





Abstract
Congenital heart disease is a major health issue, accounting for a third of all congenital defects. Improved early surgical management has led to a growing population of adults with congenital heart disease, including patients with defects affecting the right ventricle, which are often classified as severe. Defects affecting the right ventricle often cause right ventricular volume or pressure overload and affected patients are at high risk for complications such as heart failure and sudden death. Recent insights into the developmental mechanisms and distinct developmental origins of the left ventricle, right ventricle, and the outflow tract have shed light on the common features and distinct problems arising in specific defects. Here, we provide a comprehensive overview of the current knowledge on the development into the normal and congenitally malformed right heart and the clinical consequences of several congenital heart defects affecting the right ventricle.
1. Introduction
Congenital heart disease (CHD) is highly prevalent with a total birth prevalence of 9 per 1.000 live births, accounting for a third of all major congenital defects.1 Due to improved surgical management of CHD in infancy and childhood, the number of adult patients with CHD has increased rapidly. Current prevalence among adults is ∼3 per 1.000 persons,2 5% with severe lesions and 36% with moderate lesions. This includes a range of defects affecting the right ventricle (RV). In CHD, the RV is often affected by a combination of anatomical factors and acquired factors, such as residual lesions after surgery. This is in contrast to acquired heart disease, which predominantly affects the left ventricle (LV). Patients with CHD affecting the RV are at risk for serious complications such as heart failure and sudden death. In order to optimize clinical management of the adult patients with CHD affecting the RV, it is important to understand the clinical consequences associated with the developmental origin of the specific defects. In this review, we discuss the developmental origin of the RV and outflow tract (OFT), and some of the molecular and morphogenetic mechanisms that have been found to cause malformations when dysfunctional. We provide an overview of selected congenital defects to illustrate how these malformations affect the RV and clinical outcomes in adulthood. The selected defects vary considerably in symptoms, treatment, and late outcome, but share developmental features and have RV complications as one of the focal points during adult life.
2. Heart tube formation and looping; congenitally corrected transposition of the great arteries
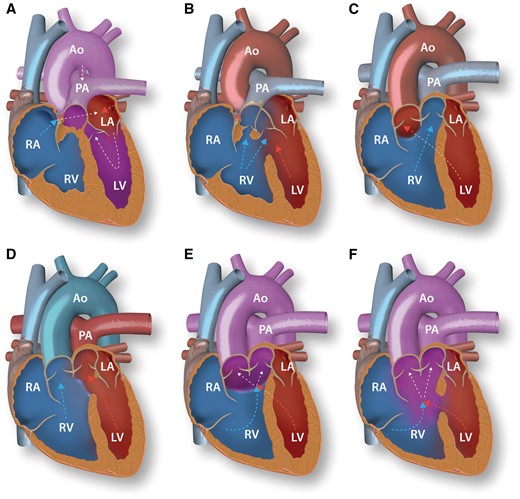
The initial heart tube is formed from mesodermal progenitors in the anterior lateral splanchnic mesoderm, the first heart field (FHF). At about 7–7.5 days after conception (embryonic day (E) 7.5) in mice, corresponding to E15 in humans, the bilateral FHF progenitors fuse at the embryonic midline to form the linear heart tube.3 This initial heart tube consists of an outer myocardial and inner endocardial layer separated by extracellular matrix known as cardiac jelly. It is connected to the dorsal body wall by the dorsal mesocardium, which during subsequent stages ruptures, leaving only the arterial and venous poles connected to the pericardial wall.4 The heart tube elongates very extensively (see below), while at the same time the chambers differentiate and dextral looping of the heart tube occurs (Figure 1). The cranial part of the elongated heart tube, which is looping to the right, gives rise to the future RV. The middle part of the looping tube, somewhat displaced to the left, forms the future LV. Looping of the heart tube in leftward direction results in a mirror-imaged position of the right and left ventricles and thus gives rise to congenitally corrected TGA (ccTGA), provided that the position of the atria and outflow channels remains unaffected. Due to the abnormal looping in ccTGA, the morphological RV is situated between the left atrium and the aorta (Figure 2). It therefore functions as the systemic ventricle. This majorly affects the RV, which has evolved to function with low sub-pulmonary pressures. While the morphological RV in ccTGA is subject to systemic high pressure overload, it does not acquire the required LV-like properties. The difference in origin and mechanism of development between the RV and LV (see Section 3) may underlie this phenomenon. As a result, the RV will develop hypertrophy, dilate, and become more spherical over time (Figure 3A). This systemic situation may not cause problems immediately after birth, but the permanent pressure overload is detrimental in adult life. Systemic RV dysfunction and symptomatic heart failure already occur in 32–56% and 25–67% of patients by the age of 45.5 The morphological RV is perfused by a single coronary artery as it would also be in the normal subpulmonary situation. Following the abnormal looping, this single coronary perfusion of the systemic morphological RV can contribute to further systemic RV failure by relative coronary insufficiency of the systemic RV.6 Frequent monitoring is needed in ccTGA patients, considering a median survival of only 66 years in patients reaching adulthood7 and risks of heart failure, tricuspid regurgitation (TR), and arrhythmias.5,8,9 Pharmaceutical renin-angiotensin-aldosterone-system inhibition may be useful in symptomatic patients.10 Other strategies to decrease heart failure burden and prevent progression of RV dysfunction include tricuspid valve replacement,11 cardiac resynchronization therapy,12 and pulmonary artery banding.13 In early diagnosis, a ‘double switch procedure’ (Table 1) can restore atrioventricular and ventriculo-arterial concordance, alleviating systemic RV burden. However, in absence of strategies that improve RV function in the continuous presence of systemic pressures, the only long-term solutions in adults with severe systemic RV dysfunction remain ventricular assist devices and heart transplantation.
