




Abstract
Polycystin-2 (or polycystic kidney disease gene 2 product, PKD2) and its homologues are calcium-regulated ion channels. Mutations in PKD2 are causative for autosomal dominant polycystic kidney disease. Alternative splicing has been documented for the ‘PKD2-like’ genes as a naturally occurring event and for PKD2 in pathologic context. Here we studied naturally occurring PKD2/Pkd2 (human/murine) splice forms on the mRNA and protein levels. Systematic scanning of PKD2/Pkd2 cDNAs obtained through RT–PCR from murine tissues and human cell lines revealed alternative splice forms that were sequenced and checked for translation. We identified three major alternative transcripts of PKD2/Pkd2, PKD2/Pkd2Δ6, PKD2/Pkd2Δ7 and PKD2/Pkd2Δ9, and one minor splice form, PKD2/Pkd2Δ12−13, numbered according to deleted exons or parts thereof. A transcript lacking exon 7 (PKD2/Pkd2Δ7) generated significantly altered protein variant. This polycystin-2Δ7 protein appeared stable, when expressed in cell culture and apparently did not interact with polycyctin-1, which should be due to the reversed topology (extracellular) of the interacting C-terminus (intracellular in polycystin-2). Pkd2Δ7 transcript was predominantly expressed in brain and amounted to 3–6.4% of Pkd2 transcripts in the relevant organ. Moreover, both Pkd2 and Pkd2Δ7 were developmentally regulated. Polycystin-2Δ7 adds on to the number of identified polycystin molecules. The predominant expression in brain indicates a function in this organ. The inability to interact with polycystin-1 expands further the PKD1-independent functions of polycystin-2 forms.
INTRODUCTION
Polycystin-2 is a protein with cation channel function (TRPP2), which can be affected in autosomal dominant polycystic kidney disease (ADPKD), a very common Mendelian disorder with a 1 : 1000 frequency worldwide. ADPKD is a systemic condition, inflicting severe damage of kidney function, which results from mutations in either of two genes, PKD1 and PKD2, and a third unknown locus ( 1 – 3 ). Molecular defects in PKD2, a gene identified through positional cloning almost a decade ago ( 4 ), are responsible for ADPKD in about one-sixth of the affected individuals. The human gene contains 15 exons, which code for a protein of 968 amino acids and has a predicted relative molecular mass of 110 kDa. Murine Pkd2 consists of 15 exons as well and encodes a 966 amino acids protein ( 5 , 6 ). Polycystin-2 is well conserved in mammals; the mouse and human proteins share 91% identity and 98% similarity at the amino acid level. Modeling data propose integral membrane proteins with six transmembrane domains and intracellular localization of the N- and C-termini. Recent evidence, including the topical ciliar expression of PKD2, directs at mechanosensory and flow-induced calcium signaling function for the protein ( 7 , 8 ). Polycystin-2 exhibits distant homology to TRP cation channels ( 9 , 10 ) and is an interacting partner of polycystin-1, the gene product of PKD1 ( 11 , 12 ). This structural and functional interaction as well as clinical manifestations of the disease and characteristics of animal models indicates that both proteins are integral parts of a common signal transduction pathway ( 13 and references therein). It has been proposed that the assembly of polycystins-1 and -2 at the plasma membrane, when co-expressed in cultured CHO cells, produces unique Ca 2+ -permeable non-selective currents ( 14 ). However, more recent studies demonstrate that the expression of polycystin-1 alone can result in such cation currents ( 15 ). In addition, calcium-permeable cation channel properties of polycystin-2 in endoplasmic reticulum have been documented convincingly by single-channel measurements ( 10 ).
Two PKD2-like genes have been identified in mouse (Pkd2L1 and Pkd2L2) and human (PKD2L1 and PKD2L2), which share significant homology with PKD2 ( 16 – 20 ). PKD2L1 is active as cation channel, permeable to calcium ions ( 21 ), and circumstantial evidence hints at a possible similar function for Pkd2L2 in rat ventricular cardiomyocytes ( 22 ). To expand further the diversity of the polycystin-2 molecules, alternative splice forms have been confirmed and characterized for murine and human PKD2L1 and PKD2L2 ( 19 , 20 ). Sequence analysis of the alternative transcripts indicated protein forms harboring internal in-frame deletions, hence altered general and transmembrane topology (for PKD2L1/Pkd2L1 and PKD2L2/Pkd2L2) and truncated C-terminus (in case of PKD2L1/Pkd2L1). Some alternative transcripts (PKD2L2/Pkd2L2) due to frameshift mutations would generate very short peptides, with no apparent functional relevance. Interestingly, a restricted tissue expression was observed for all these splice forms, which did not necessarily match the expression pattern of the master gene ( 19 , 20 ). The question about functional importance of these splice forms needs to be addressed.
In this study, we characterized alternatively spliced forms of murine and human PKD2 genes. We provided translational evidence for a PKD2 splice variant, specific for mammals, lacking exon 7, which we termed PKD2Δ7/Pkd2Δ7. We elucidated the expression pattern and relative quantity of this particular splice form in developing mouse embryo, newborn and adult organism, using quantitative RT–PCR analysis. Further, we demonstrated that polycystin-2Δ7 should be incapable of interacting with polycystin-1 when expressed in a cell culture model system, showing extracellular localization of the C-terminus. We propose a topology model of the polycystin-2Δ7 molecule, which is consistent with the obtained experimental data. Altogether, our findings add to the structural and functional diversity of polycystin molecules, certainly directing to new aspects of polycystin-2 function.
RESULTS
Alternative splicing of PKD2/Pkd2
Multiple alternative transcripts have been described for PKD2L and PKD2L2/Pkd2L2 ( 16 , 17 , 19 , 20 ). Initially, no PKD2/Pkd2 alternative splice forms has been reported in the early works describing these genes ( 4 – 6 , 23 ). The NIA/NIH Mouse Gene Index 4.9 lists a splice form for exon 10, exon 10a and additional non-coding exons 16 and 17 ( http://lgsun.grc.nia.nih.gov/geneindex4/exons/U042663.html ). However, exon 10a seems not to be in use (has never been identified in transcripts). Exon 16 variant has been found once and no exon-17-containing Pkd2 mRNAs has been described so far. The ECgene resource on gene modeling with alternative splicing ( http://genome.ewha.ac.kr/ECgene/ ) finds no alternative transcripts for the human PKD2 gene and has difficulties to generate full-length Pkd2 mRNA. However, aberrant PKD2 splicing resulting from nucleotide substitutions in exon 14 has been reported as a cause of polycystic kidney disease ( 24 ). To identify normally occurring PKD2/Pkd2 splice variants, we initially performed PCR scans on a human fetal tissue cDNA library (Clontech) and mouse embryonic cDNA with various primers derived from the PKD2/Pkd2 sequence. The splice forms identified were then confirmed in different human cell lines as well as in murine adult tissues (see Total RNA extraction from cells and tissue). Altogether we found three major and one minor splice forms for PKD2/Pkd2. Figure 1 is a graphic representation of the PKD2/Pkd2 exon structure with aligned identified splice forms. Well-detectable PKD2/Pkd2 alternative transcripts were those lacking exons 6, 7 and 9. A minor splice form, which was amplified at very low levels from the cells and tissues studied, was an mRNA molecule lacking exons 12 and 13 (Fig. 1 ).
The transcript lacking half of exon 6 is produced through a splice junction next to the A473 codon in the murine and next to the A475 codon in the human PKD2 sequence, in a 100% conserved region, fusing the first ‘t’ nucleotide of the next codon (F474/F476 and Pkd2/PKD2) to the beginning of exon 7 (Fig. 2 A). This splice variant results in a shift of the reading frame and would generate a truncated protein of 481/483 amino acids for Pkd2/PKD2. Similarly, the alternative transcript resulting from full exclusion of exon 9 (Δ9) has also a frameshift after C630/C632 (Pkd2/PKD2) and would code for a truncated Pkd2/PKD2 protein of 644/646 amino acids (Fig. 2 B). Another major splice form, lacking the entire exon 7, would result in intact Pkd2Δ7/PKD2Δ7 proteins of 910 and 912 amino acids accordingly. The minor Pkd2/PKD2 splice variant, lacking exons 12 and 13, would also generate intact proteins of 872/874 amino acids. To further characterize identified Pkd2/PKD2 splice forms, we tried to amplify these from a translated mRNA fraction (see Preparation of Translated mRNA from Polysomal Fraction), using primers located in adjacent exons. We were unable to amplify Pkd2/PKD2 splice variants, containing deletions of exons 6 and 9, and were successful in amplifying the Pkd2Δ7/PKD2Δ7 splice form. Examples of PKD2 exon 7 and exon 9 amplifications from translated mRNA and from total RNA are shown in Figure 3 D and E, respectively.
