




Abstract
Aberrant crypt foci (ACF), the earliest neoplastic lesions of the colon, have genetic and epigenetic alterations. Loss of heterozygosity (LOH) of tumor suppressor gene loci is seen in most colon cancers, but it is not known how early in tumorigenesis this takes place. Nine microsatellite markers close to specific genes, that is, APC (5q21), PTPRJ (11p11), p53 (17p13) and DCC (18q21), were analyzed in 32 ACF and samples of normal crypts from the same 28 patients. Six losses of heterozygosity were found in 5 of 32 ACF: 4 losses of heterozygosity were at 11p11, the location of the gene for protein tyrosine phosphatase receptor type J (PTPRJ) and of a second independent region of deletion; the others were at 5q21 and 18q21. Microsatellite instability (MSI) with markers for a single locus was found in 4 of 32 ACF. All the observed allelic alterations (LOH and MSI) were in 8 of 32 ACF. The finding of LOH in ACF with normal expressions of adenomatous polyposis coli (APC) and beta-catenin proteins suggests that LOH can occur very early in colon neoplasia and perhaps even before APC mutations. The finding of 3 of 4 of the losses of heterozygosity at 11p11 for PTPRJ and half of all the losses of heterozygosity in this study at PTPRJ suggest that this gene plays a role early in colon neoplasia.
Introduction
Aberrant crypt foci (ACF) are identified and defined by their microscopic appearance, usually after staining with methylene blue, in unembedded colonic mucosa that appears normal on visual inspection [( 1 ), reviewed in ref. 2 ]. Compared with normal, ACF have crypt(s) that are larger, have cryptal walls that are increased in thickness with lumens that are often oval or serrated rather than round, have increased pericryptal area, are not in the same focal microscopic plane as adjacent normal crypts and appear as a unit when more than one crypt is contained in the ACF (reviewed in ref. 2 ). The histopathology of ACF varies widely from non-dysplastic with minor alterations to severely dysplastic [( 1 ), reviewed in ref. 2 ]. An ACF can contain areas that are dysplastic and areas, often many serial sections removed, that are not dysplastic ( 3 ).
Colorectal cancer (CRC) is a malignant disease in which multiple genetic alterations of oncogenes and tumor suppressor genes occur stepwise during the tumor development ( 4 ). Two pathways of genomic instability, microsatellite instability (MSI) and chromosomal instability (CIN) have been proposed for the development of CRC ( 5 , 6 ). Mismatch repair deficiency, which is characterized by MSI, has been found in ∼10% of ACF ( 7 , 8 ) and in a similar proportion of sporadic colon cancers ( 9 , 10 ). CIN, which is characterized by multiple chromosomal abnormalities and a high frequency of loss of heterozygosity (LOH), appears to be the major pathway to colon cancer ( 5 , 11 , 12 ). Frequent allelic losses at 5q, 17p, 18q ( 4 , 11 ) and 11p ( 13 , 14 ) are seen in colonic adenomas as well as cancers; but allelic loss has not been evaluated in ACF, the earliest identified neoplastic lesions in the colon and putative precursors of CRC ( 2 , 15 ). We analyzed microdissected, paraffin-embedded sections from 32 ACF and paired normal crypts for LOH by using microsatellite markers close to the genes APC (5q21), protein tyrosine phosphatase receptor type J ( PTPRJ ) (11p11), p53 (17p13) and DCC (18q21). Our finding of LOH in 5 of 32 ACF confirms our earlier study with random primers ( 16 ) that suggests that chromosomal abnormalities can occur early in colon neoplasia. LOH of the PTPRJ gene in three of six LOH found in this study suggests that this gene may be important in the early events leading to CRC.
Materials and methods
Specimens
Human colon specimens were collected in 4°C saline by the Tissue Procurement Core Facility after approval of our study of discarded tissues by our institutional review board. Strips of macroscopically normal mucosa, previously separated from the submucosa and snap-frozen flat over liquid nitrogen (−195°C), were fixed for 30 min in 70% ethanol and stained with 0.2% methylene blue ( 16 , 17 ). ACF, marked with TBS, tissue marking dye, (Triangle Biomedical Sciences, Durham, NC) under a dissecting microscope, and histologically normal crypts from the same patient were embedded in paraffin. H&E-stained 5 µm sections were evaluated every 100 µm for the presence of the marked ACF. From 28 patients, a total of 32 ACF with 87 ± 29 (mean ± SD) crypts (range: 42–150 crypts) were microdissected.