Common repair strategies of the discussed congenital defects
| Congenital heart defect | Surgical repair |
|---|---|
| Congenitally corrected transposition of the great arteries | Double switch procedure: anatomical correction consisting of intra-atrial baffle repair to reach atrioventricular concordance and an arterial switch procedure to reach ventriculo-arterial concordance. |
| Ebstein’s anomaly |
|
| Double-chambered right ventricle | Relief of RVOT obstruction by resecting the obstructive tissue. |
| Hypoplastic right heart |
|
| Tetralogy of Fallot | VSD closure and relief of RVOT obstruction. A transannular patch across the pulmonary valve is necessary in some patients. |
| Double outlet right ventricle | Usual surgical strategy depends largely on anatomical subtype.90
|
| Congenital heart defect | Surgical repair |
|---|---|
| Congenitally corrected transposition of the great arteries | Double switch procedure: anatomical correction consisting of intra-atrial baffle repair to reach atrioventricular concordance and an arterial switch procedure to reach ventriculo-arterial concordance. |
| Ebstein’s anomaly |
|
| Double-chambered right ventricle | Relief of RVOT obstruction by resecting the obstructive tissue. |
| Hypoplastic right heart |
|
| Tetralogy of Fallot | VSD closure and relief of RVOT obstruction. A transannular patch across the pulmonary valve is necessary in some patients. |
| Double outlet right ventricle | Usual surgical strategy depends largely on anatomical subtype.90
|
RV, right ventricle; RVOT, right ventricular outflow tract; VSD, ventricular septal defect; LV, left ventricle.
Common repair strategies of the discussed congenital defects
| Congenital heart defect | Surgical repair |
|---|---|
| Congenitally corrected transposition of the great arteries | Double switch procedure: anatomical correction consisting of intra-atrial baffle repair to reach atrioventricular concordance and an arterial switch procedure to reach ventriculo-arterial concordance. |
| Ebstein’s anomaly |
|
| Double-chambered right ventricle | Relief of RVOT obstruction by resecting the obstructive tissue. |
| Hypoplastic right heart |
|
| Tetralogy of Fallot | VSD closure and relief of RVOT obstruction. A transannular patch across the pulmonary valve is necessary in some patients. |
| Double outlet right ventricle | Usual surgical strategy depends largely on anatomical subtype.90
|
| Congenital heart defect | Surgical repair |
|---|---|
| Congenitally corrected transposition of the great arteries | Double switch procedure: anatomical correction consisting of intra-atrial baffle repair to reach atrioventricular concordance and an arterial switch procedure to reach ventriculo-arterial concordance. |
| Ebstein’s anomaly |
|
| Double-chambered right ventricle | Relief of RVOT obstruction by resecting the obstructive tissue. |
| Hypoplastic right heart |
|
| Tetralogy of Fallot | VSD closure and relief of RVOT obstruction. A transannular patch across the pulmonary valve is necessary in some patients. |
| Double outlet right ventricle | Usual surgical strategy depends largely on anatomical subtype.90
|
RV, right ventricle; RVOT, right ventricular outflow tract; VSD, ventricular septal defect; LV, left ventricle.
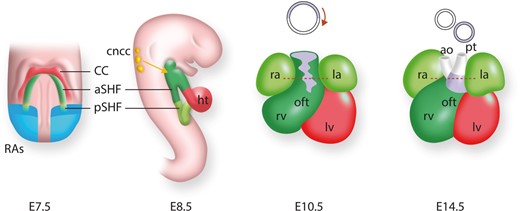
Heart development and the contribution of cardiac lineages in the mouse. At E7.5, myocardial cells form the cardiac crescent (derived from the first heart field), with the second heart field (SHF) lying medial to it. The posterior part of the SHF received retinoic acid signalling (RAs; blue). The SHF and its contributions are shown in green, with the anterior SHF (aSHF) sub-domain in dark green and posterior SHF (pSHF) in light green. At E8.5, the early cardiac tube has formed and undergoes looping; the anterior right ventricle (rv) and posterior left ventricle (lv) will be placed in their definitive positions. The cardiac neural crest cells (cncc; yellow) will migrate into the outflow tract (OFT). At E10.5 the OFT has formed and the chambers have expanded. At foetal stage (E14.5), the chambers are separated and connected to the pulmonary trunk (pt) and aorta (ao). The OFT will rotate to align the aorta (ao) and pulmonary trunk (pt). The inferior OFT (purple) will form the sub-pulmonary myocardium.
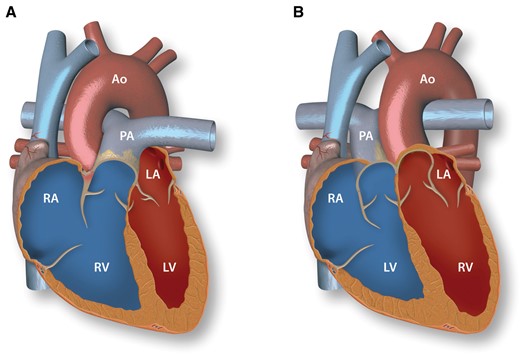
Normal heart anatomy and congenitally corrected transposition of the great arteries. A: Normal heart with concordant atrioventricular and ventriculo-arterial connections. B: Congenitally corrected transposition of the great arteries with atrioventricular and ventriculo-arterial discordance. RV, right ventricle; RA, right atrium; PA, pulmonary artery; LV, left ventricle; LA, left atrium; Ao, aorta.