Characterization of the PKD2/Pkd2Δ7 splice variant
We amplified Pkd2Δ7 from various murine tissues, using primers Pkd2ex6.F and Pkd2ex8.R located in exons 6 and 8 (Table 1 and Fig. 3 A). Such amplifications resulted in two PCR products with different amplicon sizes. The amplicon of 323 bp is indicative of the normal Pkd2 transcript containing exon 7 and an amplicon of 159 bp is specific for the Pkd2Δ7 variant. The shorter amplicon was sequenced to obtain exon/exon transitions and protein sequence of this region. Figure 2 C shows the direct transition of exon 6, ending on V514 in exon 8, starting with L571. Using the same primer set and amplification conditions, we were able to amplify adequate dog Pkd2 and Pkd2Δ7 regions from total RNA of MDCK cells (data not shown). Next, we designed specific primers, which would selectively amplify the Pkd2Δ7 or Pkd2 transcripts and used these further in our real-time RT–PCR assays (Fig. 3 B, scheme). Pkd2ex6/ex8.F/Pkd2ex8.R2 amplification results in a PCR product of 169 bp, characteristic of the Pkd2Δ7 splice form and Pkd2ex7/ex8.F/Pkd2ex8.R2 amplifies exon 7, when present in the transcript, with diagnostic amplicon of 174 bp (Fig. 3 B, gel image). To confirm the existence of the PKD2Δ7 (human) splice form, we amplified exon 7 from various cell lines using primers PKD2ex6.F and PKD2ex8.R. An amplicon of 390 bp is diagnostic for the presence of exon 7 sequence in the PCR product, whereas the shorter 222 bp amplicon is specific for the PKD2Δ7 variant (Fig. 3 C).
We wanted to verify that an alternative PKD2/Pkd2 transcript lacking exon 7 could be translated, thus having possible functional relevance. For this purpose, we amplified exon 7 and surrounding sequence again, from a translated mRNA fraction of human microvascular endothelial cell (HMEC; Fig. 3 D). Both PKD2 and PKD2Δ7 amplicons were obtained indeed using this fraction as a template. In contrast, with primers PKD2ex8.F and Pkd2ex10.R, we were able to generate an amplicon of 301 bp only, indicative of PKD2, and no 180 bp amplicon, specific for PKD2Δ9. Using same primer sets and amplification conditions, we obtained amplicons of exons 7 and 9 of PKD2 together with amplicons of their alternate splice variants, PKD2Δ7 and PKD2Δ9, from total RNA (Fig. 3 E). In additional control reactions for amplification of PKD1 and PKD1-pseudogenes ( 25 ), we were able to generate only PKD1-specific amplicons from HMEC-1 mRNA polysomal fraction and both PKD1-specific and PKD1-pseudogenes-indicative amplicons from HMEC-1 total RNA (data not shown).
The Pkd2Δ7/PKD2Δ7 protein of 910/912 amino acids has a predicted relative molecular mass of 103 kDa and p I of 5.51/5.53, which is not very different from the p I value calculated for the Pkd2/PKD2 proteins (5.47/5.49).
Tissue distribution of Pkd2Δ7
To trace the expression pattern of the Pkd2Δ7 splice variant in comparison with Pkd2, we performed quantitative real-time RT–PCR analysis on murine tissues from different developmental stages, 16.5 days post-conception (pc) old embryo, newborn and adult mice (Fig. 4 A–C). Amplifications were performed using the specific exon 7, delta exon 7 formats generated to discriminate the Pkd2Δ7 from other Pkd2 transcripts. Data sets for each tissue and developmental stage compared Pkd2 and Pkd2Δ7 expression and consisted of three independent analyses on three different animals. Only for adult tissues, Pkd2Δ7 expression, two animals were used for the analysis (Fig. 4 C). Expression values for each tissue in the corresponding panel, Pkd2 or Pkd2Δ7, were normalized to expression levels in kidney, ‘kidney=1’. Despite individual physiological differences in particular tissues of the animals analyzed, general expression patterns for Pkd2Δ7 in comparison with Pkd2 could be concluded. From all developmental stages, Pkd2 seemed to be expressed at most in kidney and second strong in lung, whereas Pkd2Δ7 peaked exclusively in brain and retained some lower expression in kidney and lung. Pkd2Δ7 was expressed generally on very low levels in thymus and virtually no expression could be detected in liver.
The embryonic expression patterns of Pkd2 and Pkd2Δ7 at day 16.5 pc almost overlapped, although the trend for comparatively high levels of Pkd2Δ7 mRNA in brain already emerged and no Pkd2Δ7 liver representation as well as very low thymus expression was evident (Fig. 4 A). Both Pkd2 and Pkd2Δ7 were better expressed in kidney at this stage, with expression of declining strengths in skeletal muscle, stomach and skin. Calculated expression of Pkd2 mRNA in kidney was 3.6×10 −4 pg/µg RNA and Pkd2Δ7 mRNA was represented in kidney at 2.7×10 −5 pg/µg RNA. Maximum of Pkd2Δ7 expression compared to Pkd2 was 6.4% (animal 1, brain).
In newborn, Pkd2 expression in kidney and lung was strongest, Pkd2 mRNA levels in skeletal muscle, stomach and skin gradually diminished and expression in thymus was very low (Fig. 4 B). Pkd2Δ7 peaked in brain; there was some expression in kidney and lung and no detectable expression in liver and thymus. Levels of Pkd2Δ7 mRNA in skeletal muscle, stomach and skin gradually diminished. Pkd2 mRNA level in kidney was about 2.9×10 −3 pg/µg RNA and Pkd2Δ7 was represented with 10 −5 pg/µg RNA. Maximum of Pkd2Δ7 expression compared to Pkd2 was 3.1% (animal 5, brain).
In adult animals (8 weeks), kidney and lung expression of Pkd2 remained comparatively high. Low expression was observed in brain and heart and basal expression levels for the other examined tissues were maintained (Fig. 4 C). Both Pkd2 and Pkd2Δ7 were not expressed in skeletal muscle. Pkd2Δ7 had some comparatively low expression in kidney and lung and higher mRNA levels in brain. A very low Pkd2Δ7 basal expression was observed for the rest of tissues. Pkd2 mRNA in kidney is 4×10 −3 pg/µg RNA and level of Pkd2Δ7 mRNA is about 6×10 −6 pg/µg RNA. Maximum of Pkd2Δ7 expression compared to Pkd2 was 3.0% (animal 7, brain).
Characterization of anti-Pkd2-Ct antibodies
We designed our (GST)–Pkd2 fusion protein in a very similar fashion to the already described (GST)–PKD2 (human) C-terminal fusion for the production of anti-PKD2 antibodies, directed to the last 280 amino acids of polycystin-2 ( 26 ). This region of polycystin-2 with predicted cytosolic location demonstrated good antigenic properties and was well detectable with the affinity-purified Pkd2-Ct antiserum as a GST fusion protein, in lysates and as glutathione sepharose-bound material (Fig. 5 A, lanes 1 and 2). No signal could be detected in bacterial lysates from culture, induced to express GST only (Fig. 5 A, lane 3). The antibodies detected a band of ∼110 kDa in mouse kidney lysate (Fig. 5 B, lane 1) and a band of same relative molecular mass in HMEC-1 cells lysate (Fig. 5 B, lane 2). In HeLa cell lysates, no detectable protein bands could be visualized with these antibodies (data not shown). Immunoprecipitation with Pkd2-Ct antibodies on lysates of PKD1/Pkd2- (Fig. 5 C, lane 2) and PKD1/Pkd2Δ7 (Fig. 5 C, lane 3)-transfected HeLa cells resulted in defined protein bands of ∼110 kDa (Pkd2) and ∼103 kDa (Pkd2Δ7), detectable with c-myc antibodies. The ∼103 kDa band was matching the expected relative molecular mass of polycystin-2Δ7. Transfection with PKD1 only (Fig. 5 C, lane 1) yielded no signal under these conditions.
