





Abstract
Objectives: This study was carried out to explore lectin-functionalized poly (lactide-co-glycolide) nanoparticles (PLG-NPs) as bioadhesive drug carriers against tuberculosis (TB), in order to reduce the drug dosage frequency of antitubercular drugs and thus improve patient compliance in TB chemotherapy.
Methods: Wheat germ agglutinin (WGA)-coated PLG-NPs were prepared by a two-step carbodiimide procedure. This formulation was administered to guinea pigs through the oral/aerosol route for a detailed pharmacokinetic and chemotherapeutic evaluation. Immunological or hepatotoxic effects of WGA lectin, if any, were also determined.
Results: WGA-functionalized PLG-NPs were in the size range of 350–400 nm, with binding of 3–3.5 μg of WGA/mg of PLG-NPs and drug encapsulation efficiency of 54%–66%. Upon administration of lectin-coated PLG-NPs through the oral/aerosol route, the presence of drugs in plasma was observed for 6–7 days for rifampicin and 13–14 days for isoniazid and pyrazinamide. However, upon administration of uncoated PLG-NPs (oral/aerosolized) rifampicin was detectable in plasma for 4–6 days, whereas isoniazid and pyrazinamide were detectable for 8–9 days. All three drugs were present in lungs, liver and spleen for 15 days. Administration of WGA-coated PLG-NPs caused a significant (P < 0.001) increase in the relative bioavailability of antitubercular drugs. Chemotherapeutic studies revealed that three doses of oral/nebulized lectin-coated nanoparticles fortnightly could yield undetectable mycobacterial colony forming units (cfu); this was achievable with 45 doses of oral free drugs.
Conclusion: WGA-functionalized PLG-NPs could be potential drug carriers for antitubercular drugs through the oral as well as aerosol route for effective TB control.
Introduction
Bioadhesive drug delivery systems enhance drug bioavailability by prolonged residence at the site of absorption owing to increased epithelial contact.1 Most bioadhesives known to date are synthetic macromolecules, in the form of micro/nanoparticles that interact with the mucosal surface and are hence referred to as mucoadhesives.2 The maximum duration of adhesion of a mucoadhesive polymer to some mucosal tissue will therefore be limited by the turnover time of the mucus gel layer, being only a few hours for most mucosal surfaces. In order to circumvent this problem, polymeric drug carriers can be attached to certain cytoadhesive ligands that bind to epithelial surfaces through specific receptor-mediated interactions. Lectins are appropriate candidates for this approach; they comprise a structurally diverse class of proteins found in organisms ranging from viruses and plants to humans.3 The specificity of lectins4 and their resistance to proteolytic degradation5 have advocated their use in pharmaceutics. Wheat germ agglutinin (WGA) is finding wide application in drug delivery research because it is one of the least immunogenic lectins.6 Also, WGA is known to have its receptors on intestinal7 as well as alveolar epithelium,8 thus potentiating its use for oral as well as aerosol drug delivery.
In the present study, WGA lectin-functionalized poly (lactide-co-glycolide) nanoparticles (PLG-NPs) have been employed with the aim of reducing the drug dosing frequency of three frontline antitubercular drugs, i.e. isoniazid, rifampicin and pyrazinamide. This method may avoid the problems with patient compliance associated with daily dosing of antitubercular drugs in conventional tuberculosis chemotherapy.
Materials and methods
Chemicals
The biodegradable polymer PLG (50:50; resomer RG 502 H; acid number, 10.7 mg KOH/g) was purchased from Boehringer Ingelheim (Germany). WGA, polyvinyl alcohol (PVA; 87–89% hydrolysed; Mr, 13 000–23 000), rifampicin, isoniazid, pyrazinamide, 1-ethyl-3,3-(dimethylaminopropyl) carbodiimide, N-hydroxysuccinimide and HEPES were purchased from Sigma (St Louis, MO, USA). Tritiated thymidine was obtained from Bhabha Atomic Research Centre, Mumbai, India. All other reagents used for the present study were of analytical grade.
Animals
Dunkin Hartley guinea pigs of either sex (300–400 g), obtained from Haryana Agricultural University, Hisar, India, were used in the study. The animals were fed a standard pellet diet and water ad libitum. The study was approved by the Institute's Ethics Committee.
