




Abstract
Epidemiologic studies have suggested that dietary intake and blood levels of folate may be inversely related to the risk of breast cancer. However, epidemiologic evidence has not been consistent nor has it provided unequivocal support for this purported inverse relationship. Recent evidence has also raised a concern that folate supplementation may promote carcinogenesis if provided after neoplastic foci are established in the target organ. This study investigated the effect of dietary folate deficiency and supplementation on the development and progression of mammary tumors in the N -methyl- N -nitrosourea (MNU) rat model. Weanling, female Sprague–Dawley rats were fed diets containing 0, 2 (control) or 8 mg folic acid/kg diet during the initiation or the promotion phase of MNU-induced mammary tumorigenesis. At necropsy, all macroscopic mammary tumors were identified and histologically confirmed. Dietary folate deficiency and supplementation provided during the initiation phase did not significantly modulate the development of mammary tumors. In contrast, dietary folate deficiency provided during the promotion phase significantly inhibited the rate of appearance, incidence, mean volume and weight of adenocarcinomas compared with the control and supplemental diets. Folate supplementation provided during the promotion phase did not significantly modulate mammary tumorigenesis compared with the control group. These data indicate that moderate folate deficiency inhibits, whereas dietary folate supplementation at four times the basal dietary requirement does not promote, the progression of MNU-induced mammary neoplastic foci in this rat model. However, the limitations associated with the route and dose of MNU administration preclude a definitive conclusion concerning the effect of folate status on the initiation of MNU-induced mammary tumorigenesis.
Introduction
Dietary intake and blood levels of folate, a water-soluble B-vitamin and an important co-factor in one-carbon metabolism ( 1 ), appear to be inversely associated with the risk of several malignancies including cancer of the colorectum, lungs, pancreas, esophagus, stomach, cervix and ovary, and neuroblastoma and leukemia ( 2 , 3 ). The precise nature and magnitude of the relationship between folate status and the risk of these malignancies, however, have not yet been clearly established ( 2 , 3 ). As an essential co-factor for the de novo biosynthesis of purines and thymidylate, folate plays an important role in DNA synthesis, stability and integrity, and repair, the aberrations of which have been implicated in carcinogenesis ( 4 , 5 ). Folate, in the form of 5-methyltetrahydrofolate, is also involved in remethylation of homocysteine to methionine, thereby ensuring the supply of S -adenosylmethionine (SAM), the primary methyl group donor for most biological methylation reactions including that of DNA ( 6 ). In this role, folate may modulate DNA methylation, which is an important epigenetic determinant in gene expression, in the maintenance of DNA integrity and stability, in chromosomal modifications and in the development of mutations ( 7 ). A growing body of evidence from in vitro , animal and human studies indicates that folate deficiency is associated with DNA strand breaks, impaired DNA repair, increased mutations and aberrant DNA methylation, and that folate supplementation can correct some of these defects induced by folate deficiency ( 2 , 4 – 6 , 8 ).
An accumulating body of epidemiologic studies also suggest an inverse association between folate status and the risk of breast cancer ( 9 – 23 ). However, epidemiologic evidence available thus far has not been consistent nor has it provided unequivocal support for the purported inverse relationship between folate status and breast cancer risk ( 24 – 29 ). Some epidemiologic studies have suggested that folate status alone may not be sufficient in modifying breast cancer risk. However, with alcohol consumption, a well-established risk factor for breast cancer development ( 30 , 31 ), folate deficiency potentiates, whereas folate supplementation reduces, the risk of breast cancer ( 12 , 14 , 15 , 19 , 20 , 22 ). Furthermore, some studies have suggested that folate status interacts with other dietary factors involved in one-carbon metabolism (e.g. methionine, vitamins B 6 and B 12 ) to modify breast cancer risk ( 15 , 19 ). Also, there is evidence that the direction and magnitude of the breast cancer risk modification associated with folate status may depend on the C677T polymorphism in the methylenetetrahydrofolate reductase (MTHFR) gene that encodes an enzyme critical for intracellular folate homeostasis ( 16 , 23 , 32 – 35 ).
Since only a few modifiable risk factors for breast cancer exist ( 31 , 36 ), — recent epidemiologic observations that suggest that folate deficiency increases, whereas supplementation reduces, —the risk of breast cancer underscore the need to further evaluate the role of folate in the development of this disease. Folate is generally regarded as safe and has long been presumed to be purely beneficial ( 37 ), and possesses biologically plausible mechanisms for cancer prevention ( 2 , 4 – 6 , 8 , 38 ). However, studies performed in animal models of colorectal cancer have shown that the dose and timing of folate intervention are critical in providing safe and effective chemoprevention; exceptionally high supplemental folate levels and folate intervention after microscopic neoplasia established in the colorectal mucosa promote rather than suppress colorectal carcinogenesis ( 39 , 40 ). These observations suggest that folate possesses dual modulatory effects on carcinogenesis depending on the timing and dose of folate intervention ( 39 , 40 ). Folate deficiency has an inhibitory effect, whereas folate supplementation has a promoting effect on the progression of established neoplasms ( 39 , 40 ). In contrast, folate deficiency in normal epithelial tissues appears to predispose them to neoplastic transformation, and modest levels of folate supplementation suppress, whereas supraphysiological doses enhance the development of tumors in normal tissues ( 39 , 40 ).
Given the dual modulatory role of folate in carcinogenesis, clarifying the potential role of folate in breast cancer prevention and progression in appropriate animal models is important because the vast majority of the US and Canadian populations are exposed to high amounts of folic acid owing to folic acid fortification and supplementation ( 40 ). One animal study employing the N -methyl- N -nitrosourea (MNU) rat model showed that a folate-deficient diet provided during the initiation phase of mammary tumorigenesis significantly reduced tumor multiplicity and increased tumor latency, but had no effect on tumor incidence, compared with a control and folate-supplemented diet ( 41 ). We have recently reported that dietary folate deficiency of a moderate degree (without anemia, growth retardation or premature death) suppresses, whereas a modest level (four times the basal dietary requirement) of folate supplementation does not significantly modulate, mammary tumorigenesis in the MNU rat model ( 42 ).
MNU is a direct acting, potent rodent mammary carcinogen that does not require metabolism ( 43 ). MNU induction is associated with a mammary tumor incidence of >90%, which are predominantly adenocarcinomas of a ductal origin with very few benign tumors ( 43 ). MNU is an alkylating agent that forms O6 -methylguaine in DNA, leading to G→A transition during DNA replication ( 44 ). More than 85% of MNU-induced mammary tumors contain a G→A transition mutation at codon 12 of the Ha- ras oncogene, and consequent Ha- ras activation has been considered the primary mechanism by which MNU induces mammary tumors in rodent ( 45 , 46 ). MNU also induces DNA hypomethylation ( 47 ), and MNU-induced mammary tumors are associated with genomic DNA hypomethylation ( 42 ). Given the role of folate in DNA synthesis, mutagenesis and DNA methylation ( 2 , 4 – 6 , 8 ), folate possesses biologically plausible mechanisms by which it can modulate MNU-induced mammary tumorigenesis.
The aim of this study was to elucidate the effects of dietary folate deficiency and supplementation on the development and progression of mammary tumors by determining the effects of folate on the initiation and promotion phases of mammary tumorigenesis separately in the well-established MNU rat model of breast cancer. Our hypothesis was that dietary folate deficiency would enhance, whereas dietary folate supplementation would suppress, the initiation of MNU-induced mammary tumorigenesis. In contrast, once microscopic mammary neoplastic foci were established, we reasoned that dietary folate deficiency would suppress, whereas dietary folate supplementation would enhance, the promotion and progression of MNU-induced mammary tumorigenesis.