Pkd2Δ7 expression in cell culture and interaction with PKD1
To analyze conceivable differences in respect to the cellular function of polycystin-2 isoforms, we cloned the Pkd2 and Pkd2Δ7 cDNAs in a eukaryotic expression vector (see Plasmid Constructs). To characterize interactions of ectopically expressed Pkd2 and Pkd2Δ7 with PKD1, we co-immunoprecipitated polycystin-1 and polycystin-2/polycystin2Δ7 from cell lysates of Pkd2/Pkd2Δ-transfected PKD1 transgenic cells with antibodies recognizing the tagging epitopes of interacting molecules (Fig. 6 ). MDCK AF-20 PKD1Zeo cells ( 27 ) (Fig. 6 , lanes 1) were transfected with Pkd2 or Pkd2Δ7 constructs for ectopic expression of cloned cDNAs (Fig. 6 , lanes 2 and 3). HEK-293 cells transfected with same cDNAs served as controls (Fig. 6 , lanes 4 and 5). PKD1 protein (full-length, FL, upper band and N-terminal cleavage fragment, NTF, lower band ( 28 )) were detected using PKD1-LRR antiserum ( 28 ) on anti-FLAG M2 immunoprecipitates in all three lanes of AF-20 PKD1Zeo cell lysates (Fig. 6 A, panel 1, lanes 1–3) as well as in c-myc Pkd2 co-immunoprecipitates (Fig. 6 A, panel 2, lane 2), but not (signal close to background) in c-myc Pkd2Δ7 co-immunoprecipitates (Fig. 6 A, panel 2, lane 3). As expected, Pkd2- and Pkd2Δ7-transfected HEK-293 cells gave no PKD1 signal under these conditions (Fig. 6 A, panels 1 and 2, lanes 4 and 5). Surprisingly, the amount of PKD1 NTF cleavage product was enhanced in Pkd2 co-immunoprecipitates (Fig. 6 A, lane 2 of panel 2). We were able to detect Pkd2 and PkdΔ7 proteins with c-myc antibodies in all anti-c-myc immunoprecipitates of Pkd2- and PkdΔ7-transfected cells (Fig. 6 B, lanes 2–5, panel 4) and in anti-FLAG M2 co-immunoprecipitates of Pkd2- and PkdΔ7-transfected AF-20 PKD1Zeo cells, reciprocal to panel 2 (Fig. 6 B, lanes 2 and 3, panel 3). A band of ∼110 kDa, characteristic of the Pkd2 protein could be observed in lane 2 of panel 3 and in lanes 2 and 4 of panel 4 (Fig. 6 B). Another band of ∼103 kDa, which would correspond to a shortened protein product lacking exon 7, was detected in lane 3, panel 3 and in lanes 3 and 5 (Fig. 6 B). As expected, no such bands could be observed in lane 1 (AF-20 PKD1Zeo cell lysates) of both panels in Figure 6 B, as well as in lanes 4 and 5 of panel 3 (anti-FLAG M2 immunoprecipitates of Pkd2-transfected HEK-293 cells). The characteristic 55 kDa band of the immunoprecipitating antibody heavy chains was seen across all lanes in Figure 6 B under these conditions. The bands close below the Pkd2 and Pkd2Δ7 protein bands in Figure 6 B, panel 4, as well as the high relative molecular mass bands above 250 kDa observed in all five lanes of panel 4 were additional bands of uncertain nature. Same blots as in Figure 6 B were stripped and immunoprecipitation products were detected with PKD1-CT antibodies ( 27 ), Figure 6 C, to exclude unspecific antibodies absorption. PKD1-CT antibodies detected the PKD1 GPS cleavage product ( 28 ), PKD1-CTF, as well as additional polycystin-2-enhanced polycystin-1 cleavage product ( 29 ), PEC, in polycystin-2 co-immunoprecipitates. In lanes 1–3 of Figure 6 C, panel 5 (polycystin-1 immunoprecipitated with anti-FLAG M2 antibodies), CTF of ∼150 kDa could be detected. Interestingly, CTF was enhanced in Pkd2 co-immunoprecipitates (lane 2) and almost absent in Pkd2Δ7 co-immunoprecipitates (lane 3). Conversely, the PEC fragment of ∼60 kDa ( 29 ), observed in Pkd2/Pkd2Δ7 co-immunoprecipitates (lanes 2 and 3 of panel 5), was enhanced in Pkd2Δ7 co-immunoprecipitates. The possibility that the observed differences in CTF and PEC cleavage products in the Pkd2 and Pkd2Δ7 lanes are artifacts of skewed antibodies sensitivity was ruled out by the c-myc co-immunoprecipitation results in panel 6 of Figure 6 C. PKD1-CT antibodies detected predominantly PKD1-CTF in lane 2 (Pkd2) and exclusively PKD1-PEC in lane 3 (Pkd2Δ7) of panel 6.
To address the situation of different molecular topologies of the Pkd2 and Pkd2Δ7 proteins, in accordance with the results of our co-immunoprecipitation experiments, we performed computer-assisted topology modeling using the PSORT ( 30 ) and TMHMM ( 31 ) servers. The models predicted for polycystin-2Δ7 loss of the third transmembrane domain, generated from parts of exons 6 and 7 and its replacement through a new transmembrane span built out of relevant exon 6 and exon 8 regions (Fig. 8 B). In comparison to polycystin-2 (Fig. 8 A), polycystin-2Δ7 had one predicted transmembrane domain less and as a consequence of this (PSORT II), reversed topologies of the third and fourth extracellular/intracellular loops, as well as predicted reversed polarity (extracellular) of the C-terminus (cytosolic in polycystin-2).
We wanted to verify experimentally the predicted extracellular C-terminus of polycystin-2Δ7, which would reconcile disputable differences to the possibility of Pkd2Δ7/PKD1 interaction, arising from the results of our co-immunoprecipitation experiments. For this purpose, we performed localization studies of ectopically expressed Pkd2 and Pkd2Δ7 in HeLa cells.
Attempts to express polycystin-2 or polycystin-2Δ7 as N- or C-terminal fusions with fluorescent proteins (YFP or dsRed) in cell culture definitely led to accumulation of protein aggregates with unnatural subcellular localizations (data not shown). Immunofluorescent detection of ectopically expressed Pkd2Δ7 from a bi-cistronic EGFP carrying construct (no protein fusion) in permeabilized HeLa cells with anti-Pkd2-Ct antibodies resulted in essentially the same staining pattern observed for Pkd2-expressing cells (compare Fig. 7 B and D). This was characteristic ER staining, documented for polycystin-2 expression also by others ( 10 , 32 ). The signal was specific, with low background staining (compare with Fig. 7 A).
However, on non-permeabilized HeLa cells, the antibodies specifically recognized external epitopes (surface staining) in patched membrane regions of Pkd2Δ7-expressing cells (Fig. 7 F) in contrast to Pkd2 transfectants (Fig. 7 H), where no such signal was to be detected. Figure 7 A, C, E and G are images of EGFP produced in permeabilized and non-permeabilized cells; this serving as transfection marker for Pkd2 (A, G) and Pkd2Δ7 (C, E)-expressing cells.
DISCUSSION
Exon skipping and in-frame exon exclusion seem to be common mechanisms for generation of alternative transcripts from the PKD2L and PKD2L2/Pkd2L2 genes ( 19 , 20 ). In the case of PKD2, alternative transcription has been mainly documented as a result of pathologic mutation change leading to ADPKD phenotype ( 24 ). In our study, we identify three major and one minor naturally occurring PKD2/Pkd2 alternative transcripts. From our results, alternative Pkd2 splicing patterns seem conserved from mouse to dog and human, thus common for mammals. We also showed that two of the major splice forms, one lacking half of exon 6, and the other, with fully excluded exon 9, are not detectable as translation template in the mRNA pool of polysomal fractions. Apparently, translationally incompetent PKD2/Pkd2 mRNA species are segregated in the process of nonsense-mediated mRNA decay ( 33 , 34 ).
We concentrated our efforts to study better the polycystin-2Δ7 splice variant, because it seems to be one of the major alternative PKD2/Pkd2 splice forms and it is also found in polysomal fractions, indicative of translational competence. Using quantitative real-time RT–PCR analysis, we were able to trace the expression pattern of Pkd2Δ7 in comparison with Pkd2 in murine tissues of different developmental stages. The observed Pkd2 expression pattern correlates with data on polycystin-2 distribution on the mRNA and protein levels obtained by others ( 5 , 35 , 36 ). Relative amounts and tissue distribution of Pkd2 and Pkd2Δ7 transcripts are developmentally regulated. At all three developmental stages studied, embryo 16.5 pc, newborn and adult organism, best representation of Pkd2 mRNA was detected in kidney, whereas the Pkd2Δ7 transcript was mostly present in brain. Maximum of recorded Pkd2Δ7 expression on the mRNA level was 3–6.4% of the Pkd2 transcripts therein. This could be related to some organ-relevant function of polycystin-2Δ7, although it is generally known that brain is a common source of alternative transcripts.