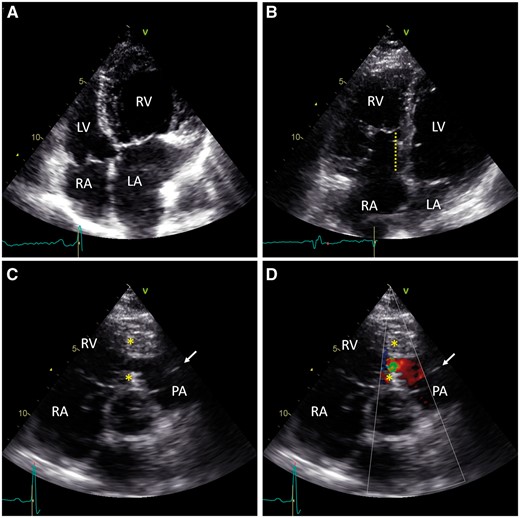
Transthoracic echocardiograms of congenitally malformed adult hearts. A: RV hypertrophy and LA dilation due to tricuspid regurgitation in congenitally corrected transposition of the great arteries. For comparison, no RV hypertrophy is seen in B. B: Apical displacement of the tricuspid leaflet from the tricuspid annulus (dotted line) causes partial RV atrialization in Ebstein’s anomaly. C: Obstructive tissue (*) divides the RV, creating a low-pressure compartment below the pulmonary valve (arrow) in double-chambered right ventricle. D: Doppler colour view shows turbulent flow across the obstruction in double-chambered right ventricle. RV, right ventricle; RA, right atrium; PA, pulmonary artery; LV, left ventricle; LA, left atrium.
3. Development of the atrioventricular canal and right ventricular expansion; Ebstein’s anomaly and double-chambered right ventricle
In the third week of human heart development (mouse E9), highly specific patterns of gene expression and cell proliferation on the outer curvature of the embryonic heart tube induce an enormous expansion of the cardiac chambers. This process is referred to as ballooning morphogenesis.14,15 By 4–5 weeks of human development (mouse E10.5), the four cardiac chambers are clearly visible(Figure 1).14,16,17 The newly differentiated chamber myocardial cells (future working myocardium) acquire properties of high conduction velocity and high contractility, in keeping with their ability to efficiently pump blood.14 In contrast, the portion of the heart tube in-between the forming ventricles and atria maintains slow proliferation rates, does not initiate chamber gene programs, and maintains its slow-conductive properties. This portion forms the atrioventricular canal (AVC), and appears as a constriction because it grows much less than the chambers. Part of the atrioventricular myocardial cells form the atrioventricular node and -bundles and the base of the LV,18 and induce valve development by the overlying endocardial cells. The AVC is initially positioned at the left side of the body axis, thus positioned over the LV cavity. It moves to the midline and expands, by which the proper connections to the ventricles are established. Subsequently, the AVC myocardium and the overlying valve primordia elongate and protrude into the ventricular cavities. At the same time, the ventricular walls expand, forming trabecula-filled cavities behind the extended AVC. This process results in valves supported by atrioventricular myocardium extending deep into the ventricular cavity. The RV base expands more extensively laterally compared to the LV, whereas the LV grows more into apical direction.19 The AVC myocardium supporting the mural valve leaflet and the underlying trabecules are initially maintained. This structure has been termed dorsal ‘gully’, a roof over the cavity that is formed as a result of the dorsal and lateral expansion of the underlying RV wall.20 The trabecular myocardium connecting the AVC myocardium below the leaflet forms the papillary muscles, the AVC myocardium below the leaflet is removed by apoptosis. In the mature heart, the moderator band and septal limb of the septo-marginal trabeculae are remnants of the trabeculae of the original (pre-expansion) embryonic RV at the anterior end of the gully. Downstream of these trabeculae, the smooth walled RV outlet myocardium (subpulmonary myocardium) is formed from the embryonic OFT (see Section 4).
Major aspects of the molecular mechanism of AVC and valve formation have been described. Bmp2 in the AVC primordium initiates expression of T-box transcription factors Tbx2 and Tbx3, which in turn suppress chamber differentiation and stimulate the pacemaker-like phenotype of the AVC.21,22 Bmp2 also stimulates signalling in the overlying endocardium to form cushions, primordia of the valve leaflets.21 Other factors controlling AVC specification and formation include Wnt- and Notch signalling,23,24 core cardiac transcription factors Tbx5, Tbx20, Gata4 and Nkx2-5, Hey1 and Hey2 in the chambers to help establish the AVC boundary, and many others.25 Mutations in genes governing these morphogenetic processes (e.g. Nkx2-526), may result in malformation of the AVC or of the associated trabeculae, or failed expansion of the RV base, hallmarks of Ebstein’s anomaly and double-chambered right ventricle (DCRV) (see below). Moreover, MYH7 mutations were found to be associated with Ebstein’s anomaly,27 suggesting that affected contractility and hemodynamics may influence AVC/RV development.