Two H&E-stained slides, within 50 µm of the microdissected sample, were evaluated for the histopathology of each ACF according to criteria we have used previously ( 3 ) that are based on the descriptions of others ( 18 , 19 ). Namely, ACF without dysplasia may have enlarged nuclei and some loss of mucin but the alterations are less than those found with dysplasia. The term atypia is used to distinguish these slightly abnormal, but not dysplastic, glands in ACF that are frequently dilated or serrated from normal glands either adjacent to ACF or in patients without CRC. ACF with mild dysplasia have enlarged nuclei, some nuclear stratification and more loss of mucin than in ACF with atypia. ACF with moderate dysplasia have pleomorphic and more stratified nuclei with greater mucin loss than those with mild dysplasia. ACF with very pleomorphic nuclei that are highly stratified, a high degree of mucin loss and sometimes increased mitotic activity are considered severely dysplastic ( 3 ). Of the 32 ACF evaluated for LOH, 25 had only atypia, that is, lacked dysplasia; 6 had mild dysplasia and 1 had moderate dysplasia. One patient had diverticulosis with polyps, another had a sigmoid volvulus. Of the 26 patients with colon cancer, 1 had Dukes' Stage A, 14 had Stage B, 9 had Stage C and 2 had Stage D disease; none were known to have a familial cancer syndrome. The patients ranged in age from 42 to 91 (70.5 ± 10.9) years old; 13 were female, 15 were male. Two of the ACF were from the right colon, 30 were from the left colon.
Laser capture microdissection (LCM)
Samples of epithelial cells from ACF and from the same number of microscopically normal crypts as in the paired ACF were microdissected from paraffin-embedded sections under direct microscopic visualization with a PixCell II laser capture microdissection apparatus (Arcturus Engineering, Mountain View, CA) ( Figure 1A–C ). From each ACF, between 70 and 122 (99 ± 11) 5 µm sections of individual crypts were obtained. The microdissected crypts were suspended in 1 µl of proteinase K solution per section of crypt. Genomic DNA was extracted with proteinase K at 42°C overnight as described previously ( 16 , 17 ) and stored over liquid nitrogen prior to PCR analysis.
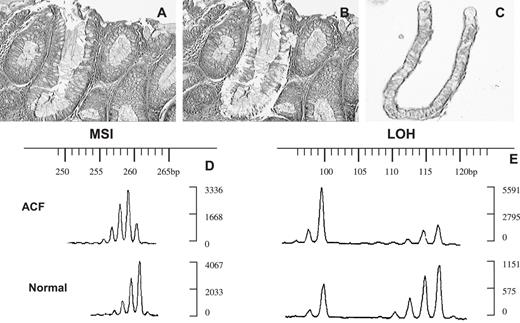
LCM and allelic alterations of ACF 4006107C2 no. 3A. ( A ) Paraffin-embedded section of ACF before microdissection; ( B ) paraffin-embedded section of ACF after microdissection; ( C) one of the crypts in the ACF microdissected with LCM. ( D and E ) Fluorescent electrophoregrams of this ACF (above) and normal crypts from the same patient (below). The sizes of the alleles in base pairs are indicated above; the intensities of the alleles are on the right. (D) MSI with marker D11S4117; (E) LOH with marker D18S35, qLOH = 0.2.
Polymerase chain reaction (PCR) and allelic analysis
Nine microsatellite markers on four different chromosomes were selected for analysis ( Table I ). Each forward primer was fluorescently labeled with FAM or TET (Invitrogen, Carlsbad, CA); PCR reactions were carried in a volume of 30 µl that contained 3 µl of genomic DNA, 125 µM dNTPs, 0.25 µM of each primer, 1.25 mM MgCl 2 , 1× PCR buffer and 0.5 U of Taq DNA polymerase (Fisher Scientific, Pittsburgh, PA). The PCR products were separated by capillary electrophoresis on an ABI 3100 Genetic Analyzer with Genescan and GeneMapper 1.1 software (Applied Biosystems, Foster City, CA) for allelic alterations. The height of the peaks produced by the DNA PCR products was quantified for each allele. The ratio of the two allelic heights was calculated for each ACF and paired normal DNA sample. When the qLOH (allelic ratio for the ACF peaks divided by the allelic ratio of paired normal sample) was ≤0.5 or ≥2.0, the case was counted as allelic imbalance and interpreted as LOH ( 20 ). An ACF with at least one definite new peak compared with normal DNA from the same patient was counted as MSI at that allele ( 21 ). All cases with allelic alterations, both LOH and MSI, were confirmed by a second amplification and analysis of both ACF and paired normal crypts.
