





Abstract
Porcine reproductive and respiratory syndrome virus (PRRSV) recently emerged in domestic pigs of Western Europe and North America. Although time of emergence was identical on the two continents, genetic composition was markedly different with a clear geographical subtype structure, indicating that subtypes diverged in separate reservoirs prior to emergence. Genetic analyses have shown that the most recent common ancestor (MRCA) of Western European isolates existed around 1980 and that these originate from Eastern European pigs. These findings are challenged by a study of Hanada et al. who place the MRCA of all PRRSV isolates around 1980 and find that no significant subtype divergence occurred before emergence. Here, I discuss problems of information content, methodology, and biological plausibility associated with this study. Using alternative methodology, I reanalyze the existing data and conclude that the MRCA of all PRRSV isolates existed around 1880, 100 years before the date estimated by Hanada et al.
Porcine reproductive and respiratory syndrome virus (PRRSV) is the causative agent of a newly emerged disease in populations of domestic pigs (Snijder and Meulenberg 1998). There was no prior knowledge in veterinary records or serum samples of this disease, until it emerged almost simultaneously in North America and Western Europe around 1990. Despite the concurrence in emergence and disease symptoms, virus strains from the two continents have proven to comprise two genetically distinct subtypes (PRRSV-EU and PRRSV-NA) with 31%–44% nucleotide differences in the different viral genes (see table 1). There is no coherent hypothesis to explain the synchrony in emergence of the subtypes, but it is commonly assumed that they diversified in separate reservoirs prior to emergence (see e.g., Nelsen, Murtaugh, and Faaberg [1999] and Plagemann [2003]).
Genetic Difference Between PRRSV Subtypes
Measure | ORF1a | ORF1b | ORF2 | ORF3 | ORF4 | ORF5 | ORF6 | ORF7 | Total |
|---|---|---|---|---|---|---|---|---|---|
| All nuc | \(\frac{2999}{(6831)}\) (0.44) | \(\frac{1551}{(4293)}\) (0.36) | \(\frac{260}{(750)}\) (0.35) | \(\frac{262}{(753)}\) (0.35) | \(\frac{174}{(534)}\) (0.33) | \(\frac{207}{(591)}\) (0.35) | \(\frac{164}{(522)}\) (0.31) | \(\frac{118}{(360)}\) (0.33) | \(\frac{5735}{(14634)}\) (0.39) |
| Position 1 | \(\frac{890}{(2277)}\) (0.39) | \(\frac{386}{(1431)}\) (0.27) | \(\frac{65}{(250)}\) (0.26) | \(\frac{81}{(251)}\) (0.32) | \(\frac{48}{(178)}\) (0.27) | \(\frac{58}{(197)}\) (0.29) | \(\frac{38}{(174)}\) (0.22) | \(\frac{34}{(120)}\) (0.28) | \(\frac{1600}{(4878)}\) (0.33) |
| Position 2 | \(\frac{690}{(2277)}\) (0.30) | \(\frac{277}{(1431)}\) (0.19) | \(\frac{66}{(250)}\) (0.26) | \(\frac{63}{(251)}\) (0.25) | \(\frac{28}{(178)}\) (0.16) | \(\frac{49}{(197)}\) (0.25) | \(\frac{27}{(174)}\) (0.16) | \(\frac{25}{(120)}\) (0.21) | \(\frac{1225}{(4878)}\) (0.25) |
| Position 3 | \(\frac{1419}{(2277)}\) (0.62) | \(\frac{888}{(1431)}\) (0.62) | \(\frac{129}{(250)}\) (0.52) | \(\frac{118}{(251)}\) (0.47) | \(\frac{98}{(178)}\) (0.55) | \(\frac{100}{(197)}\) (0.51) | \(\frac{99}{(174)}\) (0.57) | \(\frac{59}{(120)}\) (0.49) | \(\frac{2910}{(4878)}\) (0.60) |