In Ebstein’s anomaly, the septal and posterior tricuspid valve leaflets are still attached to the RV wall, resulting in apical displacement of the tricuspid valve leaflets, with anterior and apical rotation of the functional orifice28 (Figure 4A). This results in partial ‘atrialization’ of the RV (Figure 3B). The wall of the ‘atrialized’ RV is hypoplastic. Too extensive atrioventricular specification may lead to failure to initiate the chamber differentiation program in the future RV base adjacent to the AVC, diminished growth and dorsal-lateral expansion of this wall, and erroneous induction of valve development by the overlying endocardium;19 features of Ebstein’s anomaly. The degree of leaflet attachment to the hypoplastic RV wall is highly variable. The extent of valve displacement therefore varies considerably, causing moderate to severe TR. The typical rotational displacement of the tricuspid valve is associated with desynchronized RV movement and decreased RV function.29 Additionally, the right atrium and atrialized RV contract sequentially. The absence of a valve between these two, morphologically different, functionally atrial structures decreases functional output and contributes to dilation of the right atrium and atrialized RV. With increasing TR severity, the functional RV also dilates.30 Atrial arrhythmias develop in 35–50% of patients by the age of 50.31 The defect is usually restored by surgery, most importantly cone reconstruction surgery (Table 1), which restores valve function and improves RV geometry.32 Surgery decreases RV volume while effective stroke volume and LV ejection fraction increase, resulting in improved exercise capacity and functional class post-operatively.33,34 Usually, surgery also involves plication of the atrialized RV, because of concerns that this thin-walled non-contractile sac promotes thrombosis and decreases RV function.35 However, in a recent long-term patient series without plication, overall survival was 92% at 20 years after surgery without observed thrombosis or aneurysm of the area.35 The varying severity and limited patient numbers hinder research in Ebstein’s anomaly, causing guidelines to be based on observational studies and expert opinion.
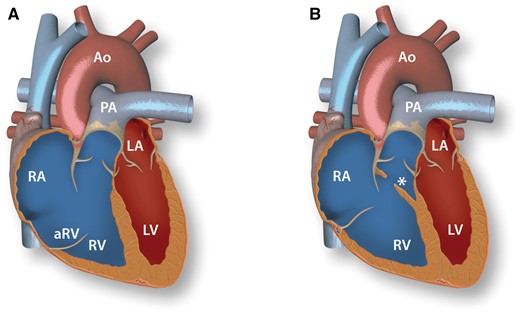
Anatomy of Ebstein’s anomaly and double-chambered right ventricle. A: Ebstein’s anomaly. Lack of delamination of the septal and posterior tricuspid valve leaflets causes apical displacement of the tricuspid annulus with a thin walled atrialized portion of the RV (aRV). B: Double-chambered right ventricle. Subinfundibular tissue (*) crosses the RV cavity and causes obstruction of outflow. RV, right ventricle; RA, right atrium; aRV, atrialized right ventricle; PA, pulmonary artery; LV, left ventricle; LA, left atrium; Ao, aorta.
When obstructive tissue crosses the RV cavity, it is functionally divided into two compartments, clinically resulting in a double-chambered right ventricle (Figure 4B). Histologic studies describe the obstructive tissue as muscular or fibromuscular.36 Most describe it as anomalous muscle bundles, but hypertrophic trabeculae and an abnormal insertion of the moderator band have also been described.37 Based on the morphogenetic process described above, we hypothesize that these obstructive tissues derive from abnormalities in the development or growth of the trabeculae of the original, pre-expansion embryonic RV. The obstructive tissue creates pressure overload in the by expansion newly formed RV cavity proximal to the obstruction, while distal pressure in the original embryonic RV cavity and the right ventricular outflow tract (RVOT) remains low (Figure 3C and D). The ensuing pressure overload proximal to the obstruction causes RV hypertrophy and progressive RVOT obstruction.38 Symptoms and age at presentation vary with severity of RVOT obstruction. Most patients present with an asymptomatic murmur, shortness of breath, or limited exercise capacity, but cyanosis may also occur in patients with severe obstruction and a ventricular septal defect (VSD), which is often present.36 Timely accurate diagnosis is essential to prevent irreversible RV failure. Surgical resection of obstructive tissue is usually performed to restore RV anatomy.