LOH and MSI in human ACF from a total of 32 ACF
| ACF | Crypts per ACF | Pathology of ACF a | Location b | Sex | Age (years) | Dukes' stage | Microsatellite markers | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D5S3 465q21 ( APC) | D11S1 31911p11 | D11S4117 11p11 (PTPRJ) | D11S4183 11p11 (PTPRJ) | TP53 17p13 (P53) | D17S960 17p13.2 (P53) | D18S35 18q21 (DCC) | D18S474 18q21.1 (DCC) | D18S487 18q21.2 (DCC) | |||||||||||||||
| 30 (94) c | 18 (56) | 23 (72) | 21 (66) | 31 (97) | 20 (63) | 23 (72) | 25 (78) | 23 (72) | |||||||||||||||
| 4002368A no. 4AB | 125 | Mild d | L | M | 65 | B | MSI | ||||||||||||||||
| 4002552C no. 1A | 67 | A | L | M | 64 | C | LOH | ||||||||||||||||
| 4003821A no. 2AD | 131 | Mild d | L | F | 66 | B | LOH | ||||||||||||||||
| 4006060B1 no. 2A | 89 | A | L | F | 69 | A | LOH | LOH | |||||||||||||||
| 4006107C2 no. 3A | 58 | A | L | F | 70 | C | MSI | LOH | |||||||||||||||
| 4006182B1 no. 2AB | 62 | A | L | M | 83 | B | MSI | ||||||||||||||||
| 4006518B1 no. 4FG | 54 | A | L | M | 67 | C | LOH | ||||||||||||||||
| 4007824B1 no. 3 | 69 | A | L | M | 83 | B | MSI | ||||||||||||||||
| ACF | Crypts per ACF | Pathology of ACF a | Location b | Sex | Age (years) | Dukes' stage | Microsatellite markers | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D5S3 465q21 ( APC) | D11S1 31911p11 | D11S4117 11p11 (PTPRJ) | D11S4183 11p11 (PTPRJ) | TP53 17p13 (P53) | D17S960 17p13.2 (P53) | D18S35 18q21 (DCC) | D18S474 18q21.1 (DCC) | D18S487 18q21.2 (DCC) | |||||||||||||||
| 30 (94) c | 18 (56) | 23 (72) | 21 (66) | 31 (97) | 20 (63) | 23 (72) | 25 (78) | 23 (72) | |||||||||||||||
| 4002368A no. 4AB | 125 | Mild d | L | M | 65 | B | MSI | ||||||||||||||||
| 4002552C no. 1A | 67 | A | L | M | 64 | C | LOH | ||||||||||||||||
| 4003821A no. 2AD | 131 | Mild d | L | F | 66 | B | LOH | ||||||||||||||||
| 4006060B1 no. 2A | 89 | A | L | F | 69 | A | LOH | LOH | |||||||||||||||
| 4006107C2 no. 3A | 58 | A | L | F | 70 | C | MSI | LOH | |||||||||||||||
| 4006182B1 no. 2AB | 62 | A | L | M | 83 | B | MSI | ||||||||||||||||
| 4006518B1 no. 4FG | 54 | A | L | M | 67 | C | LOH | ||||||||||||||||
| 4007824B1 no. 3 | 69 | A | L | M | 83 | B | MSI | ||||||||||||||||
a Mild d, mild dysplasia; A, atypia or no dysplasia.
b L, left or distal colon.
c Number (%) informative cases for each marker.
LOH and MSI in human ACF from a total of 32 ACF
| ACF | Crypts per ACF | Pathology of ACF a | Location b | Sex | Age (years) | Dukes' stage | Microsatellite markers | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D5S3 465q21 ( APC) | D11S1 31911p11 | D11S4117 11p11 (PTPRJ) | D11S4183 11p11 (PTPRJ) | TP53 17p13 (P53) | D17S960 17p13.2 (P53) | D18S35 18q21 (DCC) | D18S474 18q21.1 (DCC) | D18S487 18q21.2 (DCC) | |||||||||||||||
| 30 (94) c | 18 (56) | 23 (72) | 21 (66) | 31 (97) | 20 (63) | 23 (72) | 25 (78) | 23 (72) | |||||||||||||||
| 4002368A no. 4AB | 125 | Mild d | L | M | 65 | B | MSI | ||||||||||||||||
| 4002552C no. 1A | 67 | A | L | M | 64 | C | LOH | ||||||||||||||||
| 4003821A no. 2AD | 131 | Mild d | L | F | 66 | B | LOH | ||||||||||||||||
| 4006060B1 no. 2A | 89 | A | L | F | 69 | A | LOH | LOH | |||||||||||||||
| 4006107C2 no. 3A | 58 | A | L | F | 70 | C | MSI | LOH | |||||||||||||||
| 4006182B1 no. 2AB | 62 | A | L | M | 83 | B | MSI | ||||||||||||||||
| 4006518B1 no. 4FG | 54 | A | L | M | 67 | C | LOH | ||||||||||||||||
| 4007824B1 no. 3 | 69 | A | L | M | 83 | B | MSI | ||||||||||||||||
| ACF | Crypts per ACF | Pathology of ACF a | Location b | Sex | Age (years) | Dukes' stage | Microsatellite markers | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| D5S3 465q21 ( APC) | D11S1 31911p11 | D11S4117 11p11 (PTPRJ) | D11S4183 11p11 (PTPRJ) | TP53 17p13 (P53) | D17S960 17p13.2 (P53) | D18S35 18q21 (DCC) | D18S474 18q21.1 (DCC) | D18S487 18q21.2 (DCC) | |||||||||||||||
| 30 (94) c | 18 (56) | 23 (72) | 21 (66) | 31 (97) | 20 (63) | 23 (72) | 25 (78) | 23 (72) | |||||||||||||||
| 4002368A no. 4AB | 125 | Mild d | L | M | 65 | B | MSI | ||||||||||||||||