Materials and methods
Animals and dietary intervention
This study was approved by the Animal Care Committee of the University of Toronto. Specific pathogen-free, weanling female Sprague–Dawley rats (∼50 g; Charles River Laboratories, St Constant, Quebec, Canada) were singly housed and maintained at 24 ± 2°C at 50% humidity with a 12 h light/dark cycle. Amino acid-defined diets (Dyets, Bethlehem, PA) ( 48 ) containing 0, 2 or 8 mg folic acid/kg diet were used. These diets constitute a standard method of inducing folate deficiency or providing supplemental dietary folate in rodents ( 48 ) and have been utilized extensively in previous studies of folate and colorectal cancer ( 49 – 52 ) and breast cancer ( 42 ). The diet containing 0 mg folic acid/kg produces progressive folate deficiency of a moderate degree without anemia, growth retardation or premature death through weeks 3–5, after which systemic folate indicators stabilize ( 42 , 49 ). Although this diet is completely devoid of folate, severe folate deficiency is not induced because of de novo synthesis of folate by intestinal bacteria, some of which is incorporated into the tissue folate of the host ( 53 ). The diet containing 2 mg folic acid/kg is generally accepted as the basal dietary requirement for rodents ( 54 ). The diet containing 8 mg folic acid/kg represents folate supplementation four times the basal dietary requirement. This level of folate was chosen because the 8 mg/kg level has consistently provided a degree of chemoprevention against colorectal cancer in previous rodent studies ( 49 , 50 , 52 ). These diets contained 50 g cellulose/kg, 60% of the calories as carbohydrates, 23% as fat (or 10% by weight) and 17% as l -amino acids ( 48 ). The amount of methyl donors, methionine, choline and vitamin B 12 , were 8.2 g, 2.0 g and 50 µg per kg diet, respectively. The detailed composition of the diets has been published previously ( 48 , 52 ). Diets and water were provided ad libitum .
In the initiation study, rats ( n = 61) were randomized to receive the diet containing 0 ( n = 21), 2 ( n = 20) or 8 ( n = 20) mg folic acid/kg diet from weaning at 3 weeks of age for 5 weeks until 1 week following MNU injection, which occurred at 50 days of age. The initial diets were terminated 1 week after the MNU injection, and all the rats were placed on the control diet (2 mg folic acid/kg diet) for 22 weeks until the time of killing.
In the promotion study, rats ( n = 93) were placed on the control diet (2 mg folic acid/kg diet) from weaning at 3 weeks of age for 5 weeks until 1 week following MNU injection, which occurred at 50 days of age. One week following MNU administration, the rats were randomized to receive the diet containing 0 ( n = 33), 2 ( n = 30), or 8 ( n = 30) mg folic acid/kg diet for 22 weeks until the time of killing.
MNU administration
At 50 days of age, all rats received one intraperitoneal injection of MNU (50 mg/kg body weight; Sigma Chemical, St Louis, MO). A single injection of 50 mg MNU/kg has become the standard dosage owing to its rapid induction and high incidence of mammary tumors combined with minimal toxicity and a short latency period of 3–6 months ( 55 ). MNU has a half-life of <1 h under physiological conditions ( 56 ) and its mutagenic effects (e.g. activated Ha- ras oncogene) have been shown to occur within a very short time after its administration (i.e. during the initiation of MNU-induced tumorigenesis) ( 57 ). During initiation, MNU forms adducts with DNA and cause mutations in the Ha- ras oncogenes that become fixed upon mitosis ( 58 ). During promotion, the initiated cells are stimulated to divide by mechanical, hormonal or genetic factors and form preneoplasias ( 58 ). Progression to a malignant phenotype with acquisition of metastatic capabilities is brought on by the increasing genetic instability of the tumor, leading to mutations at key genetic sites ( 58 ). There is unequivocal evidence that MNU-induced mammary tumorigenesis is initiated within 1 week after intraperitoneal injection ( 45 , 58 , 59 ). The experimental protocol of the MNU administration and the timing of dietary intervention used in the present study is the standard protocol that is designed to clarify the effect of a dietary factor on the initiation and promotion phases of MNU-induced mammary tumorigenesis ( 59 ) and has been successfully utilized in prior animal studies by our group ( 60 , 61 ).
Observation parameters
Body weights were recorded weekly. The daily food consumption of each group was measured on a predetermined day of each week. All rats were palpated for mammary tumors once a week beginning 4 weeks after MNU administration. The number, size and location of each tumor were recorded in a manner that, after histological diagnosis, the time of appearance of the cancers could be determined. All the rats were monitored daily for clinical evidence of illness or morbidity and those approaching a moribund state were promptly killed. In addition, rats with tumor burden exceeding 10% of body weight, tumors >15–20 mm in diameter, tumors that impaired normal movement of the animals and ulcerating tumors were immediately killed during the study.
Sample collection and analysis of mammary tumors
Blood was drawn from the lateral tail vein of each rat within a week of MNU injection and from the heart at necropsy and centrifuged at 800× g for 10 min at 4°C. Serum was stored at −70°C in 0.5% ascorbic acid for serum folate assay. Given the latency period of 3–6 months associated with a single MNU injection (i.p.) and the average duration for the systemic and tissue folate levels to stabilize, the rats were killed by carbon dioxide inhalation followed by cervical dislocation at 23 weeks after MNU injection (30 weeks of age). The liver from each rat was harvested, snap-frozen and stored at −70°C for the determination of hepatic folate concentration. All macroscopic mammary tumors were counted, excised and weighed, and the diameter of each tumor was measured using a digital caliper for final tumor volume computation in a blinded fashion. One-half of each macroscopic tumor was snap-frozen in liquid nitrogen and stored at −70°C for DNA extraction. The other half of the tumor was fixed in 10% neutral-buffered formalin, processed in a standard manner for hematoxylin–eosin (H&E) staining and histologically analyzed according to Russo et al . ( 62 ) by two experienced pathologists (A.M. and R.R.) blinded to the study group independently. Normal mammary tissue was also excised at necropsy from each rat, snap-frozen in liquid nitrogen and stored at −70°C for DNA extraction and mammary tissue folate determination.
Determination of folate concentration
Serum folate concentrations were determined by a standard microbiological microtiter plate assay using Lactobacillus casei ( 63 ). Hepatic and normal mammary tissue folate concentrations were measured by the same microbiologic assay ( 63 ), utilizing a previously described method for the determination of tissue folates ( 64 ). Intraassay coefficients of variation for serum, hepatic and mammary tissue folate concentrations were 3, 5 and 6%, respectively. Interassay coefficients of variation for serum, hepatic and mammary tissue folate concentrations were 5, 5 and 7%, respectively.
DNA extraction
DNA from normal mammary tissue and mammary tumors was extracted by a standard technique using proteinase K followed by organic extraction ( 65 ). The size of DNA estimated by agarose-gel electrophoresis was >20 kb in all instances. No RNA contamination was detected on agarose-gel electrophoresis. The final preparations had a ratio of A260 to A280 between 1.8 and 2.0. The concentration of each DNA sample was determined as the mean of three independent spectrophotometric readings.
Genomic DNA methylation determination
The methylation status of cytosine–guanine (CpG) sites in genomic DNA from normal mammary tissue and mammary tumors was determined by the in vitro methyl acceptance capacity of DNA using 3 H-methyl-SAM as a methyl donor and a prokaryotic CpG DNA methyltransferase, Sss1, as previously described ( 42 , 50 , 52 ). All analyses were performed in duplicate. Both intraassay and interassay coefficients of variation of the DNA methylation assay were 5%.
Statistical analysis
Between-group comparisons of continuous variables were assessed using the Kruskal–Wallis and Mann–Whitney non-parameteric tests. For categorical response variables, differences among the groups were assessed by Pearson χ 2 -test. Differences in genomic DNA methylation between normal mammary gland and tumor in each diet group was assessed by the Wilcoxon signed ranks test. The Kaplan–Meier survival analysis and the Log rank test were used to compare the rates of tumor appearance among the three groups. Significance testing was set at P < 0.05 level (two-sided). Statistical analyses were performed using SPSS (version 10).
Results
Body weight and daily food consumption
Growth curves were not significantly different among the three dietary groups in both the initiation and promotion studies; at no time point did the mean body weights differ significantly among the three groups (data not shown). This finding indicates that folate deficiency in the rats fed 0 mg folate/kg diet was moderate; otherwise, growth retardation or premature death would have occurred ( 66 ). The mean daily food consumption, which was determined on a pre-assigned day of each week, was not significantly different among the three groups in both the initiation and promotion studies (data not shown).
Serum, liver and normal mammary gland folate concentrations
Initiation study
At the time of MNU injection (4 weeks after the start of dietary intervention), the mean serum folate concentrations were significantly different among the three groups ( P < 0.001; Table I ). The mean serum folate concentrations of the three dietary groups at this time point were comparable to those observed in rats placed on the corresponding diets for 4–5 weeks in previous studies ( 42 , 49 , 50 ). This observation indicates that a sufficient degree of systemic folate deficiency and supplementation was achieved in the folate-deficient and -supplemented rats, respectively, at the time of MNU injection for the determination of the effect of folate status on the initiation phase of MNU-induced mammary tumorigenesis. At necropsy (22 weeks after beginning the control diet), the mean serum, hepatic and mammary gland folate concentrations of the three groups were not significantly different ( Table I ), and these levels were comparable with those observed in rats placed on the same diet for 24–27 weeks in previous studies ( 42 , 49 , 67 ).
