it may perform a role in nitrogen-enrichment. Although found at low frequencies, they speculated that since Bartonella increased in wild colonies that had access to supplemented sugar water, along with other food available in their natural habitat, it may be providing nutritional benefits to the host ants. In our study we found 60 OTUs that classified as Rhizobiales but, after our feeding experiment, only five individuals, had this group of bacteria in their guts. This finding indicates that Bartonella, though possibly providing important nutrient supplementation in some cases, is more likely to be a transient member of the P. clavata gut community rather than a dependable source of nutrition.
Although our data show that the giant Neotropical bullet ant, P. clavata, has a potential stable “core” gut bacterial community, this community is likely quite simple and the abundance of community members may be impacted by their diet (Fig. 3). Although these data improve our understanding of the gut bacterial community of this species, the potential functional roles of these gut-associated bacteria remain enigmatic. Our diet manipulation experiment highlights that most of the bacteria recovered here are likely commensals with little impact on host health, a result that has relevance to many studies of gut-associated bacterial communities. We hope that these results will inspire additional studies that compare wild caught and diet manipulated individuals to better understand the stable gut microbiome and the influence of diet on the diversity, abundance, and evolution of the gut microbiota.
Acknowledgments
We thank the following people for providing specimens or access to field sites to conduct this research: Carlos Roberto F. Brandão, Alain Dejean, Winnie Hallwachs, and Daniel Janzen. For assistance in the field for the diet manipulation experiments, we thank Stefanie Kautz, Elizabeth G. Pringle, Arista Tischner, Alexandra C. Westrich, and Max E. Winston. For assistance in the molecular laboratory, we thank Brian D. Wray. We thank Kevin Kohl and Denise Dearing for the invitation to participate in the special symposium on host–microbe interactions for the 2017 annual meeting of the Society for Integrative and Comparative Biology (SICB) and the National Science Foundation for supporting this symposium (NSF IOS 1638630). Research was conducted in the Pritzker Laboratory for Molecular Systematics and Evolution at the Field Museum of Natural History, Chicago, IL, USA. We thank Bruce and René Lauer and the Lauer Foundation for Paleontology, Science and Education NFP for generous support of the Pritzker Laboratory, which made this research possible, and the Field Dreams program of The Women's Board of The Field Museum for support. We would also like to thank an anonymous donor for a generous gift to support our work.
Funding
This work was supported in part by the National Science Foundation (NSF DEB 1442316 and NSF IOS 1354193 to C.S.M.).
Supplementary data
Supplementary data available at ICB online.

{kind=link}
{kind=link}
{kind=link}