| 4002552C no. 1A | 67 | A | L | M | 64 | C | LOH | ||||||||||||||||
| 4003821A no. 2AD | 131 | Mild d | L | F | 66 | B | LOH | ||||||||||||||||
| 4006060B1 no. 2A | 89 | A | L | F | 69 | A | LOH | LOH | |||||||||||||||
| 4006107C2 no. 3A | 58 | A | L | F | 70 | C | MSI | LOH | |||||||||||||||
| 4006182B1 no. 2AB | 62 | A | L | M | 83 | B | MSI | ||||||||||||||||
| 4006518B1 no. 4FG | 54 | A | L | M | 67 | C | LOH | ||||||||||||||||
| 4007824B1 no. 3 | 69 | A | L | M | 83 | B | MSI | ||||||||||||||||
a Mild d, mild dysplasia; A, atypia or no dysplasia.
b L, left or distal colon.
c Number (%) informative cases for each marker.
Cases in which the normal sample appeared homozygous varied for the different markers from 0 cases with D5S346 to 11 cases with D17S960 (mean 6.6 ± 3.7); the amplification of samples was unsuccessful seven times, and eight samples lacked sufficient material to be assayed with D11S1319. The combination of these 3 groups resulted in 74 uninformative cases and 214 informative cases; the average frequency of informative cases per marker was 74.4 ± 13.5% (range: 56–97%) ( Table I ). The frequency of LOH was determined by dividing the number of LOHs for a particular marker by the number of informative cases found for that marker, as listed in Table I . If more than one marker identified the same gene, the results of all markers for that gene were combined.
Immunohistochemistry
Paraffin-embedded sections of the ACF that demonstrated LOH and/or MSI were evaluated for their expressions of beta-catenin and adenomatous polyposis coli (Apc) with antibodies specific for each of these proteins. The histochemical procedure for the expression of beta-catenin was carried out as described previously ( 22 ). A mouse monoclonal antibody to beta-catenin (IgG1; Transduction Laboratories, Lexington, KY) was diluted 1 : 2000 in 10% horse serum. A second section on each slide was treated with a similar concentration of non-specific mouse IgG1 (DakoCytomation, Carpinteria, CA) as a negative control. Apc was evaluated with a histochemical procedure we used previously ( 23 ) that included tyramide signal amplification (Renaissance, PerkinElmer Life Sciences, Boston, MA). A rabbit polyclonal antibody to the C-terminal portion of human Apc (C-20; sc-896; Santa Cruz Biotechnology, Santa Cruz, CA) was diluted 1 : 1000 with blocking solution; normal rabbit serum at the same concentration was used on a second section on each slide for a negative control. Paraffin sections of colon cancer and normal colonic mucosa were incubated with each antibody at the same time as the ACF for positive controls.
Results
Ten reproducible allelic alterations were identified in 8 of 32 ACF ( Figures 1 and 2 , Table I ). Of the six LOH, three ( Figure 2 ) were identified with microsatellite markers for the tumor suppressor gene PTPRJ at 11p11 ( 13 ). The other LOH were for a second independent region of deletion at 11p11 (D11S1319) ( 13 ), the chromosomal region of the DCC gene at 18q21 and for the APC gene at 5q21 ( Figure 1E , Table I ). The frequency of LOH was highest (3 out of 44) for PTPRJ , next highest (1 out of 18) for a second region at 11p11 (D11S1319), 1 out of 23 for D18S35, 1 out of 71 for the chromosomal region of DCC , and 1 out of 30 for APC ( Table I ). Interestingly, the 6 LOH were in 5 ACF from 5 of the 14 patients between 61 and 70 years old; no LOH was seen in 3 patients < 60 or in 11 patients > 71 years old. LOH in ACF was not correlated with the histopathology of the ACF, the number of crypts in the ACF or the stages of the cancers in these patients ( Table I ). The ACF (4006060B1 no. 2A) with LOH for two independent microsatellite markers on chromosome 11 contained 89 crypts but no dysplasia; it was from a 69-year-old female patient with Dukes' Stage A carcinoma ( Table I ). It is interesting to note that a second ACF from the same patient did not display LOH for these same markers or any of the markers evaluated in this study.