4. Right ventricle and outflow tract development; hypoplastic right heart, tetralogy of Fallot, and outlet malformations
The early heart tube myocardium does not proliferate.39,40 Nevertheless, it increases 4–5-fold in length in a short time span (24 h in mouse41). Growth occurs by differentiation and addition of progenitor cells at the poles. The progenitors, referred to as the second heart field (SHF) cells, are positioned medially and caudally to the FHF. During tube formation from the FHF, these cells are part of the pericardial wall positioned dorsally and caudally from the heart tube (Figure 1). The rate of proliferation of these SHF cells is very high.40 The progenitor cells bordering the heart tube decrease their proliferation rate, differentiate into myocardium and are added to the inflow tract and OFT of the tube, causing the heart tube to grow.40 Classical fate mapping, dye labelling and Cre-mediated lineage analysis and experiments have indicated that the FHF gives rise predominately to the LV and AVC of the definitive heart.42–44 A subset of SHF cells, also known as anterior SHF, which are added to the arterial pole of the elongating heart tube, go on to form the definitive RV (Figure 1). Anterior SHF cells added subsequently form the OFT myocardium, the superior part of which will become the LV outflow tract and the inferior part of which will become the subpulmonary or RVOT myocardium. The SHF cells added finally form the endothelial layer and adventitia of the aortic and pulmonary trunk, contribute to the developing semilunar valves and the smooth muscle layers of the proximal trunks.39,43,45–47 Furthermore, from mouse E9.5, neural crest-derived cells migrate into the OFT cushions. The cardiac neural crest cells contribute to the semilunar valves, and form the smooth muscle layers of the aortic trunk and contribute to the smooth muscle layer of the pulmonary trunk.47,48 Moreover, the cardiac neural crest is required for SHF deployment and for septation of the single embryonic OFT into the aortic and pulmonary trunks.49 During septation of the OFT, the largely neural crest-derived aorticopulmonary septum forms at the dorsal wall of the aortic sac between the 4th and 6th pharyngeal arch arteries, and converges with the distal OFT cushions to divide the OFT into aorta and pulmonary trunk.50 The embryonic OFT is positioned over the RV, although LV blood reaches the OFT via the inner curvature (where OFT and AVC are connected) over the forming muscular interventricular septum that has not yet closed. The OFT wall then rotates,51 the inferior hemisphere of the OFT becomes positioned superior and forms the sub-pulmonary myocardium, and the superior hemisphere of the OFT forms the subaortic myocardium (Figure 1).52 The subaortic component thus ‘moves’ from right to left and becomes positioned left to the muscular interventricular septum, over the LV cavity. The interventricular septum is often seen to be malformed in addition to the OFT defect. This could result from a primary defect in septum formation, when the mechanism that is perturbed underlies both OFT and septum development, or secondary, when LV-OFT hemodynamic communication needs to be maintained because the aorta (LV outlet) is incorrectly positioned.
The very similar morphogenetic processes between rodents and humans53 provide a firm base for our understanding of the development of particular congenital defects of the RV and OFT. Thus, defective signalling, expansion, or differentiation of the SHF progenitor population early during development may affect development of the RV, as seen in hypoplastic RV. Developmental defects of the subpopulation of anterior SHF progenitors that form the subpulmonary myocardium may result in hypoplastic subpulmonary myocardium, a hallmark of TOF and pulmonary atresia, and in common arterial trunk (persistent trucus arteriosus (PTA)).54 Errors in signalling, migration, or fate of the neural crest entering the OFT may cause OFT malformations or failure to septate the OFT, leading to PTA. Rotation defects lead to overriding aorta (partial rotation, aorta entrance positioned over ventricular septum with defect), transposition of the great arteries (TGA; severe malrotation, aorta connected to RV, pulmonary trunk to LV, ventricular septum fully formed), and to double outlet right ventricle (DORV; severe malrotation, aorta and pulmonary trunk connected to RV, accompanying VSD). Several of these defects regularly occur in combinations (e.g. TOF, pulmonary atresia and hypoplastic RV) because the progenitors of the affected components are closely associated and interact (inter-progenitor signalling). Below, we will describe some major molecular mechanisms involved in these developmental processes, and the consequences of errors in these mechanisms as a result of genetic defects. Subsequently, we will describe the clinical complications for the RV seen in patients with hypoplasic right hearts, TOF and DORV.
4.1 Molecular mechanisms of right ventricle and outflow tract development
The morphogenesis of RV and OFT described in the previous section can be summarized into following main steps: (i) specification, proliferation and migration of SHF progenitors, (ii) differentiation of SHF cells to form the RV and OFT, including the subpulmonary component, (iii) immigration of neural crest and septation of the OFT, and (iv) rotation of the OFT. When the SHF cells are specified, they proliferate and migrate, and at the border with the myocardium of the heart tube differentiate to become part of the elongating heart tube. Retinoic acid signalling defines the posterior limit of SHF specification. Wnt/b-catenin signalling stimulates a transcriptional network in the SHF, including genes for transcription factors Tbx1, Isl1 and Six1/Eya1, and FGF signalling, which regulates the undifferentiated proliferative state. SHF progenitor differentiation is induced by non-canonical Wnt signalling and by FGF signalling-stimulated BMP-Smad signalling, which in turn suppresses proliferation and induces Nkx2-5 and other genes encoding core cardiogenic transcription factors.3,55 These factors along with Gata4 and others drive expression of the cardiogenic gene program, including Mef2c, and differentiation, while suppressing the expression of Isl1 and Tbx1 encoding SHF transcription factors.
Gene modification analyses in mouse and fish models have uncovered the genes for these conserved signalling and transcriptional networks.3,56 In addition, genetic analyses of CHD patients have uncovered single gene mutations associated with RV and OFT defects, which are enriched for genes encoding the cardiogenic transcription factors and epigenetic regulatory factors such as histone modifying enzymes (TBX1, GATA4, HAND2, etc.) and signalling components (ALDH1A2, TAB2, etc.).57,58 In addition, mutations in genes involved in establishment of the left-right axis in the embryo have been identified in mouse and human (ZIC3, NODAL, and LEFTY2), implicating this process in OFT development.56 Below examples of the function of some key factors are described.