Serum, hepatic and mammary gland folate concentrations *
| Initiation study † | Promotion study ‡ | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| At the time of MNU injection (4 weeks of dietary intervention) | At necropsy (22 weeks of the 2 mg folic acid/kg diet) | At the time of MNU injection (4 weeks of the 2 mg folic acid/kg diet) | At necropsy (22 weeks of dietary intervention) | |||||||||||||||||||
| Diet group ( n ) | I (21) | II (20) | III (20) | I (21) | II (20) | III (20) | I (33) | II (30) | III (30) | I (33) | II (30) | III (30) | ||||||||||
| Serum folate (ng/ml) | 21.7 ± 1.4 a | 64.7 ± 2.5 b | 107.8 ± 3.2 c | 50.6 ± 2.2 | 50.7 ± 1.9 | 53.6 ± 2.1 | 71.1 ± 2.4 | 72.9 ± 3.1 | 76.8 ± 3.0 | 9.4 ± 1.0 a | 48.9 ± 2.0 b | 77.6 ± 2.3 c | ||||||||||
| Hepatic folate (µg/g tissue) | 6.3 ± 0.3 | 6.2 ± 0.4 | 6.8 ± 0.6 | 2.7 ± 0.4 a | 7.3 ± 0.3 b | 9.3 ± 0.3 c | ||||||||||||||||
| Mammary folate (ng/g tissue) | 125.2 ± 10.6 | 141.0 ± 18.5 | 129.9 ± 10.2 | 69.2 ± 4.2 a | 110.3 ± 12.7 b | 146.4 ± 14.3 b | ||||||||||||||||
| Initiation study † | Promotion study ‡ | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| At the time of MNU injection (4 weeks of dietary intervention) | At necropsy (22 weeks of the 2 mg folic acid/kg diet) | At the time of MNU injection (4 weeks of the 2 mg folic acid/kg diet) | At necropsy (22 weeks of dietary intervention) | |||||||||||||||||||
| Diet group ( n ) | I (21) | II (20) | III (20) | I (21) | II (20) | III (20) | I (33) | II (30) | III (30) | I (33) | II (30) | III (30) | ||||||||||
| Serum folate (ng/ml) | 21.7 ± 1.4 a | 64.7 ± 2.5 b | 107.8 ± 3.2 c | 50.6 ± 2.2 | 50.7 ± 1.9 | 53.6 ± 2.1 | 71.1 ± 2.4 | 72.9 ± 3.1 | 76.8 ± 3.0 | 9.4 ± 1.0 a | 48.9 ± 2.0 b | 77.6 ± 2.3 c | ||||||||||
| Hepatic folate (µg/g tissue) | 6.3 ± 0.3 | 6.2 ± 0.4 | 6.8 ± 0.6 | 2.7 ± 0.4 a | 7.3 ± 0.3 b | 9.3 ± 0.3 c | ||||||||||||||||
| Mammary folate (ng/g tissue) | 125.2 ± 10.6 | 141.0 ± 18.5 | 129.9 ± 10.2 | 69.2 ± 4.2 a | 110.3 ± 12.7 b | 146.4 ± 14.3 b | ||||||||||||||||
Results are expressed as mean ± SEM. Means in a row with different letters at each time point significantly differ at P < 0.005 by between-group comparisons.
In the initiation study, rats were randomized to receive the diet containing either 0 (I, folate deficiency), 2 (II, control, basal dietary requirement), or 8 (III, supplemented) mg folic acid/kg diet from weaning at 3 weeks of age for 5 weeks until 1 week following MNU injection. At 50 days of age, all the rats received an intraperitoneal injection of MNU. The initial diets were terminated 1 week after the MNU injection, and all the rats were placed on the control diet (2 mg folic acid/kg diet) for 22 weeks until the time of killing.
In the promotion study, rats were placed on the control diet (2 mg folic acid/kg diet) from weaning at 3 weeks of age for 5 weeks until 1 week following MNU injection. At 50 days of age, all the rats received an intraperitoneal injection of MNU. One week following MNU administration, the rats were randomized to receive the diet containing either 0 (I, deficient), 2 (II, control), or 8 (III, supplemented) mg folic acid/kg diet for 22 weeks until the time of killing.
Serum, hepatic and mammary gland folate concentrations *
| Initiation study † | Promotion study ‡ | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| At the time of MNU injection (4 weeks of dietary intervention) | At necropsy (22 weeks of the 2 mg folic acid/kg diet) | At the time of MNU injection (4 weeks of the 2 mg folic acid/kg diet) | At necropsy (22 weeks of dietary intervention) | |||||||||||||||||||
| Diet group ( n ) | I (21) | II (20) | III (20) | I (21) | II (20) | III (20) | I (33) | II (30) | III (30) | I (33) | II (30) | III (30) | ||||||||||
| Serum folate (ng/ml) | 21.7 ± 1.4 a | 64.7 ± 2.5 b | 107.8 ± 3.2 c | 50.6 ± 2.2 | 50.7 ± 1.9 | 53.6 ± 2.1 | 71.1 ± 2.4 | 72.9 ± 3.1 | 76.8 ± 3.0 | 9.4 ± 1.0 a | 48.9 ± 2.0 b | 77.6 ± 2.3 c | ||||||||||
| Hepatic folate (µg/g tissue) | 6.3 ± 0.3 | 6.2 ± 0.4 | 6.8 ± 0.6 | 2.7 ± 0.4 a | 7.3 ± 0.3 b | 9.3 ± 0.3 c | ||||||||||||||||
| Mammary folate (ng/g tissue) | 125.2 ± 10.6 | 141.0 ± 18.5 | 129.9 ± 10.2 | 69.2 ± 4.2 a | 110.3 ± 12.7 b | 146.4 ± 14.3 b | ||||||||||||||||
| Initiation study † | Promotion study ‡ | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| At the time of MNU injection (4 weeks of dietary intervention) | At necropsy (22 weeks of the 2 mg folic acid/kg diet) | At the time of MNU injection (4 weeks of the 2 mg folic acid/kg diet) | At necropsy (22 weeks of dietary intervention) | |||||||||||||||||||
| Diet group ( n ) | I (21) | II (20) | III (20) | I (21) | II (20) | III (20) | I (33) | II (30) | III (30) | I (33) | II (30) | III (30) | ||||||||||
| Serum folate (ng/ml) | 21.7 ± 1.4 a | 64.7 ± 2.5 b | 107.8 ± 3.2 c | 50.6 ± 2.2 | 50.7 ± 1.9 | 53.6 ± 2.1 | 71.1 ± 2.4 | 72.9 ± 3.1 | 76.8 ± 3.0 | 9.4 ± 1.0 a | 48.9 ± 2.0 b | 77.6 ± 2.3 c | ||||||||||
| Hepatic folate (µg/g tissue) | 6.3 ± 0.3 | 6.2 ± 0.4 | 6.8 ± 0.6 | 2.7 ± 0.4 a | 7.3 ± 0.3 b | 9.3 ± 0.3 c | ||||||||||||||||
| Mammary folate (ng/g tissue) | 125.2 ± 10.6 | 141.0 ± 18.5 | 129.9 ± 10.2 | 69.2 ± 4.2 a | 110.3 ± 12.7 b | 146.4 ± 14.3 b | ||||||||||||||||
Results are expressed as mean ± SEM. Means in a row with different letters at each time point significantly differ at P < 0.005 by between-group comparisons.
In the initiation study, rats were randomized to receive the diet containing either 0 (I, folate deficiency), 2 (II, control, basal dietary requirement), or 8 (III, supplemented) mg folic acid/kg diet from weaning at 3 weeks of age for 5 weeks until 1 week following MNU injection. At 50 days of age, all the rats received an intraperitoneal injection of MNU. The initial diets were terminated 1 week after the MNU injection, and all the rats were placed on the control diet (2 mg folic acid/kg diet) for 22 weeks until the time of killing.