Our cell culture expression studies of ectopic Pkd2Δ7 expression showed that polycystin-2Δ7 should be stable as a protein molecule and could be produced at levels comparable to polycystin-2 (Figs 5 C and 7 B). The interaction of Pkd2Δ7 and PKD1 proteins was difficult to judge from the co-immunoprecipitation experiments we performed. Immunoprecipitation using Pkd2-Ct antibodies followed by PKD1 detection (PKD1-LRR antibodies) (Figure 6 A, panel 2, lane 3) showed marginal (if at all) interaction of both molecules. In contrast, reciprocal immunoprecipitations revealed equal amounts of Pkd2 and Pkd2Δ7 proteins (Fig. 6 B, panel 3). Detection of the Figure 6 B blots with PKD1-CT antibodies (Fig. 6 C) gave us the possibility to control the Figure 6 B result and at the same time to evaluate conceivable differences in the interaction specificities/PKD1 cleavage events mediated by Pkd2 and Pkd2Δ7 proteins. The interaction of Pkd2 with PKD1 seemed to release more PKD1-CTF (Fig. 6 C, panel 5, lane 2, compare with lane 1), which is corroborated by the result in lane 2 of panel 2, Figure 6 A—more NTF is obviously generated and tethered to PKD1-FL as compared to lane 1 of panel 2 (PKD1 only). Conversely, interaction of Pkd2Δ7 with PKD1 seemed to ‘shield’ this cleavage (compare the amounts of CTF in lanes 2 and 3 of Fig. 6 C) and enhance the PEC cleavage reaction (PEC band intensities in same lanes of Fig. 6 C). Although potentially interesting for the molecular topology understanding of polycystin-2-mediated polycystin-1 cleavage events and topical interrelations of interacting sites, these results bear no defined physiological significance. Moreover, the possibility of Pkd2Δ7 interaction with PKD1 in our experimental conditions might have been mediated by the presence of intact coiled-coil domains in the Pkd2Δ7 C-terminus ( 11 , 12 ) and the physical vicinity of both molecules in the stochastic medium of lysed cellular compartments, which might have not reflected the real in vivo situation. This is why it was important to determine the subcellular localization of polycystin-2Δ7 C-terminus, potentially interacting with polycystin-1. Immunofluorescence detection demonstrated extracellular location of Pkd2Δ7 C-terminus in Pkd2Δ7-expressing cells (compare Fig. 7 E and F). A topology model of the polycystin-2Δ7 molecule (Fig. 8 B) shows the possibility of such location as compared to the predicted cytosolic location for the polycystin-2 C-terminus (Fig. 8 A). The incapability of polycystin-2Δ7 interaction with polycystin-1 arises then from opposite orientation of the Pkd2Δ7 C-terminus, which makes it impossible for polycystin-2Δ7 to associate with polycystin-1, after ongoing synthesis and export from the endoplasmic reticulum. Apparently a fraction of polycystin-2Δ7 molecules is able to reach the plasma membrane, independent of the PKD1 interaction indicating that such trafficking could depend on association with PACS-1 and PACS-2 proteins ( 37 ). Such a PKD1-independent travel to the plasma membrane has indeed been shown for a PKD2 C-terminal truncated mutant ( 38 ).
Polycystin-2Δ7 should have a polycystin-1-independent role, which comes in line with its distinct expression pattern. ‘Standalone’ function for polycystin-2 as a ciliar flow-induced calcium-signaling sensor ( 7 ) has been documented. It is not difficult to imagine that polycystin-2Δ7 could also possess some unknown functional relevance, in particular, when considering the high interaction potential of the extracellular C-terminus with possible ligands.
If polycystin-2Δ7 inserts in the plasma membrane correctly, as depicted in the model (Fig. 8 B), it is possible that this protein preserves TRP channel activity as it could be capable of dimerization through the intact coiled-coil domain at the extracellular C-terminus. What could be the function of a TRPP2 channel variant (the PKD2Δ7 splice form) in brain? If expressed in certain neurons, this new channel could inhibit neurite outgrowth, similar to TRPC5 in hippocampal neurons ( 39 ). Ca 2+ influx through not yet identified ion channels regulates neuronal growth cones and elevated calcium transients inversely correlate with growth cone extension rates ( 40 ). Thus, polycystin-2Δ7 in the plasma membrane of neurons could be one of the channels contributing calcium currents and its controlled availability at the cell membrane could be a factor in axon growth control.
Another attractive possibility would be that polycystin-2Δ7 participates in the process of excitatory postsynaptic conductance (EPSC) as a downstream effector of group I metabolotropic glutamine receptors (mGluRs), similar to the reported action of TRPC1 ( 41 ). Several TRP channels of the TRPC and the TRPV families and perhaps also others are discussed as potential EPSC mediators activated by mGluR1 in dopamine neurons, as identified by various TRP channel blockers ( 42 ).
As polycystin-2Δ7 expression could be well detected on the mRNA level in cultured cells of the microvasculature (HMEC-1), an interesting possibility could be that clustering of intracranial aneurysms reported for ADPKD in PKD2 families ( 43 ) is actually reflecting pathologic changes in this alternate transcript form. A finer immunohistochemical analysis is necessary to localize the sites of polycystin-2/polycystin-2Δ7 expression in brain tissue. It would be of advantage to create specific reagents for better study of PKD2Δ7/Pkd2Δ7 on the protein level. Unfortunately, PKD2/Pkd2 and PKD2Δ7/Pkd2Δ7 show very similar predicted relative molecular masses and isoelectric point values, which makes the successful distinction a rather difficult task. Generation of specific antibodies against defined PKD2/Pkd2 domains (exon 7) to discriminate polycystin-2 appears also rather difficult, as the amino acid sequence of this region exhibits a very low antigenic potential.
The minor Pkd2 splice form identified here, lacking exons 12 and 13, is also predicted not to interact with polycystin-1 on the protein level. Although transmembrane topology would not be reversed for this molecule, it would lack the coiled-coil domain encoded by exon 12 and necessary for the interaction with polycystin-1. We have identified a mutation in PKD2 in a Roma (Gypsy) ADPKD family, c.2356_2357delAG(p.R787fs), which would generate a truncated polycystin-2 protein lacking exons 14 and 15, if translated. Although the resulting protein variant is predicted to be capable of interaction with polycystin-1, the pathological expression of this mutation is a typical disease phenotype. Apparently, protein regions encoded by exons 14 and 15 could be rather important for polycystin-2 function. The minor Pkd2 splice form we identified, Pkd2Δ12–13, would have intact exons 14 and 15. Although incapable of interaction with polycystin-1, the resulting protein could have some unknown functional importance.
Altogether our study contributes to the accumulating wealth of polycystin molecules, possibly identifying some common rules for generation of other polycystin-2 species and highlighting putative new functional aspects.
MATERIALS AND METHODS
Total RNA extraction from cells and tissue
Total RNA was prepared from confluent cell cultures of HMEC-1, human umbilical vein endothelial cells (HUVECs), human lung carcinoma epithelial cells (A549), human glioblastoma cells (T98G), Madine Darby canine kidney epithelial cells (MDCK) and mouse embryo and adult and embryonic tissues using the Trizol reagent (Invitrogen, Karlsruhe, Germany) according to the provided protocol. Total RNA for real-time quantitative PCR was purified with the Qiagen RNeasy kit (Qiagen, Hilden, Germany) as per the manufacturer's instructions.
Preparation of translated mRNA from polysomal fraction
Translationally active mRNA was prepared from a polysomal fraction (membrane-bound polysomes) of 10 10 HMEC-1 cells as previously described ( 44 ). Briefly, the microsomal fraction was obtained through sucrose gradient centrifugation of an HMEC-1 cytoplasmic extract and mRNA was separated and purified from the bound polysomes.
Reverse transcription and PCR amplification of cDNA
Total RNA (∼1 µg) and mRNA (0.05–0.1 µg) were subjected to reverse transcription using the SuperScript III/II reverse transcriptase and reaction conditions specified by the manufacturer (Invitrogen, Karlsruhe, Germany). Reactions were primed with 100 µ m oligo (dT) 12–18 primer.
Single-strand cDNA synthesis products diluted 10× were used as templates for subsequent PCR amplifications. Reactions were performed in a total volume of 25 µl and the reaction mix contained the following: 50 m m Tris–Cl, pH 9.5, 20 m m (NH 4 ) 2 SO 4 , 1 m m DTT, 0.005% NP-40, 1.5 m m MgCl 2 , 1 m betaine, 5% DMSO, 20 p m of each primer, 300 µ m dNTP, 10–20 ng cDNA and 1.25 U Taq polymerase available from a local source (Institute of Molecular Pathology, ZMBE, University of Muenster, Muenster, Germany).
Mouse embryo cDNA and a human fetal tissue embryonic cDNA library (Clontech, Heidelberg, Germany) were scanned for alternative transcripts of the Pkd2/PKD2 genes with primers complementary to coding regions of exons adjacent to single exons or to exon pairs to be amplified. Exon 7 of Pkd2 (murine), for example, was amplified using primers Pkd2ex6.F and Pkd2ex8.R (Table 1 ). Similarly, PKD2 exon 7 (human) was amplified using primers PKD2ex6.F and PKD2ex8.R and exon 9 was amplified with primers PKD2ex8.F and PKD2ex10.R. Specific amplification of the Pkd2Δ7 splice variant was achieved using primers Pkd2ex6/8F and Pkd2ex8.R2. Control reaction for specific amplification of Pkd2 exon 7 was performed with primers Pkd2ex7/8.F and Pkd2ex8.R2. Both reactions were further used for a real-time RT–PCR analysis. Typical PCRs were 35 cycles of denaturation at 95°C for 30 s; annealing at the appropriate primer annealing temperature (Table 1 ) for 20 s and extension at 72°C for 30 s.