T-box transcription factor Tbx1, a central regulator of SHF specification and expansion, is a major candidate gene in 22q11 deletion syndrome, which is associated with TOF and other OFT diseases. Tbx1 mutants lack the expansion of SHF progenitor pool and exhibit reduced subpulmonary myocardium (Figure 5), which is suggested as a primary event in TOF progression.52,59,Tbx1 has been shown to regulate cell dynamics, and has been deemed responsible for proliferative defects and ectopic differentiation of SHF cells in Tbx1 mutants.60,Tbx3 mutants develop DORV whereas Tbx2 mutants show mild OFT alignment defects. However, loss of two of the three genes (Tbx1/2/3) results in severe pharyngeal hypoplasia and heart tube extension defects. These findings reveal an indispensable T-box gene network governing pharyngeal and OFT development.61,Six1/Eya1 mutants recapitulated most features of human 22q11 deletion syndrome, revealing a Tbx1-Six1/Eya1-Fgf8 genetic pathway that is crucial for OFT development.62
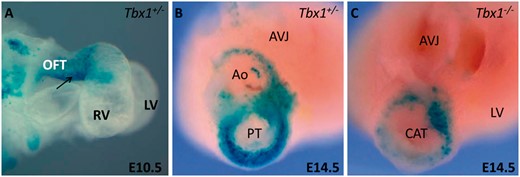
Tbx1 is required for subpulmonary myocardium formation. A: Right lateral view of an E10.5 heart in which the y96-Myf5-nlacZ-16 transgene marks the inferior OFT (arrow). B: At E14.5 the aorta and pulmonary trunk have formed. The inferior OFT forms the subpulmonary myocardium at the base of the pulmonary trunk as revealed by transgene expression. C: The subpulmonary myocardium is not formed in Tbx1−/− foetuses as revealed by the absence of transgene expression. The embryonic OFT does not undergo septation and shows the presence of a common arterial trunk. OFT, outflow tract; RV, right ventricle; LV, left ventricle; Ao, aorta; PT, pulmonary trunck; AVJ, atrioventricular junction; CAT, common arterial trunk. Modified from Théveniau-Ruissy et al.52 Images are courtesy of Robert Kelly.
Transcription factor Isl1 is expressed in the cardiogenic mesoderm. Its expression is maintained in the SHF progenitors but downregulated upon differentiation of the progenitors.39 Isl1 mutants fail to elongate the heart tube, revealing its requirement for SHF deployment. The direct transcriptional target of Isl1 and Gata factors, Mef2c, governs the development of SHF-derived structures during embryonic development.63,Mef2c null mice show defects in cardiac looping and development of the OFT and RV, ranging from VSD and overriding aorta to DORV.64,65 This finding implies that these defects are different expressions of the same underlying failed developmental mechanism.
Amongst various Fgf ligands, Fgf8 and Fgf10 have been implicated in the SHF development. Fgf8 and Fgf10 are expressed in SHF progenitors and downregulated upon the differentiation and recruitment of the progenitors to the heart tube.42,66,67 Fgf signalling increases the proliferation rate of SHF progenitors and inhibits Bmp-induced myocardial differentiation.68 Analysis of mice harbouring Fgf8 hypomorphic alleles revealed looping defects in the heart tube, RV hypoplasia, hypoplastic OFTs and alignment defects like DORV and TGA.69,70 Loss of a major Fgf10 receptor, Fgfr2-IIIb, is associated with SHF developmental defects like RV and OFT hypoplasia, VSD, overriding aorta, and DORV.71
Nkx2.5 is expressed in the entire heart. The Nkx2.5−/− mice show OFT truncation and reduced proliferation of SHF cells. These defects were rescued when Nkx2.5 hypomorphic alleles were raised in Smad1−/− genetic background, showing the significance of an Nkx2.5/Bmp2/Smad1 negative feedback loop during cardiac development.72 In support of these findings, it has also been reported that patients with mutations in NKX2.5 suffer from severe cardiac defects like TOF and DORV.73 Gata4 is crucial for regulating RV morphogenesis. Deletion of Gata4 in the Nkx2.5+ lineage earlier in development results in myocardial thinning, hypoplastic RV and smaller atrioventricular canal and OFT cushions with fewer mesenchymal cells. These Gata4-deleted hearts lack Hand2 transcripts, crucial for RV formation.74 Late deletion of Gata4 in the Myh6 (αMHC)-expressing lineage resulted in myocardial hypoplasia and DORV.74
Shh regulated Six2 marks SHF progenitors. Global ablation of Six2+ progenitors leads to hypoplastic RV and pulmonary atresia in mouse foetuses. However, when only a small subset of Six2+ progenitors are ablated during early embryogenesis, mice do not present any gross morphological defect at birth, but develop heart dysfunction due to thickening of the RV free wall and reduced RV fraction shortening, which suggests that early errors in SHF deployment may cause late onset RV dysfunction.47
Elongation of the heart tube involves a close coordination between cells of pharyngeal mesoderm and neighbouring cells like neural crest and pharyngeal endoderm cells, which makes proper functioning of neural crest cells integral to the development of SHF structures and OFT septation. Ablation of neural crest cells in chicken resulted in an increase in Fgf8 signalling leading to abnormal expansion of SHF progenitor pool, PTA and abnormalities in heart looping.68
Following heart tube formation, the formation of the aortic-pulmonary septum to divide the embryonic OFT into aorta and pulmonary trunk is regulated by various signalling molecules including BMP, FGF and Semaphorins.75 Additionally, the myocardialization of the proximal outlet septum at later stages of development is governed by PCP signalling pathways.76,77 A next step in OFT remodelling involves the rotation of the myocardial wall of the OFT, which aligns the aorta with the LV outlet and the pulmonary trunk with the RV.51,Pitx2c has been implicated in the OFT rotation process, as Pitx2c−/− show cardiac defects like DORV and PTA following disrupted rotation of the OFT.51 As Pitx2c is involved in left-right patterning, this suggests that OFT morphogenesis, including rotation, is downstream of the signalling cues required for establishing organ laterality.