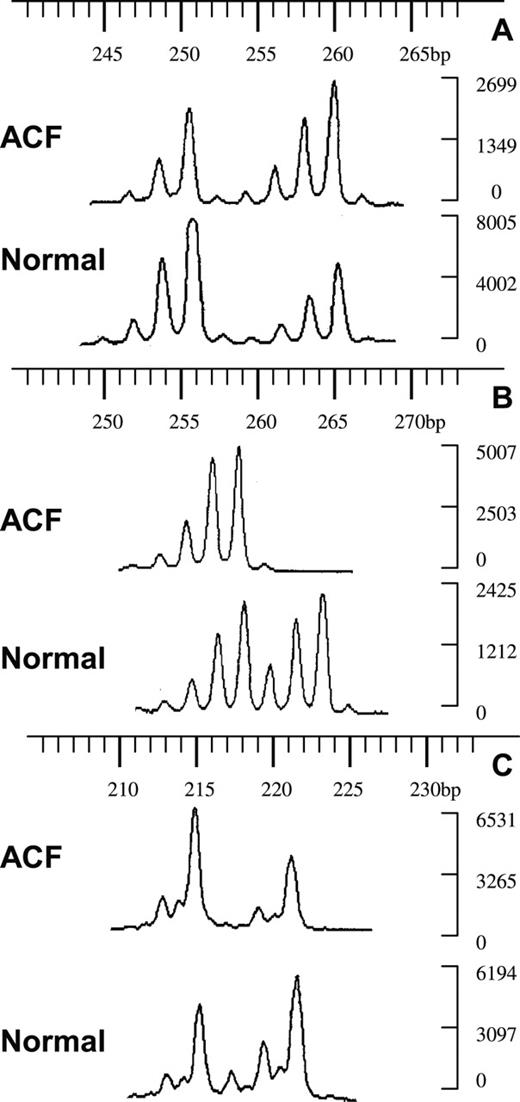
LOH in human ACF at PTPRJ (11p11). Fluorescent electrophoregrams of ACF and normal crypts from three different patients. The sizes of the alleles in base pairs are indicated above; the intensities of the alleles are on the right. ( A ) ACF 4003821A no. 2AD with marker D11S4117, qLOH = 0.5; ( B ) ACF 4006060B1 no. 2A with D11S4117, qLOH > 2; ( C ) ACF 4006518B1 no. 4FG with D11S4183, qLOH = 2.2.
A second ACF from a different patient, with 58 crypts and no dysplasia, also had two allelic alterations: MSI with a marker for 11p11 and LOH of the chromosomal region 18q21 ( Figure 1 , Table I ). MSI at single alleles was seen in 4 of 32 ACF ( Figure 1D , Table I ). The functional significance of low-level MSI is unknown ( 24 , 25 ). It is noteworthy that six of the eight ACF identified with allelic alterations lacked dysplasia; the remaining two ACF had mild dysplasia ( Table I ). Since it was not possible to assess the mutational status of the APC and/or beta-catenin genes in these very small lesions, immunohistochemistry was used to evaluate the expressions of beta-catenin and Apc proteins in the eight ACF with allelic alterations. These ACF displayed similar expressions of beta-catenin and Apc proteins as that seen in adjacent, normal colonic mucosa ( Figure 3 ) and in control normal mucosa taken a distance from ACF and cancer. The control normal colonic mucosa displayed strong cytoplasmic expression of Apc and membranous expression of beta-catenin; colonic carcinomas had a marked reduction or absence of expression of Apc with increased cytoplasmic and reduced membranous expression of beta-catenin as seen previously ( 22 , 23 ).