Sequencing analysis
Sequencing reactions were performed in both directions on approx. 100 ng of purified PCR product using the BigDye Terminator Cycle Sequencing Ready Reaction Kit (ABI/Perkin-Elmer, Weiterstadt, Germany) and the amplification primers. Sequence products were purified with the MultiScreen™-HV plates (Millipore, Eschborn, Germany), according to the manufacturer's instructions and analyzed on an ABI PRISM® 3700 DNA Analyzer (ABI/Perkin-Elmer, Weiterstadt, Germany).
Predictive computer modeling
Computer-assisted topology modeling and transmembrane region prediction for polycystin-2 and the polycystin-2Δ7 variant were performed using PSORT II ( 31 ) on the PSORT server ( http://psort.ims.u-tokyo.ac.jp/form2.html ) and TMHMM ( 32 ) on the TMHMM server, version 2.0 ( http://www.cbs.dtu.dk/services/TMHMM/ ).
Real-time RT–PCR analysis
A Rotor Gene 3000 instrument (Corbett Research, Sydney, Australia) was used for quantitative real-time RT–PCR measurements. Reverse transcription and quantitative PCR reactions were performed with One-Step RT–PCR Master Mix Reagents (ABI/Perkin-Elmer, Weiterstadt, Germany) as specified by the manufacturer. For amplification of Pkd2, exon 7 and the Pkd2Δ7 splice variant primers Pkd2ex7/8.F/Pkd2ex8.R2 and Pkd2ex6/8.F/Pkd2ex8.R2 were used in separate reactions accordingly. A Taqman probe (Table 1 ) was used for quantitative detection. The quantity of cDNA for Pkd2 and Pkd2Δ7 was normalized to different quantities (100, 10, 1 and 0.5 ng) of the cloned full-length Pkd2 and Pkd2Δ7 cDNAs, used as templates for standard reactions. Internal cDNA synthesis controls were included in each reaction using the Taqman Rodent GAPDH Control Reagents VIC dye (ABI/Perkin–Elmer, Weiterstadt, Germany). Measurements for each tissue were performed three times, on RNA from three different animals.
Generation of anti-polycystin-2 antibodies
The region encoding the last 279 amino acids of the Pkd2 C-terminus (nucleotide positions 2145–2984 in the Pkd2 cDNA) was cloned into pGEX-4T (Amersham Biosciences, Freiburg, Germany) to produce a glutathione- S -transferase (GST)–Pkd2 bacterial fusion protein, essentially as described for the production of anti-PKD2 (C-term) antibodies ( 26 ). Polyclonal antiserum was raised in New Zealand White rabbits (Imgenex, San Diego, CA, USA). The serum (Pkd2-Ct) was affinity-purified and tested positive against the GST fusion protein and negative against GST itself (Fig. 5 A).
Plasmid constructs
The PKD1-F plasmid represents full-length PKD1 cDNA, cloned in pCI-CMV and modified to include a C-terminal Flag epitope ( 14 ). Pkd2-M and Pkd2Δ7-M plasmids we prepared by cloning full-length Pkd2 cDNA and the full-length transcript lacking exon 7 accordingly, into a modified pCI-CMV vector, pCI-CMV-IRES-GFP (a kind gift from D. Trouet, Laboratory of Physiology, KU Leuven, Leuven, Belgium). The constructs were engineered to harbor a c-myc tag at their respective C-termini.
Cell culture, transfection and immunofluorescence
Transient transfections were performed on subconfluent HeLa cells, using the Effectene transfection reagent (Qiagen, Hilden, Germany) and on HEK-293 cells and transgenic MDCK PKD1-expressing clone AF-20 ( 28 ) with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) as specified by the manufacturers. The pCI-CMV vector (Promega, Madison, WI, USA and Mannheim, Germany) was used as a negative control for expression. Cells were harvested 24 h after transfection for protein immunoprecipitation, western blot analysis and immunofluorescence. For antibodies staining cells were fixed with 4% paraformaldehyde in PBS (pH 7.5) for 5 min, followed by incubation in 50 m m NH 4 Cl/PBS for 5 min. Cells to be permeabilized were treated for 20 min with 0.1% Triton X-100 and 1.5% goat serum (Sigma, Munich, Germany) in PBS and cells for external epitope staining were pre-incubated with 1.5% goat serum/PBS only for the same time. Cells were then incubated with anti-Pkd2-Ct antibodies for 30 min, washed (discussed subsequently) and stained with Texas Red-conjugated goat anti-rabbit immunoglobulin (Ig)G antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) for 20 min. We washed the specimens six times in PBS (for surface-stained cells) and with 0.1% Triton X-100 in PBS (for permeabilized cells) and mounted them for imaging. Microscopy was performed on a Leica DM-RXA epifluorescence microscope (Leica, Bensheim, Germany) and photographs were taken with a MicroMax CCD camera (Visitron Systems, Puchheim, Germany).
Immunoprecipitation and western blot analysis
Cells were lysed in 10 m m sodium phosphate buffer with 1% Triton X-100, as previously described ( 14 ). Cleared lysates were incubated with 2 µg of 9e10 monoclonal anti-myc antibody (Roche, Indianapolis IN, USA and Mannheim, Germany) or equivalent amount of Pkd2-Ct antibodies (Fig. 5 C) for 1 h at 4°C. Then 50 µl of Protein G Sepharose beads (Amersham Biosciences, Piscataway, NJ, USA and Freiburg, Germany) were added to the samples and they were incubated for another hour at 4°C. Immunoprecipitates were washed five times with 1 ml of lysis buffer and eluted in 60 µl of 1× Laemmli loading buffer. We used M2-FLAG-conjugated agarose beads (Sigma, St Louis, MO and Munich, Germany) to immunoprecipitate FLAG-tagged products. Ten to fifteen microliters of the eluted products were subjected to polyacrylamide gel electrophoresis (4% acrylamide, panels 1 and 2; 6% acrylamide, Fig. 5 C 3–8% gradient acrylamide gels (Invitrogen, Carlsbad, CA, USA), panels 3–6) and analyzed using standard western blot protocols (ECL, Amersham Biosciences, Piscataway, NJ, USA and Freiburg, Germany).
ACKNOWLEDGEMENTS
We are grateful to Hangxue Xu for the excellent technical assistance. This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG) to B.D., P.P. and J.H.; from Interdisziplinäres Zentrum für Klinische Forschung (IZKF), Universitätsklinikum Münster to B.D., J.H. and V.G.; from NIH grants P50 DK57325 to G.G. and F.Q., and R01 DK062199 to F.Q.
Conflict of Interest statement . Authors have declared no conflict of interest.
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.

Figure 1. PKD2 (human)/Pkd2 (murine) alternative transcripts identified in this study. Exons are boxed in black, drawn to scale. Alternative transcripts are named after lacking exons or parts thereof, preceded by a ‘Δ’, for deletion. Intron phases for the human/murine transcripts are indicated on respective intron positions, separated by a slash ‘/’.
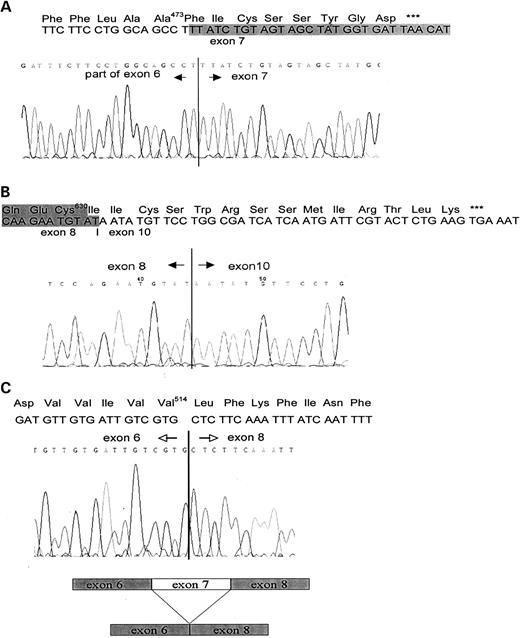
Figure 2. Sequencing analysis of Pkd2/PKD2 alternate splice forms: ( A ) Pkd2Δ6; ( B ) Pkd2Δ9; ( C ) Pkd2Δ7.