| All aa | \(\frac{1092}{(2261)}\) (0.48) | \(\frac{435}{(1429)}\) (0.30) | \(\frac{92}{(249)}\) (0.37) | \(\frac{100}{(249)}\) (0.40) | \(\frac{53}{(176)}\) (0.30) | \(\frac{81}{(195)}\) (0.42) | \(\frac{36}{(173)}\) (0.21) | \(\frac{42}{(118)}\) (0.36) | \(\frac{1931}{(4850)}\) (0.40) |
Measure | ORF1a | ORF1b | ORF2 | ORF3 | ORF4 | ORF5 | ORF6 | ORF7 | Total |
|---|---|---|---|---|---|---|---|---|---|
| All nuc | \(\frac{2999}{(6831)}\) (0.44) | \(\frac{1551}{(4293)}\) (0.36) | \(\frac{260}{(750)}\) (0.35) | \(\frac{262}{(753)}\) (0.35) | \(\frac{174}{(534)}\) (0.33) | \(\frac{207}{(591)}\) (0.35) | \(\frac{164}{(522)}\) (0.31) | \(\frac{118}{(360)}\) (0.33) | \(\frac{5735}{(14634)}\) (0.39) |
| Position 1 | \(\frac{890}{(2277)}\) (0.39) | \(\frac{386}{(1431)}\) (0.27) | \(\frac{65}{(250)}\) (0.26) | \(\frac{81}{(251)}\) (0.32) | \(\frac{48}{(178)}\) (0.27) | \(\frac{58}{(197)}\) (0.29) | \(\frac{38}{(174)}\) (0.22) | \(\frac{34}{(120)}\) (0.28) | \(\frac{1600}{(4878)}\) (0.33) |
| Position 2 | \(\frac{690}{(2277)}\) (0.30) | \(\frac{277}{(1431)}\) (0.19) | \(\frac{66}{(250)}\) (0.26) | \(\frac{63}{(251)}\) (0.25) | \(\frac{28}{(178)}\) (0.16) | \(\frac{49}{(197)}\) (0.25) | \(\frac{27}{(174)}\) (0.16) | \(\frac{25}{(120)}\) (0.21) | \(\frac{1225}{(4878)}\) (0.25) |
| Position 3 | \(\frac{1419}{(2277)}\) (0.62) | \(\frac{888}{(1431)}\) (0.62) | \(\frac{129}{(250)}\) (0.52) | \(\frac{118}{(251)}\) (0.47) | \(\frac{98}{(178)}\) (0.55) | \(\frac{100}{(197)}\) (0.51) | \(\frac{99}{(174)}\) (0.57) | \(\frac{59}{(120)}\) (0.49) | \(\frac{2910}{(4878)}\) (0.60) |
| All aa | \(\frac{1092}{(2261)}\) (0.48) | \(\frac{435}{(1429)}\) (0.30) | \(\frac{92}{(249)}\) (0.37) | \(\frac{100}{(249)}\) (0.40) | \(\frac{53}{(176)}\) (0.30) | \(\frac{81}{(195)}\) (0.42) | \(\frac{36}{(173)}\) (0.21) | \(\frac{42}{(118)}\) (0.36) | \(\frac{1931}{(4850)}\) (0.40) |
NOTE.—The number of differences recorded in all ORFs and in total when comparing the Lelystad reference strain of PRRSV-EU (GenBank accession number M96262) with the VR2332 reference strain of PRRSV-NA (GenBank accession number AF030244). Measures are given for all nucleotides (nuc), all amino acids (aa), and the three codon positions separately. Results are listed as:
Genetic Difference Between PRRSV Subtypes
Measure | ORF1a | ORF1b | ORF2 | ORF3 | ORF4 | ORF5 | ORF6 | ORF7 | Total |
|---|---|---|---|---|---|---|---|---|---|
| All nuc | \(\frac{2999}{(6831)}\) (0.44) | \(\frac{1551}{(4293)}\) (0.36) | \(\frac{260}{(750)}\) (0.35) | \(\frac{262}{(753)}\) (0.35) | \(\frac{174}{(534)}\) (0.33) | \(\frac{207}{(591)}\) (0.35) | \(\frac{164}{(522)}\) (0.31) | \(\frac{118}{(360)}\) (0.33) | \(\frac{5735}{(14634)}\) (0.39) |
| Position 1 | \(\frac{890}{(2277)}\) (0.39) | \(\frac{386}{(1431)}\) (0.27) | \(\frac{65}{(250)}\) (0.26) | \(\frac{81}{(251)}\) (0.32) | \(\frac{48}{(178)}\) (0.27) | \(\frac{58}{(197)}\) (0.29) | \(\frac{38}{(174)}\) (0.22) | \(\frac{34}{(120)}\) (0.28) | \(\frac{1600}{(4878)}\) (0.33) |
| Position 2 | \(\frac{690}{(2277)}\) (0.30) | \(\frac{277}{(1431)}\) (0.19) | \(\frac{66}{(250)}\) (0.26) | \(\frac{63}{(251)}\) (0.25) | \(\frac{28}{(178)}\) (0.16) | \(\frac{49}{(197)}\) (0.25) | \(\frac{27}{(174)}\) (0.16) | \(\frac{25}{(120)}\) (0.21) | \(\frac{1225}{(4878)}\) (0.25) |