In the promotion study, rats were placed on the control diet (2 mg folic acid/kg diet) from weaning at 3 weeks of age for 5 weeks until 1 week following MNU injection. At 50 days of age, all the rats received an intraperitoneal injection of MNU. One week following MNU administration, the rats were randomized to receive the diet containing either 0 (I, deficient), 2 (II, control), or 8 (III, supplemented) mg folic acid/kg diet for 22 weeks until the time of killing.
Promotion study
At the time of MNU injection (4 weeks after beginning the control diet) the mean serum folate concentrations were not significantly different among the three groups ( Table I ). The mean serum folate concentrations of the three dietary groups at this time point were comparable to those observed in rats placed on the same diet for 4–5 weeks in previous studies ( 42 , 49 , 50 ). At necropsy (22 weeks after the start of dietary intervention), the mean serum and hepatic folate concentrations were significantly different among the three dietary groups ( P < 0.001; Table I ) and were comparable to those observed in rats placed on corresponding diets for 24–27 weeks in previous studies ( 42 , 49 , 50 ). This observation indicates that a sufficient degree of systemic folate deficiency and supplementation was induced in the folate-deficient and -supplemented rats, respectively, after MNU injection for the determination of the effect of folate status on the promotion phase of MNU-induced mammary tumorigenesis. The mean mammary gland folate concentration of the folate-deficient group was significantly lower than the control and folate-supplemented groups ( P ≤ 0.004; Table I ). Although the mean mammary gland folate concentration of the folate-supplemented group was higher than that of the control group, this difference did not reach statistical significance ( P = 0.063; Table I ).
Effects of dietary folate on MNU-induced mammary tumorigenesis
No rat died prematurely nor was killed before necropsy in the three dietary groups for reasons other than the presence of large and/or ulcerating tumors as defined in the Materials and methods section. The prevalence of euthanized rats was similar among the three groups (data not shown). Consistent with previous observations made in the MNU-Sprague–Dawley rat model of mammary tumorigenesis ( 42 , 43 , 58 , 59 , 62 ), >90% of macroscopic mammary tumors in the present study were identified histologically as either adenomas (15%) or adenocarcinomas (85%). There was an excellent agreement in histological diagnosis of either adenoma or adenocarcimona between the two study pathologists (kappa statistic = 0.95). The analyses pertaining to mammary tumors were performed for the combination of adenocarcinomas and adenomas and for adenocarcinomas alone. The number of adenomas for independent analysis was not sufficient.
Initiation study
The rates of appearance of either adenocarcinomas or adenomas among the three dietary groups were not significantly different ( P = 0.83; Figure 1A ). When the analysis was confined to adenocarcinomas alone, no significant difference was observed among the three groups ( P = 0.81; Figure 1B ). As shown in Table II , there was no significant difference in the final incidence, mean tumor latency (mean time for the appearance of the first palpable tumor), multiplicity (mean number of tumors per tumor-bearing rat), volume or weight of adenocarinomas and adenomas at necropsy among the three groups. A similar observation was made when the analyses were confined to adenocarcinomas alone ( Table II ).
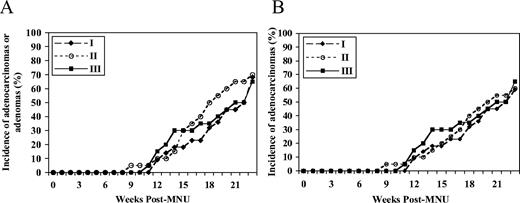
The rate of appearance of either mammary adenomas or adenocarcinomas ( A ) or adenocarcinomas alone ( B ) among the three dietary groups ( P -overall = 0.83 and P -overall = 0.81, respectively, by the Kaplan–Meier survival analysis) in the initiation study. In the initiation study, groups I, II and III received the 0 (deficient), 2 (control) and 8 (supplemented) mg folic acid/kg diet, respectively, from weaning at 3 weeks of age for 5 weeks until 1 week following MNU injection (at 50 days of age), followed by the 2 mg folic acid/kg diet for 22 weeks until the time of killing (30 weeks of age).
Effects of dietary folate on the incidence, latency, multiplicity, volume and weight of mammary tumors in the initiation study *
| Adenocarcinomas + adenomas | Adenocarcinomas | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diet group † | P -value ANOVA | Diet group † | P -value ANOVA | |||||||||||
| I | II | III | I | II | III | |||||||||
| Incidence (%) | 68 | 70 | 70 | 0.99 | 59 | 60 | 70 | 0.73 | ||||||
| Mean latency (weeks post-MNU injection) | 18.4 ± 1.1 | 16.6 ± 1.0 | 17.5 ± 1.3 | 0.54 | 17.7 ± 1.2 | 16.8 ± 1.2 | 17.5 ± 1.3 | 0.88 | ||||||
| Mean multiplicity | 1.6 ± 0.3 | 3.2 ± 1.8 | 1.1 ± 0.1 | 0.52 | 1.3 ± 0.2 | 3.6 ± 2.2 | 1.1 ± 0.1 | 0.39 | ||||||
| Mean volume (cm 3 ) | 4.0 ± 1.6 | 4.2 ± 1.8 | 2.0 ± 0.9 | 0.50 | 5.5 ± 2.1 | 3.8 ± 1.8 | 2.0 ± 0.9 | 0.23 | ||||||
| Mean weight (g) | 1.5 ± 0.6 | 1.4 ± 0.5 | 1.0 ± 0.3 | 0.70 | 2.0 ± 0.8 | 1.2 ± 0.4 | 1.0 ± 0.3 | 0.58 | ||||||
| Adenocarcinomas + adenomas | Adenocarcinomas | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diet group † | P -value ANOVA | Diet group † | P -value ANOVA | |||||||||||
| I | II | III | I | II | III | |||||||||
| Incidence (%) | 68 | 70 | 70 | 0.99 | 59 | 60 | 70 | 0.73 | ||||||
| Mean latency (weeks post-MNU injection) | 18.4 ± 1.1 | 16.6 ± 1.0 | 17.5 ± 1.3 | 0.54 | 17.7 ± 1.2 | 16.8 ± 1.2 | 17.5 ± 1.3 | 0.88 | ||||||
| Mean multiplicity | 1.6 ± 0.3 | 3.2 ± 1.8 | 1.1 ± 0.1 | 0.52 | 1.3 ± 0.2 | 3.6 ± 2.2 | 1.1 ± 0.1 | 0.39 | ||||||
| Mean volume (cm 3 ) | 4.0 ± 1.6 | 4.2 ± 1.8 | 2.0 ± 0.9 | 0.50 | 5.5 ± 2.1 | 3.8 ± 1.8 | 2.0 ± 0.9 | 0.23 | ||||||
| Mean weight (g) | 1.5 ± 0.6 | 1.4 ± 0.5 | 1.0 ± 0.3 | 0.70 | 2.0 ± 0.8 | 1.2 ± 0.4 | 1.0 ± 0.3 | 0.58 | ||||||
Results are expressed as mean ± SEM.
Groups I, II and III received the 0 (deficient), 2 (control) and 8 (supplemented) mg folic acid/kg diet, respectively, from weaning at 3 weeks of age for 5 weeks until 1 week following MNU injection (at 50 days of age), followed by the 2 mg folic acid/kg diet for 22 weeks until the time of killing (30 weeks of age).