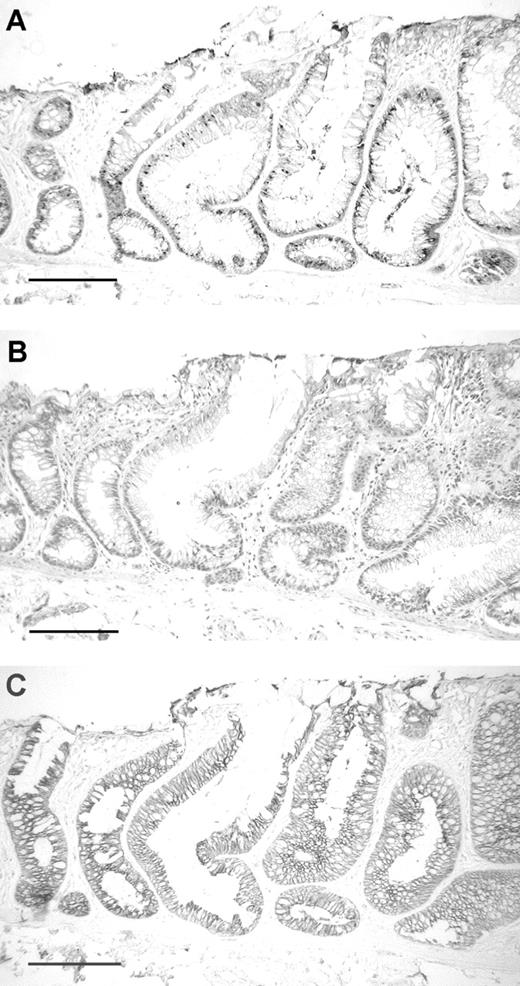
Paraffin-embedded sections of ACF 4006107C2 no. 3A. ( A ) Immunohistochemical demonstration of Apc expression in the cytoplasm of the ACF (large crypts to the right) and in two adjacent normal crypts (left); ( B ) nearby section stained with H&E shows the ACF (large crypts to the right with yellow ink above) with atypia but no dysplasia; ( C ) immunohistochemical demonstration of beta-catenin expression in the membranes of the same ACF and adjacent normal crypts; bar = 200 µm.
Discussion
The finding of LOH of tumor suppressor loci in ACF, the earliest neoplastic lesions identified in human colon ( 2 , 15 ), demonstrates that large segments of chromosomes can be lost very early in colon neoplasia. While we demonstrate a low frequency of LOH in ACF, the real frequency of LOH and/or chromosomal abnormalities in general and with regard to specific markers or genes can only be known when a much larger sample of ACF is evaluated. It is likely that the frequency of LOH would be higher if a larger number of markers were used; if the markers had a higher frequency of heterozygosity, that is, less homozygosity, in our population; and especially if we had a better way to identify those regions most susceptible to loss early in the tumorigenic process. Our choice of areas to search for LOH in ACF was based on the findings of LOH in adenomas on 5q ( 4 , 26 ), 11p ( 14 ), 17p ( 4 , 26 – 28 ) and 18q ( 4 , 26 – 30 ). While alterations on 17p and 18q are thought to be late events in colon tumorigenesis ( 4 ), the reported frequency of loss of regions on 17p and 18q in adenomas is close to or exceeds that of 5q ( 4 , 26 , 27 , 29 , 30 ).
The identification of LOH in ACF confirms our earlier finding of aberrant DNA fingerprints in 23% of ACF amplified with random primers ( 16 ). These LOH studies now identify specific regions of the genome that appear to be lost early in colon neoplasia. Recently, aneusomy was detected for 1p, 17p and 18q in macroscopically normal mucosa from patients with colon cancer ( 31 ). In that study ( 31 ), we do not know if the mucosal areas detected with aneuploidy were histologically normal or the sites of unidentified ACF since ACF were not evaluated in those tissues. Are these genetic alterations that we are reporting in ACF independent genetic events or merely a field effect due to CRC in the same patient? Several previous studies ( 32 – 34 ), as well as this study, have demonstrated that different ACF collected from the same surgical resections have different phenotypes and/or genotypes from each other and/or the synchronous cancer. In the present study, as in our previous work, the genotype of the ACF was compared with that seen in the same number of normal crypts isolated from mucosa within the same 10 cm 2 specimen. If an area of crypts had undergone mutation prior to the development of cancer and ACF, the same mutations would be seen throughout the area. In LOH studies, those ACF and adjacent normal crypts would appear the same and be indistinguishable from normal, that is, would give a ratio of 1.
The occurrence of allelic imbalance in 90% of 32 small (1 to 3 mm) adenomas provided evidence that CIN occurs in early precursors of CRC ( 35 ). The lower frequency of allelic imbalance detected in our studies is probably due to the facts that ACF are even earlier precursors than grossly identifiable small adenomas, fewer markers were used, microsatellite analysis is less sensitive than digital single-nucleotide polymorphism analysis ( 35 ) and we lacked material to repeat or do some assays. Many of these drawbacks are directly related to the very limited size of ACF; for example, the lack of template DNA from eight ACF contributed to the low percentage of informative cases (18 out of 32 or 56%) for marker D11S1319. The method used by Shih et al . ( 35 ) detected genetic gains as well as losses while LOH with microsatellite markers is thought to detect mostly the loss of genetic material. Like Shih et al . ( 35 ), we found allelic imbalance at chromosomal arms 5q and 18q, the only loci evaluated in both studies. As noted above, several other investigators have reported LOH at 5q, 11p, 17p and 18q in adenomas including some with low-grade dysplasia ( 4 , 26 – 30 ). In those studies the frequency of LOH in adenomas rarely reached 50% even for large adenomas or those that had areas progressed to carcinoma ( 4 , 14 , 28 ).