| Position 3 | \(\frac{1419}{(2277)}\) (0.62) | \(\frac{888}{(1431)}\) (0.62) | \(\frac{129}{(250)}\) (0.52) | \(\frac{118}{(251)}\) (0.47) | \(\frac{98}{(178)}\) (0.55) | \(\frac{100}{(197)}\) (0.51) | \(\frac{99}{(174)}\) (0.57) | \(\frac{59}{(120)}\) (0.49) | \(\frac{2910}{(4878)}\) (0.60) |
| All aa | \(\frac{1092}{(2261)}\) (0.48) | \(\frac{435}{(1429)}\) (0.30) | \(\frac{92}{(249)}\) (0.37) | \(\frac{100}{(249)}\) (0.40) | \(\frac{53}{(176)}\) (0.30) | \(\frac{81}{(195)}\) (0.42) | \(\frac{36}{(173)}\) (0.21) | \(\frac{42}{(118)}\) (0.36) | \(\frac{1931}{(4850)}\) (0.40) |
Measure | ORF1a | ORF1b | ORF2 | ORF3 | ORF4 | ORF5 | ORF6 | ORF7 | Total |
|---|---|---|---|---|---|---|---|---|---|
| All nuc | \(\frac{2999}{(6831)}\) (0.44) | \(\frac{1551}{(4293)}\) (0.36) | \(\frac{260}{(750)}\) (0.35) | \(\frac{262}{(753)}\) (0.35) | \(\frac{174}{(534)}\) (0.33) | \(\frac{207}{(591)}\) (0.35) | \(\frac{164}{(522)}\) (0.31) | \(\frac{118}{(360)}\) (0.33) | \(\frac{5735}{(14634)}\) (0.39) |
| Position 1 | \(\frac{890}{(2277)}\) (0.39) | \(\frac{386}{(1431)}\) (0.27) | \(\frac{65}{(250)}\) (0.26) | \(\frac{81}{(251)}\) (0.32) | \(\frac{48}{(178)}\) (0.27) | \(\frac{58}{(197)}\) (0.29) | \(\frac{38}{(174)}\) (0.22) | \(\frac{34}{(120)}\) (0.28) | \(\frac{1600}{(4878)}\) (0.33) |
| Position 2 | \(\frac{690}{(2277)}\) (0.30) | \(\frac{277}{(1431)}\) (0.19) | \(\frac{66}{(250)}\) (0.26) | \(\frac{63}{(251)}\) (0.25) | \(\frac{28}{(178)}\) (0.16) | \(\frac{49}{(197)}\) (0.25) | \(\frac{27}{(174)}\) (0.16) | \(\frac{25}{(120)}\) (0.21) | \(\frac{1225}{(4878)}\) (0.25) |
| Position 3 | \(\frac{1419}{(2277)}\) (0.62) | \(\frac{888}{(1431)}\) (0.62) | \(\frac{129}{(250)}\) (0.52) | \(\frac{118}{(251)}\) (0.47) | \(\frac{98}{(178)}\) (0.55) | \(\frac{100}{(197)}\) (0.51) | \(\frac{99}{(174)}\) (0.57) | \(\frac{59}{(120)}\) (0.49) | \(\frac{2910}{(4878)}\) (0.60) |
| All aa | \(\frac{1092}{(2261)}\) (0.48) | \(\frac{435}{(1429)}\) (0.30) | \(\frac{92}{(249)}\) (0.37) | \(\frac{100}{(249)}\) (0.40) | \(\frac{53}{(176)}\) (0.30) | \(\frac{81}{(195)}\) (0.42) | \(\frac{36}{(173)}\) (0.21) | \(\frac{42}{(118)}\) (0.36) | \(\frac{1931}{(4850)}\) (0.40) |
NOTE.—The number of differences recorded in all ORFs and in total when comparing the Lelystad reference strain of PRRSV-EU (GenBank accession number M96262) with the VR2332 reference strain of PRRSV-NA (GenBank accession number AF030244). Measures are given for all nucleotides (nuc), all amino acids (aa), and the three codon positions separately. Results are listed as:
Two genetic studies have revealed the nature of this reservoir for PRRSV-EU (Forsberg et al. 2001; Stadejek et al. 2002). In the former, a large sample of sequences from open reading frame (ORF) 3 was used to estimate that the local most recent common ancestor (MRCA) of Western European isolates existed in 1980 ∼10 years before emergence. Furthermore, it was shown that initiation of the Western European epidemic involved the transfer of several lineages from the original reservoir, indicating