Effects of dietary folate on the incidence, latency, multiplicity, volume and weight of mammary tumors in the initiation study *
| Adenocarcinomas + adenomas | Adenocarcinomas | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diet group † | P -value ANOVA | Diet group † | P -value ANOVA | |||||||||||
| I | II | III | I | II | III | |||||||||
| Incidence (%) | 68 | 70 | 70 | 0.99 | 59 | 60 | 70 | 0.73 | ||||||
| Mean latency (weeks post-MNU injection) | 18.4 ± 1.1 | 16.6 ± 1.0 | 17.5 ± 1.3 | 0.54 | 17.7 ± 1.2 | 16.8 ± 1.2 | 17.5 ± 1.3 | 0.88 | ||||||
| Mean multiplicity | 1.6 ± 0.3 | 3.2 ± 1.8 | 1.1 ± 0.1 | 0.52 | 1.3 ± 0.2 | 3.6 ± 2.2 | 1.1 ± 0.1 | 0.39 | ||||||
| Mean volume (cm 3 ) | 4.0 ± 1.6 | 4.2 ± 1.8 | 2.0 ± 0.9 | 0.50 | 5.5 ± 2.1 | 3.8 ± 1.8 | 2.0 ± 0.9 | 0.23 | ||||||
| Mean weight (g) | 1.5 ± 0.6 | 1.4 ± 0.5 | 1.0 ± 0.3 | 0.70 | 2.0 ± 0.8 | 1.2 ± 0.4 | 1.0 ± 0.3 | 0.58 | ||||||
| Adenocarcinomas + adenomas | Adenocarcinomas | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diet group † | P -value ANOVA | Diet group † | P -value ANOVA | |||||||||||
| I | II | III | I | II | III | |||||||||
| Incidence (%) | 68 | 70 | 70 | 0.99 | 59 | 60 | 70 | 0.73 | ||||||
| Mean latency (weeks post-MNU injection) | 18.4 ± 1.1 | 16.6 ± 1.0 | 17.5 ± 1.3 | 0.54 | 17.7 ± 1.2 | 16.8 ± 1.2 | 17.5 ± 1.3 | 0.88 | ||||||
| Mean multiplicity | 1.6 ± 0.3 | 3.2 ± 1.8 | 1.1 ± 0.1 | 0.52 | 1.3 ± 0.2 | 3.6 ± 2.2 | 1.1 ± 0.1 | 0.39 | ||||||
| Mean volume (cm 3 ) | 4.0 ± 1.6 | 4.2 ± 1.8 | 2.0 ± 0.9 | 0.50 | 5.5 ± 2.1 | 3.8 ± 1.8 | 2.0 ± 0.9 | 0.23 | ||||||
| Mean weight (g) | 1.5 ± 0.6 | 1.4 ± 0.5 | 1.0 ± 0.3 | 0.70 | 2.0 ± 0.8 | 1.2 ± 0.4 | 1.0 ± 0.3 | 0.58 | ||||||
Results are expressed as mean ± SEM.
Groups I, II and III received the 0 (deficient), 2 (control) and 8 (supplemented) mg folic acid/kg diet, respectively, from weaning at 3 weeks of age for 5 weeks until 1 week following MNU injection (at 50 days of age), followed by the 2 mg folic acid/kg diet for 22 weeks until the time of killing (30 weeks of age).
Promotion study
There was a non-significant trend toward a difference in the rates of appearance of either adenocarcinomas or adenomas among the three dietary groups ( P = 0.10) with the folate-deficient group demonstrating a trend toward a decrease in the rates compared with the control and folate-supplemented groups ( Figure 2A ). When the analysis was confined to adenocarcinomas alone, a significant difference in the rates of appearance of adenocarcinomas among the three groups was observed ( P = 0.02; Figure 2B ). The rate of appearance of adenocarcinomas in the folate-deficient group was significantly lower than those in the control ( P = 0.01) and folate-supplemented ( P = 0.02) groups ( Figure 2B ). There was no significant difference between the control and folate-supplemented groups ( P = 0.87) ( Figure 2B ).
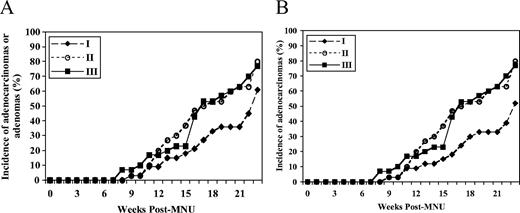
( A ) The rate of appearance of either mammary adenomas or adenocarcinomas among the three dietary groups ( P -overall = 0.10 by the Kaplan–Meier survival analysis) in the promotion study. ( B ) The rate of appearance of mammary adenocarcinomas among the three dietary groups ( P -overall = 0.02 by the Kaplan–Meier survival analysis; P = 0.01 between groups I and II, P = 0.02 between groups I and III and P = 0.87 between groups II and III by the Log rank test) in the promotion study. In the promotion study, groups I, II and III received the 2 mg folic acid/kg diet (control) from weaning at 3 weeks of age for 5 weeks until 1 week following MNU injection (at 50 days of age), followed by the 0 (deficient), 2 (control) and 8 (supplemented) mg folic acid/kg diet, respectively, for 22 weeks until the time of killing (30 weeks of age).
There was a non-significant trend toward lower final incidence and multiplicity of adenocarcinomas and adenomas at necropsy in the folate-deficient group compared with the control and folate-supplemented groups ( P = 0.18 and P = 0.13, respectively; Table III ). The mean tumor volume of adenocarcinomas and adenomas in the folate-deficient group was significantly lower than that of the control and folate-supplemented groups ( P < 0.02), whereas no difference was observed between the control and folate-supplemented groups ( Table III ). The mean tumor weight of the adenocarcinomas and adenomas in the folate-deficient group was significantly lower than that of the folate-supplemented ( P = 0.005) group. There was a non-significant trend toward lower tumor weight in the folate-deficient group compared with the control group ( P = 0.18). No significant difference was observed between the control and folate-supplemented groups ( Table III ). The mean tumor latency was not significantly different among the three groups ( Table III ). When the analyses were confined to adenocarcinomas alone, a similar pattern was observed ( Table III ) and a clearer picture emerged with respect to the final incidence of adenocarcinomas; the final incidence of adenocarcinomas was significantly lower in the folate-deficient group than in the control and folate-supplemented groups ( P < 0.04), whereas no significant difference was observed between the control and folate-supplemented groups ( Table III ).
Effects of dietary folate on the incidence, latency, multiplicity, volume and weight of mammary tumors in the promotion study *
| Adenocarcinomas + adenomas | Adenocarcinomas | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diet group † | P -value ANOVA | Diet group † | P -value ANOVA | |||||||||||
| I | II | III | I | II | III | |||||||||
| Incidence (%) | 61 | 80 | 77 | 0.18 | 52 a | 80 b | 77 b | 0.027 | ||||||
| Mean latency (weeks post-MNU injection) | 17.9 ± 1.1 | 16.5 ± 0.9 | 16.2 ± 0.9 | 0.40 | 17.6 ± 1.2 | 16.5 ± 0.9 | 16.2 ± 0.9 | 0.53 | ||||||
| Mean multiplicity | 1.9 ± 0.3 | 2.9 ± 0.4 | 2.7 ± 0.5 | 0.13 | 1.8 ± 0.3 | 2.8 ± 0.4 | 3.2 ± 0.7 | 0.18 | ||||||
| Mean volume (cm 3 ) | 0.9 ± 0.3 a | 3.0 ± 0.6 b | 2.9 ± 0.6 b | 0.002 | 1.0 ± 0.4 a | 3.0 ± 0.7 b | 2.9 ± 0.6 b | 0.01 | ||||||
| Mean weight (g) | 0.5 ± 0.1 a | 1.1 ± 0.2 a,b | 1.3 ± 0.2 b | 0.019 | 0.6 ± 0.2 a | 1.1 ± 0.3 a,b | 1.3 ± 0.2 b | 0.069 | ||||||
| Adenocarcinomas + adenomas | Adenocarcinomas | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diet group † | P -value ANOVA | Diet group † | P -value ANOVA | |||||||||||
| I | II | III | I | II | III | |||||||||
| Incidence (%) | 61 | 80 | 77 | 0.18 | 52 a | 80 b | 77 b | 0.027 | ||||||
| Mean latency (weeks post-MNU injection) | 17.9 ± 1.1 | 16.5 ± 0.9 | 16.2 ± 0.9 | 0.40 | 17.6 ± 1.2 | 16.5 ± 0.9 | 16.2 ± 0.9 | 0.53 | ||||||
| Mean multiplicity | 1.9 ± 0.3 | 2.9 ± 0.4 | 2.7 ± 0.5 | 0.13 | 1.8 ± 0.3 | 2.8 ± 0.4 | 3.2 ± 0.7 | 0.18 | ||||||
| Mean volume (cm 3 ) | 0.9 ± 0.3 a | 3.0 ± 0.6 b | 2.9 ± 0.6 b | 0.002 | 1.0 ± 0.4 a | 3.0 ± 0.7 b | 2.9 ± 0.6 b | 0.01 | ||||||
| Mean weight (g) | 0.5 ± 0.1 a | 1.1 ± 0.2 a,b | 1.3 ± 0.2 b | 0.019 | 0.6 ± 0.2 a | 1.1 ± 0.3 a,b | 1.3 ± 0.2 b | 0.069 | ||||||
Results are expressed as mean ± SEM. Means in a row with different letters at each time point significantly differ at P < 0.04 by between-group comparisons.