Culture
The culture of Mycobacterium tuberculosis H37Rv, originally obtained from the National Collection of Type Cultures (NCTC), London, UK, was maintained on Youman's modified medium.
Preparation of WGA-coated PLG-NPs
Drug-loaded PLG-NPs were prepared by a multistep emulsion procedure, as reported previously by our laboratory.9 Briefly, isoniazid and pyrazinamide dissolved in 3 mL of distilled water, and rifampicin and PLG dissolved in 30 mL of dichloromethane (DCM) were mixed in the ratio of 1:1 and sonicated for 1 min. Primary emulsion so formed was poured into 24 mL of 1% PVA and sonication was carried out for 3 min. The resulting nanoparticle suspension was kept on a magnetic stirrer overnight for evaporation of DCM, and nanoparticles were collected by centrifugation at 8000 rpm. These were bound to WGA lectin by the two-step carbodiimide method of Ertl et al.10 with few modifications. Briefly, 50 mg of PLG-NPs was suspended in 20 mM HEPES/NaOH buffer. Activation of carboxyl groups of PLG-NPs was carried out by adding 1 mL of 0.1 M 1-ethyl-3,3-(dimethylaminopropyl) carbodiimide and 0.11 M N-hydroxysuccinimide in HEPES/NaOH buffer. After 3 h of incubation, excess coupling agents were removed by washing the PLG-NPs three to four times. The latter were suspended in 1 mL of HEPES/NaOH buffer (pH 7.0) containing 250 μg of WGA, and after incubation for 12 h lectin-coated PLG-NPs were obtained by centrifugation at 8000 rpm, followed by three to four washings with HEPES/NaOH buffer.
Characterization of WGA-conjugated PLG-NPs
WGA-coated PLG-NPs were analysed for their size by transmission electron microscopy. The amount (μg) of lectin bound to PLG-NPs (mg) was estimated by quantifying the amount of unbound lectin recovered upon centrifugation and washings of PLG-NPs, and subtracting it from the initial amount by using a microBCA kit.1 The coupling efficiency was expressed as the percentage of lectin bound to PLG-NPs, as compared with the initial amount of lectin. The amount of drugs entrapped in WGA lectin-coated PLG-NPs was estimated as the amount of unentrapped drugs recovered in the supernatant after centrifugation and subsequent washings of the formulation. The drug encapsulation efficiency was calculated as the percentage of drugs entrapped in PLG-NPs, as compared with the initial amounts added. The encapsulation was confirmed by lysing the formulation in 5% SDS/0.1 M NaOH. The in vitro release of drugs from lectin-coated PLG-NPs containing antitubercular drugs was determined by suspending the formulation in 2 mL of simulated gastric fluid (SGF) or simulated intestinal fluid (SIF) for 1 month to evaluate the gastrointestinal stability of the formulation.11
The lectin-coated PLG-NPs were aerosolized by using a compressor nebulizer system (Medel Aerofamily, Italy; 250 kPa pressure, nebulizer airflow 5.5 L/min). Nebulized lectin-coated PLG-NPs were sized on a 7-stage Anderson Cascade Impactor (Anderson Samplers, Inc., Atlanta, GA, USA) and evaluated for mass median aerodynamic diameter (MMAD), i.e. the aerodynamic diameter above and below which 50% of the particles in the aerosol are contained, and geometric standard deviation (GSD), i.e. the range of distribution of aerosol particle size around the median. For in vivo studies, each animal received drug formulations (containing dose of PLG-NPs coated with WGA for each animal) suspended in 4 mL of 0.9% sodium chloride via a suitable facemask connected to a nebulizer, with an exposure time of 4 min/animal.
In vivo drug disposition studies
Lectin-functionalized PLG-NPs encapsulating antitubercular drugs at therapeutic dosage (isoniazid, 10 mg/kg body weight; rifampicin, 12 mg/kg body weight; and pyrazinamide, 25 mg/kg body weight) were administered to guinea pigs through the oral/aerosol route. For comparison, a separate set of animals was given free drugs orally. Animals were bled at different time intervals to study the plasma profile of drugs, and sacrificed at various time points to assess tissue (lungs, liver and spleen) drug levels. The drugs were also estimated in 20% tissue homogenates prepared in normal saline (w/v). Isoniazid, rifampicin and pyrazinamide were estimated by spectrofluorimetric,12 microbiological13 and spectrophotometric14 methods, respectively.