4.2 Clinical complications of hypoplastic right hearts, tetralogy of Fallot, and double outlet right ventricle
Underdevelopment of the right heart is seen in a range of phenotypes, mostly in patients with severe obstructive valvar malformations such as pulmonary atresia, where the RV cavity gets obliterated by hypertrophic muscle during foetal life (Figure 6A). Isolated RV hypoplasia with only an atrial septal defect or patent foramen ovale also occurs. The main factor defining presenting symptoms is the extent of limited RV capacity, causing RV inflow restriction. This results in right atrial enlargement and raised right atrial pressure. If an interatrial communication is present, right-to-left shunting will arise as escape route. In patients with only mild hypoplasia, biventricular repair may be feasible depending on associated defects (Figure 7A), but long-term function of these small RVs remains unknown. In severe hypoplasia, RV preload reduction is necessary. Adults with severe hypoplasia will therefore generally present with a 1.5-ventricle repair or Fontan circulation (Table 1). Although Fontan circulation has improved survival in patients with severe RV hypoplasia and thus functionally univentricular hearts, late mortality in Fontan patients remains at 2.1% per year,78 with a median survival of patients reaching 50 years of age.7 The complete lack of a subpulmonary ventricle in Fontan circulation makes the pulmonary circulation dependent on changes in intrathoracic pressure and systemic venous hypertension. Furthermore, the systemic ventricle suffers from chronically decreased preload. Optimal treatment strategies are uncertain and recommendations are based on small studies with short follow-up.79
Anatomy of pulmonary atresia, tetralogy of Fallot and double outlet right ventricle. A: RV hypoplasia in pulmonary atresia. Underdevelopment of the RV outflow tract results in pulmonary atresia with a small hypertrophic RV and right-to-left shunting through an interatrial communication as escape route. Blood flows to the pulmonary circulation via the patent arterial duct. B: Tetralogy of Fallot. The aorta overrides the interventricular septum. Due to infundibular pulmonary stenosis there is RV hypertrophy and blood from both ventricles flows to the aorta. C–F: Subtypes of double outlet right ventricle (DORV). Arrows show the predominant direction of blood flow. C: DORV with subaortic VSD. D: DORV with subpulmonary VSD. E: DORV with doubly committed VSD. F: DORV with non-committed VSD. RV, right ventricle; RA, right atrium; PA, pulmonary artery; LV, left ventricle; LA, left atrium; Ao, aorta; VSD, ventricular septal defect.
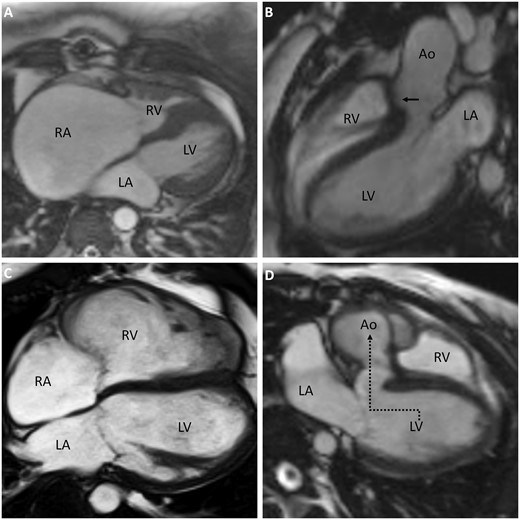
Magnetic resonance images of congenitally malformed adult hearts after surgical repair. A: RA dilation due to limited capacity of the small RV after biventricular repair in hypoplastic RV in context of severe valvar pulmonary stenosis. B: Overriding aorta and closed ventricular septal defect (arrow) in tetralogy of Fallot. C: Right ventricular dilation in tetralogy of Fallot. D: Conduit guiding blood from the LV to the aorta (dotted arrow) in double outlet right ventricle. LV, left ventricle; LA, left atrium; RV, right ventricle; RA, right atrium, Ao, aorta.