Groups I, II and III received the 2 mg folic acid/kg diet (control) from weaning at 3 weeks of age for 5 weeks until 1 week following MNU injection (at 50 days of age), followed by the 0 (deficient), 2 (control) and 8 (supplemented) mg folic acid/kg diet, respectively, for 22 weeks until the time of killing (30 weeks of age).
Effects of dietary folate on the incidence, latency, multiplicity, volume and weight of mammary tumors in the promotion study *
| Adenocarcinomas + adenomas | Adenocarcinomas | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diet group † | P -value ANOVA | Diet group † | P -value ANOVA | |||||||||||
| I | II | III | I | II | III | |||||||||
| Incidence (%) | 61 | 80 | 77 | 0.18 | 52 a | 80 b | 77 b | 0.027 | ||||||
| Mean latency (weeks post-MNU injection) | 17.9 ± 1.1 | 16.5 ± 0.9 | 16.2 ± 0.9 | 0.40 | 17.6 ± 1.2 | 16.5 ± 0.9 | 16.2 ± 0.9 | 0.53 | ||||||
| Mean multiplicity | 1.9 ± 0.3 | 2.9 ± 0.4 | 2.7 ± 0.5 | 0.13 | 1.8 ± 0.3 | 2.8 ± 0.4 | 3.2 ± 0.7 | 0.18 | ||||||
| Mean volume (cm 3 ) | 0.9 ± 0.3 a | 3.0 ± 0.6 b | 2.9 ± 0.6 b | 0.002 | 1.0 ± 0.4 a | 3.0 ± 0.7 b | 2.9 ± 0.6 b | 0.01 | ||||||
| Mean weight (g) | 0.5 ± 0.1 a | 1.1 ± 0.2 a,b | 1.3 ± 0.2 b | 0.019 | 0.6 ± 0.2 a | 1.1 ± 0.3 a,b | 1.3 ± 0.2 b | 0.069 | ||||||
| Adenocarcinomas + adenomas | Adenocarcinomas | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diet group † | P -value ANOVA | Diet group † | P -value ANOVA | |||||||||||
| I | II | III | I | II | III | |||||||||
| Incidence (%) | 61 | 80 | 77 | 0.18 | 52 a | 80 b | 77 b | 0.027 | ||||||
| Mean latency (weeks post-MNU injection) | 17.9 ± 1.1 | 16.5 ± 0.9 | 16.2 ± 0.9 | 0.40 | 17.6 ± 1.2 | 16.5 ± 0.9 | 16.2 ± 0.9 | 0.53 | ||||||
| Mean multiplicity | 1.9 ± 0.3 | 2.9 ± 0.4 | 2.7 ± 0.5 | 0.13 | 1.8 ± 0.3 | 2.8 ± 0.4 | 3.2 ± 0.7 | 0.18 | ||||||
| Mean volume (cm 3 ) | 0.9 ± 0.3 a | 3.0 ± 0.6 b | 2.9 ± 0.6 b | 0.002 | 1.0 ± 0.4 a | 3.0 ± 0.7 b | 2.9 ± 0.6 b | 0.01 | ||||||
| Mean weight (g) | 0.5 ± 0.1 a | 1.1 ± 0.2 a,b | 1.3 ± 0.2 b | 0.019 | 0.6 ± 0.2 a | 1.1 ± 0.3 a,b | 1.3 ± 0.2 b | 0.069 | ||||||
Results are expressed as mean ± SEM. Means in a row with different letters at each time point significantly differ at P < 0.04 by between-group comparisons.
Groups I, II and III received the 2 mg folic acid/kg diet (control) from weaning at 3 weeks of age for 5 weeks until 1 week following MNU injection (at 50 days of age), followed by the 0 (deficient), 2 (control) and 8 (supplemented) mg folic acid/kg diet, respectively, for 22 weeks until the time of killing (30 weeks of age).
In the initiation study, 100% of the tumors were adenocarcinomas in the folate-supplemented group, whereas only 87 and 86% of the tumors in the folate-deficient and control groups, respectively, were adenocarcinomas ( Table II ). Similarly, in the promotion study, 100% of the tumors were adenocarcinomas in both the folate-supplemented and control groups, whereas only 85% of the tumors were adenocarcinomas in the folate-deficient group ( Table III ). Although these observations were not statistically significant, these data suggest that folate deficiency might have retarded the progression of adenoma to adenocarcinoma.
Effect of dietary folate on genomic DNA methylation in mammary tumors and non-neoplastic mammary tissues
Given the role of folate in DNA methylation ( 6 ), an important epigenetic determinant in carcinogenesis ( 7 ), we investigated whether dietary folate modulates genomic DNA methylation in MNU-induced mammary tumorigenesis. The manner in which this assay is performed produces an inverse relationship between the endogenous DNA methylation status and the exogenous 3 H-methyl incorporation. The degree of 3 H-methyl incorporation into DNA of the mammary adenocarcinomas was 3-fold–5-fold higher than that of non-neoplastic mammary tissue within each dietary group ( P < 0.04; Figure 3A and B ), indicating a significantly lower degree of genomic DNA methylation in adenocarcinomas compared with normal mammary tissue. However, for both the initiation ( Figure 3A ) and promotion ( Figure 3B ) studies, the degree of 3 H-methyl incorporation into DNA of the mammary adenocarcinoma and into DNA from the pair-matched non-neoplastic mammary tissue was not significantly different among the three dietary groups.
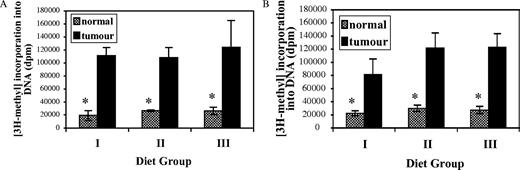
Effects of dietary folate on genomic DNA methylation in mammary adenocarcinomas and non-neoplastic mammary tissues as determined by the in vitro methyl acceptance assay in the initiation ( A ) and promotion ( B ) studies. The manner in which this assay is performed produces a reciprocal relationship between the endogenous DNA methylation status and the exogenous 3 H-methyl incorporation into DNA. Asterisk denotes significant differences by the Wilcoxon signed ranks test at P < 0.04 compared with adenocarcinomas within each dietary group. Values are mean ± SD.
Discussion
Our data from the initiation study indicate that dietary folate deficiency and supplementation do not significantly modulate the development of MNU-induced mammary tumors in this rodent model. The lack of effect of dietary folate deficiency on the initiation of MNU-induced mammary tumorigenesis is not entirely surprising because prior epidemiologic observations have suggested that folate deficiency alone may not be sufficient to modulate the development of breast cancer; folate deficiency increases the risk of breast cancer in those who consume alcohol regularly ( 12 , 14 , 15 , 19 , 20 , 22 ) and in those with genetic predispositions (e.g. MTHFR C677T polymorphism) ( 16 , 23 , 32 – 35 ). The lack of inhibitory effect associated with folate supplementation four times the basal dietary requirement may be related to the observation that this level of folate supplementaton failed to significantly increase mammary gland folate concentrations compared with the control diet. It is, therefore, possible that higher levels of dietary folate supplementation may be necessary to increase mammary gland folate concentrations compared with the control diet in order to exert an inhibitory effect on tumor initiation in this model. Another possible explanation for the lack of effect is that the conventional dose and route of MNU administration employed in the present study may be too overwhelmingly carcinogenic for folate to modulate the initiation of mammary tumorigenesis. Regardless of the levels of dietary folate, MNU probably induced and established neoplastic foci in mammary tissues. Therefore, the effect of dietary folate on initiation of MNU-induced neoplastic foci may not be clearly determined in this model using the current protocol. Nevertheless, our data differ from the promoting and protective effect of folate deficiency and supplementation, respectively, on intestinal tumorigenesis observed in the chemical carcinogen and genetically engineered rodent models utilizing the same diets employed in the present study ( 49 – 52 ). This may be related to differences in folate uptake, retention and metabolism between the intestine and breast, to possible tissue-specific effect of folate on carcinogenesis and to inherent differences in carcinogens used and methods of cancer induction.