that these represent intraspecies transmissions. The likely source of transmission was located in the second study, which showed that Eastern European pigs harbor highly diverse PRRSV-EU lineages that diversified well before the lineages found in Western Europe. Thus, a plausible hypothesis is that PRRSV-EU originates in Eastern European pigs from where it was transferred to Western Europe around 1990 through the increased east-west contact, which followed the dramatic political changes in Europe at that time.
These results are challenged by a study of Hanada et al. (2005). Here, data from ORFs 3, 4, and 5 of PRRSV-NA are used to estimate that the global MRCA of both PRRSV-EU and PRRSV-NA existed as recently as 1980 (see fig. 1). Based on this, the authors propose that PRRSV was transferred from another species to pigs around 1980 and subsequently spread to the two continents. This hypothesis is appealing as it circumvents the need to explain how the virus could diversify undetected in isolated reservoirs before its simultaneous emergence on the two continents. However, it is in obvious conflict with the time of the local MRCA of the PRRSV-EU subtype estimated by Forsberg et al. (2001), and it is important to resolve this issue in order to focus future studies which aim at elucidating the causal factors behind the emergence of PRRSV.
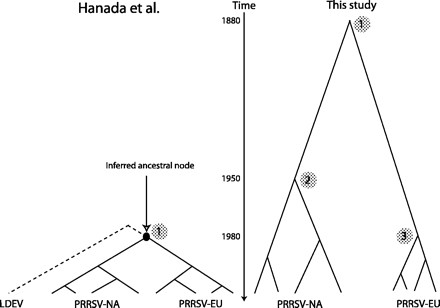
Schematic phylogeny of the North American (NA) and European (EU) subtypes of PRRSV. A time line is shown in the middle of the figure. The left side of the figure shows the subtype divergence time (global MRCA) (1) estimated by Hanada et al. (2005) by using the closest relative, the LDEV, to infer the nucleotide sequence of the subtype ancestor. On the right-hand side are shown estimates of the divergence time of subtypes (global MRCA) (1) and of lineages within subtypes (local MRCAs) (2 and 3), as estimated in this study and in Forsberg et al. (2001). See text for CIs on estimates.
I find three major reasons to question the estimates of Hanada et al. The first is that the estimated time of the global MRCA implies an evolutionary process in PRRSV, radically different from any previously observed. Besides a large number of insertions and deletions, 1,931 amino acid differences exist between the reference strains for PRRSV-EU and PRRSV-NA, both isolated around 1991 (table 1). Not correcting for multiple substitutions, this number provides the most conservative estimate of the number of amino acid changes that have undergone fixation since PRRSV-EU and PRRSV-NA diverged. Assuming that subtypes diverged in 1980, this would imply that amino acid changes have accumulated at an average rate of one fixation event per ∼4.2 days. At this rate, the average time for a new beneficial amino acid change to arise and fix in the ancestral PRRSV populations would have been on the order of only a few generations, and furthermore, PRRSV genes, including nonstructural and usually conserved genes encoding the RNA polymerase etc., would have evolved more than twice as fast as the most rapidly evolving viral genes known, such as the Influenza A hemagglutinin gene (Fitch et al. 1997).