The plasma profile of isoniazid, rifampicin and pyrazinamide was used to evaluate pharmacokinetic parameters. Peak plasma concentration (Cmax) and time to reach Cmax (Tmax) were calculated directly from the time–concentration curves for drugs. Elimination rate constant (kel) was calculated by regression analysis and elimination half-life (t½) was calculated as 0.693/kel. Relative bioavailability was calculated as the ratio of AUC0–∞ of WGA-coated PLG-NPs and AUC0–∞ of free drugs.
Experimental infection and chemotherapy
Guinea pigs were infected intramuscularly with 1.5 × 105 viable bacilli of M. tuberculosis H37Rv in 0.1 mL of sterile normal saline. Infection was confirmed after 20 days by sacrificing two guinea pigs and performing Ziehl–Neelsen staining of lung and spleen homogenates. For chemotherapy, guinea pigs were divided into different groups of 6–8 animals, i.e. I, untreated controls; II, drug-loaded PLG-NPs coated with WGA administered every 15 days (three doses) orally for 45 days; III, nebulized drug-loaded PLG-NPs coated with WGA, every 15 days (three doses) for 45 days; IV, oral free drugs, daily for 45 days; V, empty PLG-NPs coated with WGA (three doses), every 15 days for 45 days through the oral route. Animals were sacrificed 46 days post-chemotherapy. The caudal lobe of the right lung and the spleen of each animal were removed aseptically and homogenized in 3 mL of normal saline under sterile conditions, and 1:100 dilutions of tissue homogenates were plated onto Middlebrook 7H10 agar plates and colony forming units (cfu) were enumerated 30 days post-inoculation.
Lungs of animals subjected to chemotherapy were evaluated for histopathological changes compared with controls. Hepatotoxicity induced (if any) during the course of chemotherapy was studied by assaying serum alanine transaminase (ALT), alkaline phosphatase (ALP) and total bilirubin employing standard kits. Immunological responses to lectin were studied by carrying out a T cell proliferation assay.15 The animals were administered lectin-coated PLG-NPs every 15 days (three doses) or equivalent WGA lectin alone (three doses) for 6 weeks. Upon sacrifice, spleens were isolated and splenocytes (2 × 105 cells) were plated on 96-well plates in RPMI 1640 medium. These were challenged with 5–10 μg of WGA lectin, WGA lectin-coated PLG-NPs or concanavalin A as positive control, and incubated at 37°C in 5% CO2. Tritiated thymidine was added in the last 22 h of incubation. The cells were harvested on fibreglass paper and incorporated radioactivity was measured using a liquid scintillation counter. Stimulation index, i.e. the ratio of incorporated radioactivity in test group to that in controls, was calculated.
Statistical analysis
The pharmacokinetic parameters and cfu data were analysed by Student's t-test.
Results
Characterization of WGA-coated PLG-NPs
Nanoparticles prepared from PLG-H and conjugated to WGA lectin were in the size range of 350–400 nm, as compared with 180–290 nm for unconjugated NPs.9 The binding of WGA to empty or drug-loaded PLG-NPs was observed to be 3–3.5 μg of WGA/mg of NPs, yielding a coupling efficiency of ∼60%–70%. Drug encapsulation efficiency was recorded as 54% ± 2% for rifampicin, 64.3% ± 7.4% for isoniazid and 66.8% ± 7.7% for pyrazinamide. Aerodynamic characterization revealed that ∼88% of WGA-coated PLG-NPs were in the respirable range (≤6 μm), MMAD being 2.8 ± 1.4 μm with a GSD of 2.1 ± 0.3. The in vitro drug release studies showed a minimal release of drugs in SGF (rifampicin, 332 mg/L; isoniazid, 335 mg/L; and pyrazinamide, 464 mg/L) and SIF (rifampicin, 448 mg/L; isoniazid, 404 mg/L; and pyrazinamide, 345 mg/L) within the first 7 days, followed by a release of < 100 mg/L over the subsequent 3 weeks.