Inactivation of the APC gene also occurs very early in colon tumorigenesis, but it is observed in only 5% of sporadic ACF (reviewed in refs 2 , 36 , 37 ). ACF with APC mutations are dysplastic while four of five of our ACF with LOH lack dysplasia and the fifth ACF has only mild dysplasia. On the basis of the frequency of APC mutations reported in ACF and the lack of dysplasia in 80% of our ACF with LOH, it seems unlikely that these ACF have APC mutations. While we were not able to directly test these ACF for APC mutations owing to lack of material, we were able to evaluate the immunohistochemical expression of both the Apc and beta-catenin proteins in them. We used an antibody to the C-terminal portion of the Apc protein that detects only full-length or wild-type Apc protein and not a truncated form that is made from a mutated APC gene. All of our ACF with LOH showed the same expression of Apc as normal colonic mucosa that contain at least one copy of wild-type APC and very different from the loss of expression seen in colonic cancers, over 80% of which have inactivated APC genes ( 38 ). When APC is mutated, it loses its binding site for beta-catenin that generally results in an increased expression of beta-catenin in the cytoplasm and possibly the nucleus with decreased membranous expression ( 22 ). Again, the ACF with LOH showed a similar expression of beta-catenin as seen in normal mucosa and not the aberrant expression seen in colon cancers evaluated at the same time. These beta-catenin results are further evidence that our ACF with LOH lack APC mutations. While mutation of one allele of APC is thought to precede the deletion of the second allele, we had one ACF (4002552C no. 1A) with LOH at 5q21 that appears to have retained a wild-type copy of APC similar to that reported recently ( 39 ).
As noted above, there is evidence that CIN occurs as early as the small adenoma stage in colon tumorigenesis ( 35 ). CIN is proposed to be the major pathway for the development of CRCs ( 5 ), but the requirement of CIN to drive tumorigenesis is still controversial ( 40 ). CIN, as evaluated by the frequency of anaphase bridges, did result in increased tumor formation in compound mutant mice ( 41 ). Likewise, it is interesting to consider the role of CIN proposed by Rajagopalan et al . ( 6 ). According to their model ( 6 ), ‘if a genetic pathway of tumorigenesis requires the inactivation of two or more tumour-suppressor genes, then it becomes increasingly likely that CIN precedes the inactivation of the first tumour-suppressor gene in most cancers.’ Since even small polyps have inactivated APC genes, their ( 6 ) hypothesis would predict that CIN occurs even before the polyp stage. While it is clear that LOH can occur in ACF and perhaps even in ACF without APC mutations, the frequency of LOH and/or chromosomal abnormalities that we found is not sufficiently high to be regarded as CIN ( 5 ). Only future studies will determine if the frequency of LOH in ACF is sufficiently high to be regarded as CIN and whether this model is correct. The placement of CIN before APC mutations parallels the similar placement of MSI ( 6 ) that also occurs in ACF ( 7 , 8 ).
The presence of 67% of the LOH at 11p11 in this study is particularly interesting. Three of those LOH are within the PTPRJ gene, and the fourth is in a second independent region 3 Mb distal to it ( 13 ). PTPRJ was found to be the candidate gene for the colon cancer susceptibility locus Scc1 in mice, and both regions show frequent LOH in several human cancers including CRC ( 13 ). Our finding of more LOH for PTPRJ than the chromosomal region 18q21 in our ACF is consistent with the finding of Ruivenkamp et al . ( 14 ) that loss of PTPRJ occurs earlier than the chromosomal region 18q21 in colon tumorigenesis. The presence of LOH in ACF with normal expressions of Apc and beta-catenin provides experimental evidence that chromosomal abnormalities and LOH can occur very early in colon neoplasia and maybe even before APC mutations. The role of PTPRJ in early colon neoplasia awaits further studies.
We thank Katherine Dunmire and Adam Kresak for their expert technical assistance. This study would not have been possible without the generous support of the faculty and staff of the Tissue Procurement Core Facility of the Comprehensive Cancer Center of Case Western Reserve University and University Hospitals of Cleveland (P30 CA43703) who procured all the tissues used in this study. This work was funded in part by NIH grants CA66725 (T.P.P.), HL65630 and HL66251 (Q.K.W.).