Consider two temporally spaced sequences. When estimating the substitution rate from such a pair, the number of substitutions (length of branches) occurring in the sampling interval (time between samples) constitutes signal and the number of substitutions occurring in the interval from the root to the start of the sampling interval constitutes noise (see also example in Drummond et al. [2003]). This argument cannot be immediately generalized to the case of more than two taxons because the ability to resolve the tree topology introduces some additional complexity. However, when the rate of substitution is estimated on a fixed topology, as is the case in the paper by Hanada et al., we can conjecture that the optimal ratio of signal to noise is obtained by maximizing the total branch length of the tree spanning the sampling interval and by minimizing the length of the branches found in the time interval from the root to the start of the sampling interval. Hanada et al. used 10 isolates, 3 from 1991, 6 from 1992, and 1 from 1994. The best case scenario would thus be that the isolates were related by a star tree with an MRCA in 1991, yielding a total of 6 × 1 + 1 × 3 = 9 years of independent evolutionary information. However, in the PRRSV-NA phylogeny, the earliest isolates are placed far from the root. The amount of independent evolutionary information available for rate estimation is therefore substantially less than ideal, and the signal to noise ratio is expected to be low. Hence, my second reservation is that the information content of the data may be insufficient for the purpose.
Thirdly, the method of data analysis employed by Hanada et al. seems problematic. The rate of substitution is estimated by first constructing a phylogenetic tree from an alignment of 10 PRRSV-NA isolates, 1 PRRSV-EU isolate, and 1 isolate of the lactate dehydrogenase–elevating virus (LDEV) and then estimating the ancestral sequence at the node connecting LDEV with the two PRRSV subtypes (fig. 1). The data points used then consist of the isolation time of each PRRSV-NA isolate and its genetic distance to the ancestral sequence. To these points, a linear function is fitted, and the rate of synonymous substitution is estimated as the slope of the line and the time of the global MRCA of PRRSV-NA and PRRSV-EU is estimated as the intercept. An obvious drawback with this approach is that LDEV is a highly divergent out-group (table 2). Only synonymous substitutions are used in the analysis, essentially corresponding to third codon positions, where LDEV, PRRSV-NA, and PRRSV-EU are approximately equidistant at 55% nucleotide differences (table 2). By applying a Jukes-Cantor correction (Li 1997), I estimate that the expected number of substitutions per third codon position between any two viruses is ∼1. A three-leaf tree with equal branch lengths connecting the three viruses then has about 0.5 expected substitutions per site per branch. Accurate reconstruction of the ancestral nucleotide state requires that at most one branch has experienced substitution. The probability of simultaneous substitutions on more than one branch is (1 − P0.5(s = 0))3 + 3P0.5(s = 0)(1 − P0.5(s = 0))2 = 0.34, where P0.5(s) is the Poisson probability of s substitutions on a branch in the tree. The simple calculations above therefore lead me to conclude that about one-third of the ancestral states, which are assumed known in the estimation procedure used by Hanada et al., can in fact not be accurately inferred and that this uncertainty is expected to bias estimates when unaccounted for.
Genetic Difference Between LDEV and PRRSV Subtypes
Measure | LDEV Versus Lely. | LDEV Versus VR2332 | VR2332 Versus Lely. |
|---|---|---|---|
| All nuc | \(\frac{703}{(1533)}\) (0.46) | \(\frac{697}{(1584)}\) (0.44) | \(\frac{643}{(1878)}\) (0.34) |
| Position 1 | \(\frac{217}{(511)}\) (0.42) | \(\frac{208}{(528)}\) (0.39) | \(\frac{187}{(626)}\) (0.30) |