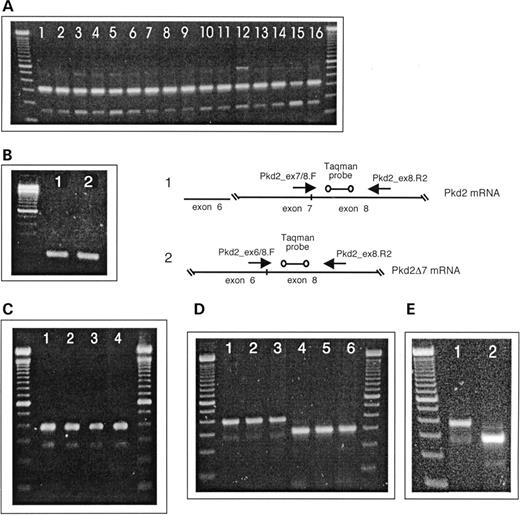
Figure 3. RT–PCR amplifications of Pkd2/PKD2 exon 7 and PKD2 exon 9 formats. DNA size standard is the 100 bp DNA ladder (Invitrogen). ( A ) Pkd2 exon7/Pkd2Δ7 amplification from total RNA of murine tissues. Lane 1, skeletal muscle; lane 2, intestines; lane 3, spleen; lane 4, kidney; lane 5, skin; lane 6, lung; lane 7, testes; lane 8, pancreas; lane 9, eye; lane 10, adrenal gland; lane 11, heart; lane 12, brain; lane 13, stomach; lane 14, thymus; lane 15, liver; lane 16, embryo, ∼16.5 pc. ( B) Specific amplification of Pkd2, exon 7 and the Pkd2Δ7 variant from total RNA (mouse embryo). Lane 1, 174 bp PCR product characteristic of exon 7; lane 2, 169 bp PCR product indicative of Pkd2Δ7. A real-time PCR approach scheme based on specific amplification of Pkd2 exon 7 and lack of exon 7 diagnostic prdoducts. ( C ) PKD2 exon7/Pkd2Δ7 amplification from total RNA of human cell lines. Lane 1, T98G; lane 2, HMEC-1; lane 3, HUVEC; lane 4, A549. ( D ) PKD2 exon7/Pkd2Δ7 (lanes 1–3) and Pkd2 exon 9/Pkd2Δ9 (lanes 4–6) amplifications from polysomal fraction mRNA of HMEC-1 cells. Amplicon bands indicative of Pkd2 exon 7 (390 bp) and Pkd2Δ7 (222 bp) can be seen in lanes 1–3. In contrast, only a PCR product of 301 bp characteristic for exon 9 and no Pkd2Δ9 diagnostic band of 180 bp is to be seen in lanes 4–6. ( E ) PKD2 exon7/Pkd2Δ7 (lane 1) and Pkd2 exon 9/Pkd2Δ9 (lane 2) amplifications from total RNA of HMEC-1 cells.
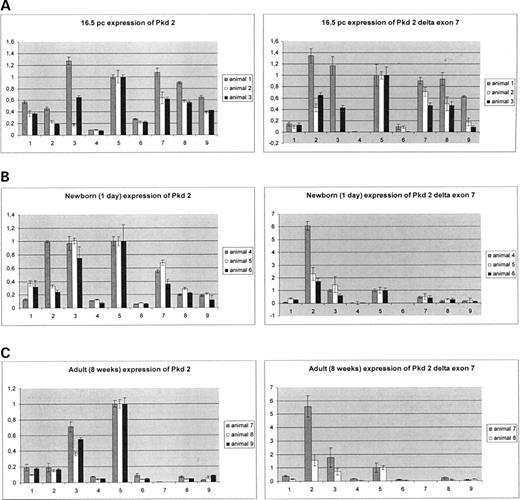
Figure 4. Quantitative real-time RT–PCR analysis of Pkd2 (left panels) and Pkd2Δ7 (right panels) transcripts in nine murine tissues. 1, heart; 2, brain; 3, lung; 4, liver; 5, kidney; 6, thymus; 7, skeletal muscle; 8, stomach; 9, skin, different developmental stages: ( A ) embryo 16.5 pc; ( B ) newborn (1 day); ( C ) adult (8 weeks).
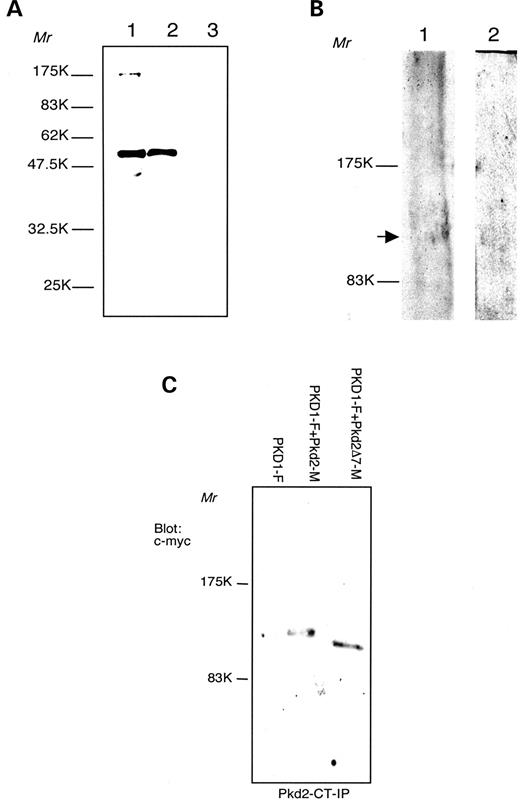
Figure 5. Characterization of the anti-Pkd2-Ct antibodies. ( A ) Bacterial expression of (GST)–Pkd2-Ct fsuion protein, predicted relative molecular mass 57.9 kDa. Western blot detection with Pkd2-Ct antibodies, 12% polyacrylamide gel (PAAG). Lane 1, glutathione sepharose 4B beads, loaded with (GST)–Pkd2-Ct; lane 2, culture lysate of (GST)–Pkd2-Ct expressing Escheirichia coli XL-1; lane 3, culture lysate of GST-expressing E. coli XL-1 (negative control). ( B ) Western blot analysis with Pkd2-Ct antibodies, 6% PAAG. Lane 1, mouse kidney lysate; lane 2, HMEC-1 cells lysate. Arrow points at polycystin-2 band. ( C ) Immunoprecipitation of polycystin-2 and polycystin-2Δ7. Cell lysates are obtained from HeLa cells transfected with PKD1-F (lane 1); PKD1-F+Pkd2-M (lane 2) and PKD1+Pkd2Δ7−M (lane 3). Products immunoprecipitated (IP) with anti-Pkd2-Ct antibodies and detected with anti c-myc antibodies.
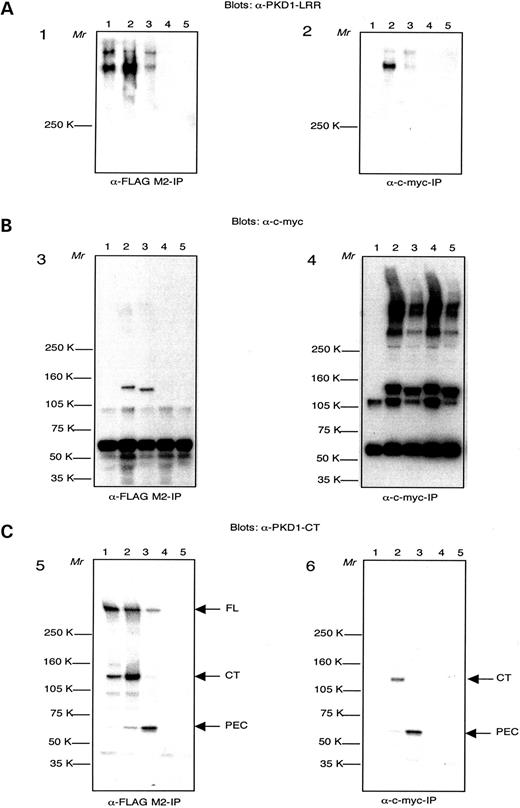
Figure 6. Co-immunoprecipitation of polycystin-1 and -2. Cell lysates are obtained from a transgenic PKD1-expressing MDCK cell clone AF-20 (lanes 1); AF-20 transfected with Pkd2-M (lanes 2); AF-20 transfected with Pkd2Δ7-M (lanes 3); HEK-293 cells transfected with Pkd2-M (lanes 4); HEK-293 cells transfected with Pkd2Δ7-M (lanes 5). Panels 1, 3 and 5: products immunoprecipitated (IP) with anti-FLAG M2 antibodies. Panels 2, 4 and 6: products immunoprecipitated (IP) with anti-c-myc antibodies. ( A ) IP products detected with anti-PKD1-LRR antsierum. ( B ) IP products detected with anti-c-myc antibodies. ( C ) Blot from (B) was stripped and IP products were detected with anti-PKD1-CT antiserum. Arrows point at full-length polycystin-1 (FL), polycystin-1 C-terminal fragment (CTF) and polycystin-1 polycystin-2 enhanced cleavage (PEC) product.