Our data from the promotion study clearly indicate that dietary folate deficiency of a moderate degree significantly suppressed the progression of or caused regression of established mammary neoplastic foci in rats. This explanation is supported by the observed significant inhibitory effect of folate deficiency on the rate of appearance, final incidence, mean volume and weight of mammary tumors as well as a trend toward decreased multiplicity of tumors in the promotion study. Several prior observations made in animal models support the inhibitory effect of folate deficiency on established neoplastic foci. In Apc Min and Apc +/− Msh2 −/− murine models of intestinal tumorigenesis, if folate intervention was started before the establishment of neoplastic foci in the intestine, moderate folate deficiency (0 mg folic acid/kg diet) enhanced, whereas modest levels (4–10 times the basal dietary requirement) of folate supplementation suppressed, the development of intestinal tumors ( 51 , 52 ). If, however, folate intervention was started after the establishment of neoplastic foci, dietary folate had opposite effects on the progression of intestinal tumors ( 51 , 52 ). Folate deficiency has also been shown to induce regression and suppress the progression of preexisting neoplasms in experimental models ( 68 – 70 ). The inhibitory effect of folate deficiency on established neoplastic foci is also consistent with the known biochemical function of folate. As an essential cofactor for the de novo biosynthesis of purines and thymidylate, folate plays an important role in DNA synthesis and replication ( 1 ). Folate deficiency in tissues with rapidly replicating cells results in ineffective DNA synthesis. In neoplastic cells where DNA replication and cell division are occurring at an accelerated rate, interruption of folate metabolism causes ineffective DNA synthesis, resulting in the inhibition of tumor growth. This has been the basis for cancer chemotherapy using antifolate agents ( 71 ).
We hypothesized that once neoplastic foci are established with MNU administration, folate supplementation would enhance the promotion and progression of MNU-induced mammary tumorigenesis. This was based on the observed promoting effect of folate supplementation on established intestinal neoplastic foci in Apc Min and Apc +/− Msh −/− mice ( 51 , 52 ) and on the previously reported accelerated progression of leukemia in children with acute leukemia treated with folate supplementation ( 72 ). In the present study, dietary folate supplementation at four times the basal dietary requirement did not significantly promote the progression of MNU-induced mammary neoplastic foci. This lack of promoting effect associated with folate supplementation on established mammary neoplastic foci in the present study may be a tissue-specific observation limited to mammary tumors. Another possible explanation is again related to the failure of folate supplementation to significantly increase mammary gland concentrations. Therefore, it is possible that higher levels of dietary folate supplementation may be necessary to increase mammary gland folate concentrations compared with the control diet in order to exert any effect on tumor progression.
Our data corroborate the findings from our previous work that demonstrated that dietary folate deficiency of moderate degree (0 mg folic acid/kg diet) provided from weanling through MNU injection for 27 weeks signifincantly inhibited, whereas dietary folate supplementation four times the basal requirement did not modulate, mammary tumorigenesis in this rat model ( 42 ). Our data clearly indicate that the inhibitory effect of folate deficiency on MNU-induced mammary tumorigenesis in this rat model is primarily on promotion and progression of established mammary neoplastic foci. The lack of effect of folate status on the initiation of MNU-induced mammary tumorigenesis in our study, however, differ from that of a previous study that has demonstrated an inhibitory effect of folate deficiency on both the initiation and early promotion phases of MNU-induced mammary tumorigenesis in Fischer 344 rats ( 41 ). The latter study is, however, significantly different from our study in several important aspects including the use of non-standard dietary means to modulate folate status, possible growth retardation of animals, the concomitant use of antibiotics that may independently affect folate concentrations and the use of animals that are of intermediate susceptibility to chemically-induced mammary tumorigenesis. Furthermore, because of the dose and route of MNU employed in the latter study, it is possible that the observed inhibitory effect of folate deficiency was actually on promotion/progression and not on initiation.
Three animal studies ( 41 , 42 ), including the present study, all conducted in the standard MNU rodent model of mammary tumorigenesis, collectively suggest that dietary folate deficiency inhibits, whereas folate supplementation (4–20 times above the basal dietary requirement) does not significantly modulate, the development and progression of mammary tumorigenesis. Epidemiologic evidence available thus far has not been consistent nor has it provided unequivocal support for the purported inverse relationship between folate status and the risk of breast cancer. Among 14 published case–control studies that investigated the relationship between dietary folate intake and breast cancer risk, 11 showed either a significant or equivocal inverse relationship that was not statistically significant, became nonsignificant after adjustment or could not be distinguished from other factors in their relation to risk ( 9 – 18 , 23 ), whereas 3 showed an unequivocal null association ( 24 – 26 ). In some studies, the observed inverse association was further modified by the intake of alcohol and other folate cofactors (e.g. methionine, vitamins B 6 and B 12 ) ( 12 , 14 , 15 ). Four large prospective studies showed either a weak inverse association that became significant when alcohol intake was taken into consideration or no relationship between folate intake and breast cancer risk ( 19 , 20 , 27 , 28 ). Similarly, the relationship between blood measurements of folate and breast cancer risk has been equivocal in epidemiologic studies ( 22 , 29 ). Overall, the portfolio of epidemiologic evidence supporting the relationship between folate status and breast cancer risk is tenuous at best, although a clearer picture emerges when studies examining the joint effects of folate and alcohol are considered.
One interesting finding in the present study is that the extent of genomic DNA methylation is significantly lower in mammary adenocarcinomas than in non-neoplastic mammary tissues regardless of folate status. Neoplastic cells simultaneously harbor widespread genomic DNA hypomethylation and more specific regional areas of hypermethylation ( 7 ). Genomic hypomethylation is an early, and consistent, event in carcinogenesis and is associated with genomic instability and increased mutations ( 7 ). Site-specific hypermethylation at the promoter region of tumor suppressor and mismatch repair genes is an important mechanism in gene silencing in carcinogenesis ( 7 ). Although hypermethylation of promoter CpG islands and consequent inactivation of several tumor suppressor genes have been observed in human breast cancer ( 73 ), very few studies have reported genomic hypomethylation in human breast cancer ( 74 , 75 ). Our study demonstrates that genomic DNA hypomethylation is an epigenetic phenomenon associated with MNU-induced mammary tumorigenesis in rats. The extent of genomic DNA methylation in mammary adenocarcinomas and in non-neoplastic mammary tissues was not significantly modulated by folate status. This observation suggests that altered genomic DNA methylation was not a likely mechanism by which folate deficiency suppressed mammary tumorigenesis in our study. Previous animal and in vitro studies have suggested that the effect of folate deficiency on DNA methylation depends on cell type, target organ and stage of transformation ( 6 ). The same degree of dietary folate deficiency and supplementation used in the present study also failed to induce significant changes in genomic DNA methylation in rodent liver and colon in previous studies ( 6 ). Since folate may modulate DNA methylation in a site-specific manner ( 6 ), however, the possibility that folate status may affect site-specific methylation of critical genes implicated in mammary tumorigenesis cannot be ruled out in the present study.
MNU-induced mammary tumorigenesis in rat is different from the human disease in several important aspects: (i) the exposure of the mammary tissue to high dosages of the genotoxic chemical carcinogen as opposed to the natural etiologic cause involved in most cases of human breast cancer; (ii) the lack of p53 mutations, which is a common molecular event in human breast cancer development; and (iii) the primary carcinogenic mechanism via the ras -mediated signal transduction pathway, which is rarely involved in human breast cancer ( 43 , 58 , 59 ). Nonetheless, the MNU rat model is widely used to determine the effects of dietary factors on mammary tumorigenesis for the following reasons: (i) histological similarities of adenocarcinoma to human breast cancer; (ii) local invasiveness and metastatic potential; (iii) a clear operational distinction between initiation and promotion stages; and (iv) hormonally dependent mammary tumorigenesis ( 43 , 58 , 59 ).
In summary, our data suggest that dietary folate deficiency of a moderate degree suppresses MNU-induced mammary tumorigenesis in rats, and this effect appears to be primarily via inhibiton of the progression of established mammary neoplastic foci. In contrast, dietary folate supplementation at four times the basal dietary requirement does not significantly modulate mammary tumorigenesis in this model. However, the limitations associated with the route and dose of MNU administration preclude a definitive conclusion concerning the effect of folate status on the initiation of MNU-induced mammary tumorigenesis. Notwithstanding the limitations associated with this model, our data, in conjunction with the lack of convincing epidemiologic evidence for the protective effect of folate supplementation on breast cancer risk and with emerging evidence for the dual modulatory role of folate in carcinogenesis depending on the timing and dose of folate intervention ( 39 , 40 ), suggest that the role of folate in mammary tumorigenesis needs to be clarified in future studies.