| Position 2 | \(\frac{179}{(511)}\) (0.35) | \(\frac{197}{(528)}\) (0.37) | \(\frac{140}{(626)}\) (0.22) |
| Position 3 | \(\frac{307}{(511)}\) (0.60) | \(\frac{292}{(528)}\) (0.55) | \(\frac{316}{(626)}\) (0.50) |
| All aa | \(\frac{283}{(501)}\) (0.56) | \(\frac{296}{(518)}\) (0.57) | \(\frac{234}{(620)}\) (0.38) |
Measure | LDEV Versus Lely. | LDEV Versus VR2332 | VR2332 Versus Lely. |
|---|---|---|---|
| All nuc | \(\frac{703}{(1533)}\) (0.46) | \(\frac{697}{(1584)}\) (0.44) | \(\frac{643}{(1878)}\) (0.34) |
| Position 1 | \(\frac{217}{(511)}\) (0.42) | \(\frac{208}{(528)}\) (0.39) | \(\frac{187}{(626)}\) (0.30) |
| Position 2 | \(\frac{179}{(511)}\) (0.35) | \(\frac{197}{(528)}\) (0.37) | \(\frac{140}{(626)}\) (0.22) |
| Position 3 | \(\frac{307}{(511)}\) (0.60) | \(\frac{292}{(528)}\) (0.55) | \(\frac{316}{(626)}\) (0.50) |
| All aa | \(\frac{283}{(501)}\) (0.56) | \(\frac{296}{(518)}\) (0.57) | \(\frac{234}{(620)}\) (0.38) |
NOTE.—The number of pairwise differences recorded in ORFs 3, 4, and 5 when comparing an isolate of LDEV (GenBank accession number L13298), the Lelystad (Lely.) reference strain of PRRSV-EU (GenBank accession number M96262), and the VR2332 reference strain of PRRSV-NA (GenBank accession number AF030244). Measures are given for all nucleotides (nuc), all amino acids (aa), and the three codon positions separately. Results are listed as:
Genetic Difference Between LDEV and PRRSV Subtypes
Measure | LDEV Versus Lely. | LDEV Versus VR2332 | VR2332 Versus Lely. |
|---|---|---|---|
| All nuc | \(\frac{703}{(1533)}\) (0.46) | \(\frac{697}{(1584)}\) (0.44) | \(\frac{643}{(1878)}\) (0.34) |
| Position 1 | \(\frac{217}{(511)}\) (0.42) | \(\frac{208}{(528)}\) (0.39) | \(\frac{187}{(626)}\) (0.30) |
| Position 2 | \(\frac{179}{(511)}\) (0.35) | \(\frac{197}{(528)}\) (0.37) | \(\frac{140}{(626)}\) (0.22) |
| Position 3 | \(\frac{307}{(511)}\) (0.60) | \(\frac{292}{(528)}\) (0.55) | \(\frac{316}{(626)}\) (0.50) |
| All aa | \(\frac{283}{(501)}\) (0.56) | \(\frac{296}{(518)}\) (0.57) | \(\frac{234}{(620)}\) (0.38) |
Measure | LDEV Versus Lely. | LDEV Versus VR2332 | VR2332 Versus Lely. |
|---|---|---|---|
| All nuc | \(\frac{703}{(1533)}\) (0.46) | \(\frac{697}{(1584)}\) (0.44) | \(\frac{643}{(1878)}\) (0.34) |
| Position 1 | \(\frac{217}{(511)}\) (0.42) | \(\frac{208}{(528)}\) (0.39) | \(\frac{187}{(626)}\) (0.30) |
| Position 2 | \(\frac{179}{(511)}\) (0.35) | \(\frac{197}{(528)}\) (0.37) | \(\frac{140}{(626)}\) (0.22) |
| Position 3 | \(\frac{307}{(511)}\) (0.60) | \(\frac{292}{(528)}\) (0.55) | \(\frac{316}{(626)}\) (0.50) |
| All aa | \(\frac{283}{(501)}\) (0.56) | \(\frac{296}{(518)}\) (0.57) | \(\frac{234}{(620)}\) (0.38) |
NOTE.—The number of pairwise differences recorded in ORFs 3, 4, and 5 when comparing an isolate of LDEV (GenBank accession number L13298), the Lelystad (Lely.) reference strain of PRRSV-EU (GenBank accession number M96262), and the VR2332 reference strain of PRRSV-NA (GenBank accession number AF030244). Measures are given for all nucleotides (nuc), all amino acids (aa), and the three codon positions separately. Results are listed as:
To clarify these points, I reanalyzed the data used by Hanada et al. Sequences downloaded from GenBank (http://www.ncbi.nlm.nih.gov/) were aligned with ClustalX using default settings (Thompson et al. 1997). Using the isolation dates and the phylogeny provided by Hanada et al., I performed maximum likelihood estimation of the rate of substitution and the branching time of the internal nodes in the PRRSV-NA tree using the BASEML program of the PAML program package (Yang 2000) with the TipDate clock model (Rambaut 2000) and a Hasegawa-Kishino-Yano + Γ model of nucleotide substitution with eight discrete categories of sites (Hasegawa, Kishino, and Yano 1985; Yang 1993). Approximative confidence intervals (CIs) were obtained from a normal approximation to the distribution of maximum likelihood estimates and should mainly be considered indicative.