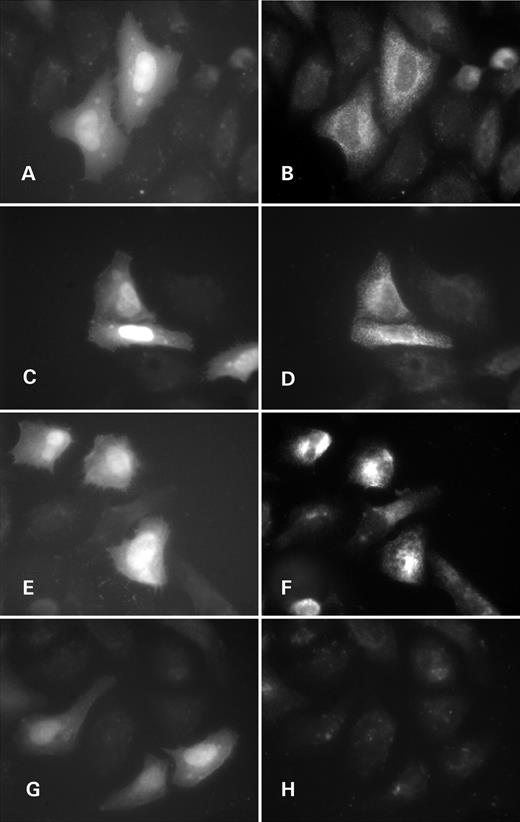
Figure 7. Subcellular localization of polycystin-2 and polycystin-2Δ7 in HeLa cells by immunofluorescence. ( A, B, G and H ) Pkd2-transfected cells; ( C – F ) Pkd2Δ7-transfected cells; (A, C, E and G) EGFP marks transfected cells (bi-cistronic expression of Pkd2/Pkd2Δ7 and EGFP); (B, D, F, andH) Pkd2 C-term antibodies staining; (A–D) permeabilized cells; (E–H) non-permeabilzed cells.
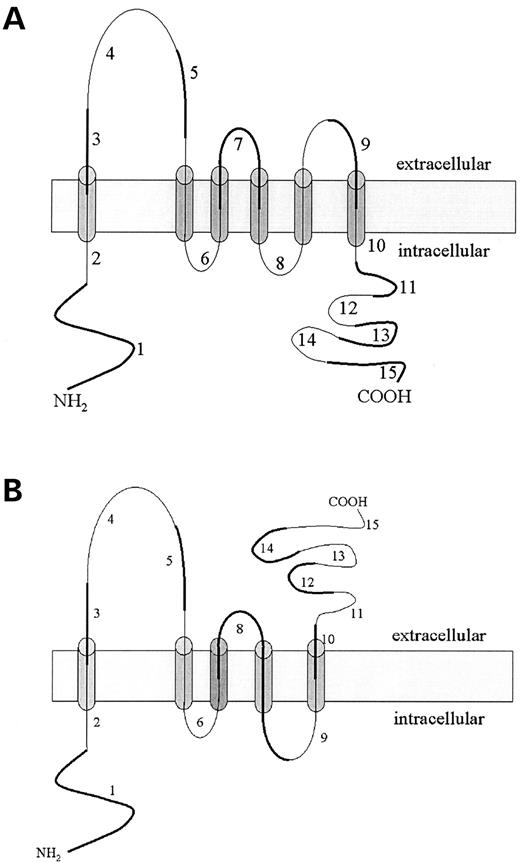
Figure 8. Predicted topologies of polycystin-2 ( A ) and polycystin-2Δ7 ( B ). The new transmembrane segment built out of exon 6 and exon 8 parts in polycystin-2Δ7 is shaded in darker gray. Exon numbering is next to corresponding protein segments. Odd numbered exons are highlighted with a thicker line and even numbered exons are marked thinner.
Oligonucleotides cited in this work
| Oligo name | Sequence (5′→3′) | Annealing temperature (°C) | Location (exon) | Amplicon size (bp) | Note |
|---|---|---|---|---|---|
| Murine | |||||
| Pkd2ex1.F | GGGGATGCAGCAGGCCGACTCCGG | 68 | 1 | 3010 | Full coding region |
| Pkd2ex15.R | ACACGTGTGGATTATTAGGCATGGACG | 15 | |||
| Pkd2ex6.F | TTACTATGTGGTGGAAGAGATATTGG | 57 | 6 | 323 | |
| Pkd2ex8.R | GGCACATCGAGACATGGTTGTGG | 8 | |||
| Pkd2ex7/8.F | CATGGTATTTTTGGTCTGGATTAAGCTC | 60 | 7/8 | 174 | Taqman assay, exon 7 specific |
| Pkd2ex8.R2 | GGGTGCCGAAGACAAGGTGTGC | 8 | |||
| Pkd2ex6/8.F | CTGGATGTTGTGATTGTCGTGCTC | 60 | 6/8 | 169 | Taqman assay Δexon7-specific |
| Pkd2ex8.R2 | GGGTGCCGAAGACAAGGTGTGC | 8 | |||
| Taqman probe | CCATGAGCCAGCTCTCCACAACCATGTC | 8 | 28 | 5′:6-FAM 3′: Darquencher | |
| Human | |||||
| PKD2ex1.F | GATGGTGAACTCCAGTCGCGTGC | 68 | 1 | 3183 | Full coding region |
| PKD2ex15.R | TGGTGGCAACTGGGCGTACCG | 15 | |||
| PKD2ex6.F | TGGTGGCAACTGGGCGTACCG | 57 | 6 | 390 | |
| Pkd2ex8.R | CCAACTGAGCATACGCTAGG | 8 | |||
| PKD2ex8.F | CCTAGCGTATGCTCAGTTGG | 60 | 8 | 301 | |
| PKD2ex10.R | GTAGCCCTTTCTGATAAGATCTGAG | 10 |
| Oligo name | Sequence (5′→3′) | Annealing temperature (°C) | Location (exon) | Amplicon size (bp) | Note |
|---|---|---|---|---|---|
| Murine | |||||
| Pkd2ex1.F | GGGGATGCAGCAGGCCGACTCCGG | 68 | 1 | 3010 | Full coding region |
| Pkd2ex15.R | ACACGTGTGGATTATTAGGCATGGACG | 15 | |||
| Pkd2ex6.F | TTACTATGTGGTGGAAGAGATATTGG | 57 | 6 | 323 | |
| Pkd2ex8.R | GGCACATCGAGACATGGTTGTGG | 8 | |||
| Pkd2ex7/8.F | CATGGTATTTTTGGTCTGGATTAAGCTC | 60 | 7/8 | 174 | Taqman assay, exon 7 specific |
| Pkd2ex8.R2 | GGGTGCCGAAGACAAGGTGTGC | 8 | |||
| Pkd2ex6/8.F | CTGGATGTTGTGATTGTCGTGCTC | 60 | 6/8 | 169 | Taqman assay Δexon7-specific |
| Pkd2ex8.R2 | GGGTGCCGAAGACAAGGTGTGC | 8 | |||
| Taqman probe | CCATGAGCCAGCTCTCCACAACCATGTC | 8 | 28 | 5′:6-FAM 3′: Darquencher | |
| Human | |||||
| PKD2ex1.F | GATGGTGAACTCCAGTCGCGTGC | 68 | 1 | 3183 | Full coding region |
| PKD2ex15.R | TGGTGGCAACTGGGCGTACCG | 15 | |||
| PKD2ex6.F | TGGTGGCAACTGGGCGTACCG | 57 | 6 | 390 | |
| Pkd2ex8.R | CCAACTGAGCATACGCTAGG | 8 | |||
| PKD2ex8.F | CCTAGCGTATGCTCAGTTGG | 60 | 8 | 301 | |
| PKD2ex10.R | GTAGCCCTTTCTGATAAGATCTGAG | 10 |
Oligonucleotides cited in this work
| Oligo name | Sequence (5′→3′) | Annealing temperature (°C) | Location (exon) | Amplicon size (bp) | Note |
|---|---|---|---|---|---|
| Murine | |||||
| Pkd2ex1.F | GGGGATGCAGCAGGCCGACTCCGG | 68 | 1 | 3010 | Full coding region |
| Pkd2ex15.R | ACACGTGTGGATTATTAGGCATGGACG | 15 | |||
| Pkd2ex6.F | TTACTATGTGGTGGAAGAGATATTGG | 57 | 6 | 323 | |
| Pkd2ex8.R | GGCACATCGAGACATGGTTGTGG | 8 | |||
| Pkd2ex7/8.F | CATGGTATTTTTGGTCTGGATTAAGCTC | 60 | 7/8 | 174 | Taqman assay, exon 7 specific |
| Pkd2ex8.R2 | GGGTGCCGAAGACAAGGTGTGC | 8 | |||
| Pkd2ex6/8.F | CTGGATGTTGTGATTGTCGTGCTC | 60 | 6/8 | 169 | Taqman assay Δexon7-specific |
| Pkd2ex8.R2 | GGGTGCCGAAGACAAGGTGTGC | 8 | |||
| Taqman probe | CCATGAGCCAGCTCTCCACAACCATGTC | 8 | 28 | 5′:6-FAM 3′: Darquencher | |
| Human | |||||
| PKD2ex1.F | GATGGTGAACTCCAGTCGCGTGC | 68 | 1 | 3183 | Full coding region |
| PKD2ex15.R | TGGTGGCAACTGGGCGTACCG | 15 | |||
| PKD2ex6.F | TGGTGGCAACTGGGCGTACCG | 57 | 6 | 390 | |
| Pkd2ex8.R | CCAACTGAGCATACGCTAGG | 8 | |||
| PKD2ex8.F | CCTAGCGTATGCTCAGTTGG | 60 | 8 | 301 | |
| PKD2ex10.R | GTAGCCCTTTCTGATAAGATCTGAG | 10 |
| Oligo name | Sequence (5′→3′) | Annealing temperature (°C) | Location (exon) | Amplicon size (bp) | Note |
|---|---|---|---|---|---|
| Murine | |||||
| Pkd2ex1.F | GGGGATGCAGCAGGCCGACTCCGG | 68 | 1 | 3010 | Full coding region |
| Pkd2ex15.R | ACACGTGTGGATTATTAGGCATGGACG | 15 | |||
| Pkd2ex6.F | TTACTATGTGGTGGAAGAGATATTGG | 57 | 6 | 323 | |