OFT malalignment towards the RV, with an aorta overriding the inter-ventricular septum, combined with hypertrophied septoparietal trabeculations that cause RVOT obstruction, are the developmental basis of the TOF phenotype.80 RV hypertrophy resulting as hemodynamic consequence of the aforementioned anatomical features concludes the tetralogy of typical TOF features (Figure 6B). Patients usually present during infancy with a murmur and a varying degree of cyanosis caused by right-to-left shunting via the VSD due to the RVOT obstruction. Currently, surgical correction of the malalignment, consisting of VSD closure (Figure 7B) and relief of RVOT obstruction, is performed within the first year in most patients.81 This has improved survival significantly: over 90% now survive into adulthood82 with a median survival of 70 years.7 In repaired adults, complications affecting the RV mostly arise from the now surgically altered situation, with pulmonary regurgitation after relief of RVOT obstruction. This causes RV volume overload, dilation, and eventually dysfunction (Figure 7C). Right bundle branch block, regional fibrosis and RVOT dyskinesis after surgery contribute to RV dysfunction and are associated with decreased exercise capacity, ventricular tachycardia, and heart failure.83,84 Additionally, RV volume overload is associated with increased diffuse myocardial fibrosis.85 Patients with a dilated RV due to pulmonary regurgitation often have TR, contributing to further dilation.86 Pulmonary valve replacement is considered to decrease volume overload, but optimal timing remains controversial.87 Whether concomitant tricuspid valve surgery or established treatment regimens for acquired heart failure should be applied to TOF patients with RV failure remains unclear.88
Malrotation of the OFT with failure to transfer one of the outflow tracts to the LV results in various phenotypes of DORV, with both great arteries arising predominantly from the RV (Figure 6C–F). The incomplete rotation causes lack of fusion between the outlet septum and septal band or septomarginal trabeculation,89 ensuing in interventricular communication. The arrangement of the great arteries varies due to various degrees of abnormal rotation. In 60% of patients, the aorta is located over the interventricular communication90 (Figure 6C). Physiology in these patients is similar to patients with a large VSD, presenting with left-to-right shunting, causing pulmonary hypertension and heart failure. Half of the patients with a subaortic interventricular communication have additional pulmonary stenosis, which leads to symptoms, treatment, and outcome similar to TOF. In 30–35% of DORV patients, the pulmonary artery is related to the interventricular communication (Figure 6D). In these patients, oxygenated blood from the LV flows into the pulmonary artery, while the aorta, related completely to the RV, receives systemic venous blood. Physiologically resembling complete TGA, these patients present with cyanosis in infancy and need surgery to restore concordance of the ventriculo-arterial connections (Figure 7D). In patients with doubly committed VSD, both great arteries arise above the interventricular communication (Figure 6E). In just 5% of patients,90 the interventricular communication is not between the limbs of the septal band, but located more apically, not committed to one of the great arteries (Figure 6F). In this situation blood flows into the ventricular cavity instead of into one of the arteries. Surgical strategies should be discussed per case, especially in patients presenting with doubly committed or non-committed VSD types, since their physiology varies greatly and is dependent on the exact location of the inter-ventricular communication, blood flow through the heart, and associated lesions as outflow tract stenosis (Table 1). Of the patients surviving into adulthood, about 50% reach the age of 50.7 Late complications are mostly determined by post-repair physiology. Since DORV presents a range of malaligned ventriculo-arterial connections, opposed to a specific anatomical defect, long-term outcome studies after DORV repair are difficult to interpret. Residual hemodynamic lesions regularly induce progressive ventricular dilation, dysfunction, and hypertrophy. Throughout adulthood, patients have a high risk for arrhythmias and sudden cardiac death.91 Frequent follow-up is mandatory to assess RV function and to determine the need for reinterventions to address residual hemodynamic lesions.
5. Collective issues in congenital heart defects affecting the right ventricle
Congenital heart defects affecting the RV are diverse and stem from various processes during heart development. As ccTGA results from inadequate looping, the RV is malpositioned in the systemic circulation, illustrating the complications arising from RV pressure overload in a morphologically normal RV. Furthermore, concomitant defects arising from different processes in heart development, such as Ebstein’s anomaly, are common in ccTGA. As Ebstein’s anomaly may result from over-specified AVC phenotype and failed expansion of the RV base, it involves defective RV formation, as are defects such as DCRV and the range of defects leading to a hypoplastic RV. As in ccTGA, the RV is subject to pressure overload in some of these defects, such as DCRV and hypoplastic RV in the context of severe pulmonary stenosis or pulmonary atresia. Patients with Ebstein’s anomaly may also have small RV cavities, but these patients primarily face volume overload due to TR as a result of the malformed tricuspid valve. In patients with OFT defects, resulting from malalignment or malrotation of the OFT, RV overload is also seen. Defects as TOF and DORV with RVOT obstruction result in combined volume and pressure overload due to the combination of interventricular communication and RVOT obstruction. Other OFT malformations such as DORV with subaortic interventricular communication and TGA have a subaortic RV due to altered OFT rotation. Therefore, the RV in unrepaired patients acts as the systemic ventricle, as described in ccTGA. However, these defects are currently mostly repaired with arterial switch operations, restoring ventriculo-arterial concordance, limiting the negative effects of the original congenital anatomy for the RV. Although these defects represent diverse heart structures affected by several developmental flaws, each with their own treatment strategies and distinct complications, they all stem from a continuum of processes during RV development. The overlapping morphological features in these defects can be explained by shared developmental origins of the affected components. Furthermore, the collective problem in these diseases is RV (volume and/or pressure) overload with related late complications, which are the main healthcare issues in the growing population of adults with CHD affecting the RV. Surgical scarring and altered hemodynamics result in myocardial fibrosis.85 Furthermore, conduction disturbances, excessive neurohormonal activation, and adverse ventricular–ventricular interactions may all influence the failure of these RVs. The multitude of factors contributing to RV failure in these CHD patients make treatment strategies for acquired heart failure difficult to incorporate in this population. Moreover, those strategies are often not as effective in CHD patients. Insight in the molecular development of CHD and the inherent complications may lead to alternative treatment options for these patients.
6. Conclusion
Novel insights in identifying major players in the regulation of SHF-derived structures have helped to understand the origin of the differences between the RV and LV, and the origin of congenital defects of the RV and OFT. Meanwhile, human genetic data have linked ∼30 developmentally relevant genes to CHD.57 This is an encouraging development, as such genetic knowledge can help improve prenatal diagnosis of CHD and genetic counselling. In the increasing population of adults with CHD, molecular and genetic background of late complications is increasingly needed to improve clinical care. With the still increasing insight into the molecular and pathophysiological mechanisms of CHD development, we may ultimately be able to provide better prognosis and prevention of disease progression in the setting of CHD affecting the RV.
Acknowledgements
We thank Antoon Moorman for helpful discussions.
Conflict of interest: none declared.
Funding
Netherlands Heart Foundation consortium COBRA3 and CVON 2014-18 CONCOR-genes to V.M.C.
References
Author notes
The first two authors contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}