This methodology, which does not rely on the estimation of a distant ancestral sequence and which uses all available sites in the data, yielded highly different results from those obtained by Hanada et al. The estimated overall rate of nucleotide substitution was 1.8 × 10−3 per site per year, and the local MRCA of lineages within only the PRRSV-NA subtype was estimated to have occurred in 1950, i.e., 30 years before the time of the global MRCA estimated by Hanada et al. (fig. 1). However, the sparse information content of the data was highlighted by wide CIs on these estimates (±1.5 × 10−3 and ±35). To this data set, I added a sequence from the 1991 Lelystad isolate of PRRSV-EU (accession number M96262). Alignment and analysis were performed as above. This analysis placed the global MRCA in 1593, 313 years before the time estimated by Hanada et al., but with a CI that spans the period 1219–1967 and thus severely questions the suitability of the data set of Hanada et al. for estimating the time of subtype divergence.
A more appropriate data set for addressing this question may be that used by Forsberg et al. (2001). This includes 44 ORF3 sequences spanning a sampling interval of 8 years and has previously been shown to display an accurate molecular clock (Forsberg et al. 2001). To this set was added an ORF3 sequence from the PRRSV-NA isolate VR2332 (for alignment see table 1, phylogenetic estimation as in Forsberg et al. [2001]). Analysis was done as above and also with the M2a model of codon substitution implemented in the CODEML program of PAML, yielding essentially identical results (not shown). This analysis placed the MRCA of the two subtypes around 1880 (fig. 1) with a narrow CI (±15 years). Caution should be taken in interpreting this result as the analysis implicitly assumes that the rate can be extrapolated over a period of time where the virus evolved in an unknown host environment and over large genetic distances, where the inadequacies of the substitution models are bound to be inflated. However, because the estimated rate of substitution is among the highest recorded in RNA virus glycoproteins (Forsberg et al. 2001), it seems unlikely that the global MRCA of the two subtypes existed later than year 1900.
In conclusion, I find the genetic evidence to disagree with the proposal of Hanada et al., namely that the two PRRSV subtypes diverged around 1980, following a species-transfer event. A more plausible hypothesis has been proposed by Plagemann (2003) who speculated that PRRSV originates in European wild boars and that it was transferred to North America via an import of wild boars in 1912. This hypothesis can explain the long period of independent evolution on the two continents and implies a divergence time which is in agreement with the genetic analysis performed here. There is, however, still no satisfactory explanation as to how the two subtypes could emerge in near synchrony on the two continents.
This, and other epidemiological problems, may eventually also be solved using genetic data analysis. But as illustrated here, this requires the use of statistical methodologies which incorporate as much of the uncertainty in the data as possible (for a recent review of rate-estimation methods and considerations see Drummond et al. [2003]). Furthermore, it is crucial that the results of such analyses are tested for their sensitivity to the choice of data and methodology and related to the existing knowledge of biological processes, especially when controversial hypotheses are being tested.
Mark Springer, Associate Editor
I thank Freddy Bugge Christiansen and Alexei Drummond for comments and suggestions.
References
Drummond, A. J., O. G. Pybus, A. Rambaut, R. Forsberg, and A. G. Rodrigo.
Fitch, W. M., R. M. Bush, C. A. Bender, and N. J. Cox.
Forsberg, R., M. B. Oleksiewicz, A. M. Petersen, J. Hein, A. Botner, and T. Storgaard.
Hanada, K., Y. Suzuki, T. Nakane, O. Hirose, and T. Gojobori.
Hasegawa, M., H. Kishino, and T. Yano.
Nelsen, C. J., M. P. Murtaugh, and K. S. Faaberg.
Pedersen, C. N. S., R. B. Lyngsø, and J. Hein.
Plagemann, P. G. W.
Rambaut, A.
Snijder, E. J., and J. J. Meulenberg.
Stadejek, T., A. Stankevicius, T. Storgaard, M. B. Oleksiewicz, S. Belak, T. W. Drew, and Z. Pejsak.
Thompson, J., T. Gibson, F. Plewniak, F. Jeanmougin, and D. Higgins.
Yang, Z.

{kind=link}