Conflict of Interest Statement : None declared.
References
Pretlow,T.P., Barrow,B.J., Ashton,W.S., O'Riordan,M.A., Pretlow,T.G., Jurcisek,J.A. and Stellato,T.A. (
Pretlow,T.P. and Pretlow,T.G. (
Siu,I.M., Pretlow,T.G., Amini,S.B. and Pretlow,T.P. (
Vogelstein,B., Fearon,E.R., Hamilton,S.R., Kern,S.E., Preisinger,A.C., Leppert,M., Nakamura,Y., White,R., Smits,A.M. and Bos,J.L. (
Lengauer,C., Kinzler,K.W. and Vogelstein,B. (
Rajagopalan,H., Nowak,M.A., Vogelstein,B. and Lengauer,C. (
Augenlicht,L.H., Richards,C., Corner,G. and Pretlow,T.P. (
Heinen,C.D., Shivapurkar,N., Tang,Z., Groden,J. and Alabaster,O. (
Aaltonen,L.A., Peltomaki,P., Leach,F.S. et al . (
Thibodeau,S.N., Bren,G. and Schaid,D. (
Vogelstein,B., Fearon,E.R., Kern,S.E., Hamilton,S.R., Preisinger,A.C., Nakamura,Y. and White,R. (
Anderson,G.R., Brenner,B.M., Swede,H. et al . (
Ruivenkamp,C.A., van Wezel,T., Zanon,C. et al . (
Ruivenkamp,C., Hermsen,M., Postma,C., Klous,A., Baak,J., Meijer,G. and Demant,P. (
Siu,I.-M., Robinson,D.R., Schwartz,S., Kung,H.-J., Pretlow,T.G., Petersen,R.B. and Pretlow,T.P. (
Luo,L., Li,B. and Pretlow,T.P. (
Luo,L., Chen,W.D. and Pretlow,T.P. (
Konishi,F. and Morson,B.C. (
Riddell,R.H., Goldman,H., Ransohoff,D.F. et al . (
Sawyer,E.J., Cerar,A., Hanby,A.M., Gorman,P., Arends,M., Talbot,I.C. and Tomlinson,I.P. (
Mueller,J.D., Haegle,N., Keller,G., Mueller,E., Saretzky,G., Bethke,B., Stolte,M. and Hofler,H. (
Hao,X.P., Pretlow,T.G., Rao,J.S. and Pretlow,T.P. (
Pretlow,T.P., Edelmann,W., Kucherlapati,R., Pretlow,T.G. and Augenlicht,L.H. (
Laiho,P., Launonen,V., Lahermo,P. et al . (
Mori,Y., Selaru,F.M., Sato,F. et al . (
Ried,T., Knutzen,R., Steinbeck,R., Blegen,H., Schrock,E., Heselmeyer,K., du Manoir,S. and Auer,G. (
Bomme,L., Bardi,G., Pandis,N., Fenger,C., Kronborg,O. and Heim,S. (
Hermsen,M., Postma,C., Baak,J. et al . (
Bomme,L., Bardi,G., Pandis,N., Fenger,C., Kronborg,O. and Heim,S. (
Muleris,M., Zafrani,B., Validire,P., Girodet,J., Salmon,R.J. and Dutrillaux,B. (
Cianciulli,A., Cosimelli,M., Marzano,R. et al . (
Pretlow,T.P., Brasitus,T.A., Fulton,N.C., Cheyer,C. and Kaplan,E.L. (
Hao,X.P., Pretlow,T.G., Rao,J.S. and Pretlow,T.P. (
Suzuki,H., Watkins,D.N., Jair,K.-W. et al . (
Shih,I.M., Zhou,W., Goodman,S.N., Lengauer,C., Kinzler,K.W. and Vogelstein,B. (
Jen,J., Powell,S.M., Papadopoulos,N., Smith,K.J., Hamilton,S.R., Vogelstein,B. and Kinzler,K.W. (
Takayama,T., Ohi,M., Hayashi,T. et al . (
Powell,S.M., Zilz,N., Beazer-Barclay,Y., Bryan,T.M., Hamilton,S.R., Thibodeau,S.N., Vogelstein,B. and Kinzler,K.W. (
Rowan,A., Halford,S., Gaasenbeek,M. et al . (
Sieber,O.M., Heinimann,K. and Tomlinson,I.P. (
Author notes
Institute of Pathology, Case Western Reserve University, Cleveland, OH 44106, USA and 1Department of Molecular Cardiology, Lerner Research Institute, Cleveland Clinic Foundation, Cleveland, OH 44195, USA

{kind=link}
{kind=link}
{kind=link}