| Pkd2ex8.R | GGCACATCGAGACATGGTTGTGG | 8 | |||
| Pkd2ex7/8.F | CATGGTATTTTTGGTCTGGATTAAGCTC | 60 | 7/8 | 174 | Taqman assay, exon 7 specific |
| Pkd2ex8.R2 | GGGTGCCGAAGACAAGGTGTGC | 8 | |||
| Pkd2ex6/8.F | CTGGATGTTGTGATTGTCGTGCTC | 60 | 6/8 | 169 | Taqman assay Δexon7-specific |
| Pkd2ex8.R2 | GGGTGCCGAAGACAAGGTGTGC | 8 | |||
| Taqman probe | CCATGAGCCAGCTCTCCACAACCATGTC | 8 | 28 | 5′:6-FAM 3′: Darquencher | |
| Human | |||||
| PKD2ex1.F | GATGGTGAACTCCAGTCGCGTGC | 68 | 1 | 3183 | Full coding region |
| PKD2ex15.R | TGGTGGCAACTGGGCGTACCG | 15 | |||
| PKD2ex6.F | TGGTGGCAACTGGGCGTACCG | 57 | 6 | 390 | |
| Pkd2ex8.R | CCAACTGAGCATACGCTAGG | 8 | |||
| PKD2ex8.F | CCTAGCGTATGCTCAGTTGG | 60 | 8 | 301 | |
| PKD2ex10.R | GTAGCCCTTTCTGATAAGATCTGAG | 10 |
References
Bogdanova, N., Dworniczak, B., Dragova, D., Todorov, V., Dimitrakov, D., Kalinov, K., Hallmayer, J. and Horst, J. (
de Almeida, S., de Almeida, E., Peters, D., Pinto, J.R., Tavora, I., Lavinha, J., Breuning, M. and Prata, M.M. (
Daoust, M.C., Reynolds, D.M., Bichet, D.G. and Somlo, S. (
Mochizuki, T., Wu, G., Hayashi, T., Xenophontos, S.L., Veldhuisen, B., Saris, J.J., Reynolds, D.M., Cai, Y., Gabow, P.A., Pierides, A. et al. (
Wu, G., Mochizuki, T., Le, T.C., Cai, Y., Hayashi, T., Reynolds, D.M. and Somlo, S. (
Pennekamp, P., Bogdanova, N., Wilda, M., Markoff, A., Hameister, H., Horst, J. and Dworniczak, B. (
McGrath, J., Somlo, S., Makova, S., Tian, X. and Brueckner, M. (
Nauli, S.M., Alenghat, F.J., Luo, Y., Williams, E., Vassilev, P., Li, X., Elia, A.E., Lu, W., Brown, E.M., Quinn, S.J. et al. (
Montell, C. (
Koulen, P., Cai, Y., Geng, L., Maeda, Y., Nishimura, S., Witzgall, R., Ehrlich, B.E. and Somlo, S. (
Qian, F., Germino, F.J., Cai, Y., Zhang, X., Somlo, S. and Germino, G.G. (
Tsiokas, L., Kim, E., Arnould, T., Sukhatme, V.P. and Walz, G. (
Wu, G. (
Hanaoka, K., Qian, F., Boletta, A., Bhunia, A.K., Piontek, K., Tsiokas, L., Sukhatme, V.P., Guggino, W.B. and Germino, G.G. (
Babich, V., Zeng, W.Z., Yeh, B.I., Ibraghimov-Beskrovnaya, O., Cai, Y., Somlo, S. and Huang, C.L. (
Nomura, H., Turco, A.E., Pei, Y., Kalaydjieva, L., Schiavello, T., Weremowicz, S., Ji, W., Morton, C.C., Meisler, M., Reeders, S.T. and Zhou, J. (
Wu, G., Hayashi, T., Park, J.H., Dixit, M., Reynolds, D.M., Li, L., Maeda, Y., Cai, Y., Coca-Prados, M. and Somlo, S. (
Veldhuisen, B., Spruit, L., Dauwerse, H.G., Breuning, M.H. and Peters, D.J. (
Guo, L., Chen, M., Basora, N. and Zhou, J. (
Guo, L., Schreiber, T.H., Weremowicz, S., Morton, C.C., Lee, C. and Zhou, J. (
Chen, X.Z., Vassilev, P.M., Basora, N., Peng, J.B., Nomura, H., Segal, Y., Brown, E.M., Reeders, S.T., Hediger, M.A. and Zhou, J. (
Volk, T., Schwoerer, A.P., Thiessen, S., Schultz, J.H. and Ehmke, H. (
Hayashi, T., Mochizuki, T., Reynolds, D.M., Wu, G., Cai, Y. and Somlo, S. (
Reynolds, D.M., Hayashi, T., Cai, Y., Veldhuisen, B., Watnick, T.J., Lens, X.M., Mochizuki, T., Qian, F., Maeda, Y., Li, L. et al. (
Bogdanova, N., Markoff, A., Gerke, V., McCluskey, M., Horst, J. and Dworniczak, B. (
Foggensteiner, L., Bevan, A.P., Thomas, R., Coleman, N., Boulter, C., Bradley, J., Ibraghimov-Beskrovnaya, O., Klinger, K. and Sandford, R. (
Boletta, A., Qian, F., Onuchic, L.F., Bhunia, A.K., Phakdeekitcharoen, B., Hanaoka, K., Guggino, W., Monaco, L. and Germino, G.G. (
Qian, F., Boletta, A., Bhunia, A.K., Xu, H., Liu, L., Ahrabi, A.K., Watnick, T.J., Zhou, F. and Germino, G.G. (
Lan, Z., Xu, H., He, X., Zhou, Q., Germino, G.G., Watnick, T. and Qian, F. (
Nakai, K. and Horton, P. (
Krogh, A., Larsson, B., von Heijne, G. and Sonnhammer, E.L. (
Grimm, D.H., Cai, Y., Chauvet, V., Rajendran, V., Zeltner, R., Geng, L., Avner, E.D., Sweeney, W., Somlo, S. and Caplan, M. (
Maquat, L.E. (
Ishigaki, Y., Li, X., Serin, G. and Maquat, L.E. (
Guillaume, R. and Trudel, M. (
Markowitz, G.S., Cai, Y., Li, L., Wu, G., Ward, L.C., Somlo, S. and D'Agati, V.D. (
Köttgen, M., Benzing, T., Simmen, T., Tauber, R., Buchholz, B., Feliciangeli, S., Huber, T.B., Schermer, B., Kramer-Zucker, A., Höpker, K. et al . (
Chen, X.Z., Segal, Y., Basora, N., Guo, L., Peng, J.B., Babakhanlou, H., Vassilev, P.M., Brown, E.M., Hediger, M.A. and Zhou, J. (
Bezzerides, V.J., Ramsey, I.S., Kotecha, S., Greka, A. and Clapham, D.E. (
Spitzer, N.C. (
Kim, S.J., Kim, Y.S., Yuan, J.P., Petralia, R.S., Worley, P.F. and Linden, D.J. (
Bengston, C.P., Tozzi, A., Bernardi, G. and Mercuri, N.B. (
van Dijk, M.A., Chang, P.C., Peters, D.J. and Breuning, M.H. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}