Presented in part at the 2002 American Association for Cancer Research Meeting, April 2002, San Francisco, CA and published in abstract form in Proceedings of the American Association for Cancer Research 2002; 43: 512 (abst #2539).
We thank the veterinary technologists of the Division of Comparative Medicine, Faculty of Medicine, University of Toronto for the care of the rats used in this study. This project has been supported in part by the Department of Defense Breast Cancer Research Program of the US Army Medical Research and Materiel Command's Office (Award Number DAMD17-01-1-0428; to Y.-I.K.) and by the Canadian Institutes of Health Research (to Y.-I.K.). Y.-I.K. is a recipient of a scholarship from the Canadian Institutes of Health Research.
Conflict of Interest Statement : The content of this publication does not necessarily reflect the position or the policy of the Government, and no official endorsement should be inferred.
References
Shane,B. (
Kim,Y.I. (
Mason,J.B. and Levesque,T. (
Duthie,S.J. (
Ames,B.N. (
Kim,Y.I. (
Jones,P.A. and Baylin,S.B. (
Choi,S.W. and Mason,J.B. (
Graham,S., Hellmann,R., Marshall,J., Freudenheim,J., Vena,J., Swanson,M., Zielezny,M., Nemoto,T., Stubbe,N. and Raimondo,T. (
Freudenheim,J.L., Marshall,J.R., Vena,J.E., Laughlin,R., Brasure,J.R., Swanson,M.K., Nemoto,T. and Graham,S. (
Ronco,A., De Stefani,E., Boffetta,P., Deneo-Pellegrini,H., Mendilaharsu,M. and Leborgne,F. (
Rohan,T.E., Jain,M.G., Howe,G.R. and Miller,A.B. (
Levi,F., Pasche,C., Lucchini,F. and La Vecchia,C. (
Negri,E., La Vecchia,C. and Franceschi,S. (
Shrubsole,M.J., Jin,F., Dai,Q., Shu,X.O., Potter,J.D., Hebert,J.R., Gao,Y.T. and Zheng,W. (
Sharp,L., Little,J., Schofield,A.C. et al . (
Adzersen,K.H., Jess,P., Freivogel,K.W., Gerhard,I. and Bastert,G. (
Freudenheim,J.L., Bonner,M., Krishnan,S., Ambrosone,C.B., Graham,S., McCann,S.E., Moysich,K.B., Bowman,E., Nemoto,T. and Shields,P.G. (
Zhang,S., Hunter,D.J., Hankinson,S.E., Giovannucci,E.L., Rosner,B.A., Colditz,G.A., Speizer,F.E. and Willett,W.C. (
Sellers,T.A., Kushi,L.H., Cerhan,J.R., Vierkant,R.A., Gapstur,S.M., Vachon,C.M., Olson,J.E., Therneau,T.M. and Folsom,A.R. (
Sellers,T.A., Vierkant,R.A., Cerhan,J.R., Gapstur,S.M., Vachon,C.M., Olson,J.E., Pankratz,V.S., Kushi,L.H. and Folsom,A.R. (
Zhang,S.M., Willett,W.C., Selhub,J., Hunter,D.J., Giovannucci,E.L., Holmes,M.D., Colditz,G.A. and Hankinson,S.E. (
Chen,J., Gammon,M.D., Chan,W. et al . (
Thorand,B., Kohlmeier,L., Simonsen,N., Croghan,C. and Thamm,M. (
Potischman,N., Swanson,C.A., Coates,R.J., Gammon,M.D., Brogan,D.R., Curtin,J. and Brinton,L.A. (
Zhu,K., Davidson,N.E., Hunter,S., Yang,X., Payne-Wilks,K., Roland,C.L., Phillips,D., Bentley,C., Dai,M. and Williams,S.M. (
Cho,E., Spiegelman,D., Hunter,D.J., Chen,W.Y., Zhang,S.M., Colditz,G.A. and Willett,W.C. (
Feigelson,H.S., Jonas,C.R., Robertson,A.S., McCullough,M.L., Thun,M.J. and Calle,E.E. (
Wu,K., Helzlsouer,K.J., Comstock,G.W., Hoffman,S.C., Nadeau,M.R. and Selhub,J. (
Smith-Warner,S.A., Spiegelman,D., Yaun,S.S. et al . (
Key,T.J., Allen,N.E., Spencer,E.A. and Travis,R.C. (
Campbell,I.G., Baxter,S.W., Eccles,D.M. and Choong,D.Y. (
Beilby,J., Ingram,D., Hahnel,R. and Rossi,E. (
Semenza,J.C., Delfino,R.J., Ziogas,A. and Anton-Culver,H. (
Shrubsole,M.J., Gao,Y.T., Cai,Q., Shu,X.O., Dai,Q., Hebert,J.R., Jin,F. and Zheng,W. (
Coyle,Y.M. (
Campbell,N.R. (
Mason,J.B. and Choi,S.W. (
Kim,Y.I. (
Kim,Y.I. (
Baggott,J.E., Vaughn,W.H., Juliana,M.M., Eto,I., Krumdieck,C.L. and Grubbs,C.J. (
Kotsopoulos,J., Sohn,K.J., Martin,R., Choi,M., Renlund,R., McKerlie,C., Hwang,S.W., Medline,A. and Kim,Y.I. (
Russo,J., Gusterson,B.A., Rogers,A.E., Russo,I.H., Wellings,S.R. and van Zwieten,M.J. (
Richardson,K.K., Richardson,F.C., Crosby,R.M., Swenberg,J.A. and Skopek,T.R. (
Sukumar,S., Notario,V., Martin-Zanca,D. and Barbacid,M. (
Zarbl,H., Sukumar,S., Arthur,A.V., Martin-Zanca,D. and Barbacid,M. (
Boehm,T.L. and Drahovsky,D. (
Walzem,R.L. and Clifford,A.J. (
Cravo,M.L., Mason,J.B., Dayal,Y., Hutchinson,M., Smith,D., Selhub,J. and Rosenberg,I.H. (
Kim,Y.I., Salomon,R.N., Graeme-Cook,F., Choi,S.W., Smith,D.E., Dallal,G.E. and Mason,J.B. (
Song,J., Medline,A., Mason,J.B., Gallinger,S. and Kim,Y.I. (
Song,J., Sohn,K.J., Medline,A., Ash,C., Gallinger,S. and Kim,Y.I. (
Rong,N., Selhub,J., Goldin,B.R. and Rosenberg,I.H. (
Reeves,P.G., Nielsen,F.H. and Fahey,G.C.Jr (
Thompson,H.J. and Adlakha,H. (
Druckrey,H., Preussmann,R., Ivankovic,S. and Schmahl,D. (
Kumar,R., Sukumar,S. and Barbacid,M. (
Sukumar,S., McKenzie,K. and Chen,Y. (
Mehta,R.G. (
Lu,S., Lee,W.M. and Archer,M.C. (
El-Sohemy,A. and Archer,M.C. (
Russo,J., Russo,I.H., Rogers,A.E., Van Zwienten,M.J. and Gusterson,B. (
Tamura,T. (
Varela-Moreiras,G. and Selhub,J. (
Laird,P.W., Zijderveld,A., Linders,K., Rudnicki,M.A., Jaenisch,R. and Berns,A. (
Clifford,A.J., Wilson,D.S. and Bills,N.D. (
Kim,Y.I., Christman,J.K., Fleet,J.C., Cravo,M.L., Salomon,R.N., Smith,D., Ordovas,J., Selhub,J. and Mason,J.B. (
Little,P.A., Sampath,A. and Paganelli,V. (
Rosen,F. and Nichol,C.A. (
Bills,N.D., Hinrichs,S.H., Morgan,R. and Clifford,A.J. (
Farber,S. (
Huang,T.H., Perry,M.R. and Laux,D.E. (
Soares,J., Pinto,A.E., Cunha,C.V., Andre,S., Barao,I., Sousa,J.M. and Cravo,M. (
Author notes
1Department of Nutritional Sciences, 2Department of Medicine, 3Division of Comparative Medicine and 4Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, Ontario, Canada, 5Inner City Health Research Unit and 6Department of Medicine, St Michael's Hospital, Toronto, Ontario, Canada

{kind=link}
{kind=link}
{kind=link}