In vivo drug disposition studies
Upon oral administration of PLG-NPs coated with WGA, the presence of isoniazid, rifampicin and pyrazinamide was observed in plasma for 13, 7 and 13 days, respectively. When this formulation was administered through nebulization, isoniazid was observed up to 13, rifampicin up to 6 and pyrazinamide up to 14 days in plasma. In contrast, free drugs were cleared from the circulation within 12–24 h (Figure 1). All the three drugs were detectable in tissues, i.e. lungs, liver and spleen, up to 15 days above the minimum inhibitory concentration (MIC) through the oral/aerosol route. Upon administration of uncoated PLG-NPs loaded with antitubercular drugs through the oral/aerosol route, rifampicin was detected in plasma for 4–6 days, whereas isoniazid and pyrazinamide were observed for 8–9 days. The presence of isoniazid, rifampicin and pyrazinamide was observed to be up to 10 days above the MIC in the tissues. As shown by plasma profile, Cmax of drugs encapsulated in WGA-coated PLG-NPs administered through the oral/aerosol route was comparable with that of free drugs, but the time to reach Cmax (Tmax) was increased in the former. kel of drugs loaded in lectin-coated PLG-NPs exhibited a decrease as compared with that for free drugs, resulting in a subsequent increase in t½. Area under the drug concentration versus time curve (AUC0–∞) and mean residence time (MRT) were found to be higher for WGA-coated PLG-NPs upon administration through the oral/aerosol route, resulting in a significant (P < 0.001) increase in relative bioavailability of encapsulated drugs in comparison with free drugs (Table 1).
Chemotherapeutic effects
Administration of oral/nebulized lectin-coated PLG-NPs fortnightly (three doses) for 45 days and oral free drugs daily for 45 days to M. tuberculosis-infected guinea pigs resulted in undetectable mycobacterial cfu in lung and spleen homogenates (1:100 dilution), as presented in Table 2. Guinea pigs administered empty WGA-coated PLG-NPs and untreated controls, however, exhibited comparable cfu counts. Histopathology revealed that lungs of untreated infected animals showed complete involvement by the disease process. Large inflamed epithelioid cell granulomas were seen to involve peribronchial areas extensively, in places destroying the wall and abutting into the lumen of airways. However, the lungs of guinea pigs administered WGA lectin-coated PLG-NPs orally/aerosolized, did not exhibit such areas of necrosis. No hepatotoxicity was observed in any of the groups subjected to chemotherapy with respect to activity of ALP/ALT and levels of bilirubin (Table 3). The T cell proliferation assay indicated that upon administration of WGA-grafted PLG-NPs or WGA alone, no immunological response was elicited, as indicated by a stimulation index of < 3 as compared with that for concanavalin A (stimulation index = 9 ± 1.5).
Discussion
Lectin-functionalized PLG-NPs constitute a drug delivery system bearing the benefits of sustained release properties of PLG-NPs and the cytoadhesive character of WGA lectin. The present study explores for the first time the therapeutic potential of WGA-functionalized PLG-NPs encapsulating antitubercular drugs against experimental tuberculosis through the oral and aerosol route in guinea pigs.
For binding of lectins to particulate matter, different strategies including avidin–biotin technology,16 carbodiimide binding10 and glutaraldehyde binding17 have been used. Avidin–biotin technology generates macromolecular spacers, whereas glutaraldehyde is a polymeric cross-linker labile in an acidic environment. Carbodiimide simply catalyses the formation of amide bonds between the carboxylic acid groups of PLG-H and the amines of lectins. Carbodiimide was used in this study yielding binding of 3–3.5 μg lectin/mg of PLG with a coupling efficiency of 60%–70%. The activation and coupling of PLG-NP with lectin had a negligible effect on the drug encapsulation, as the drug content of coated as well as uncoated PLG nanoparticles was found to be similar. In vitro drug release studies showed a release of 3%–5% of drugs in SGF and SIF from WGA-coated PLG-NPs, indicating the stability of the formulation in the gastrointestinal tract.
The prolonged presence of drugs in plasma encapsulated in WGA-coated PLG-NPs, as compared with free drugs, might be attributed to the fact that lectins immobilize the drug-loaded formulation on the intestinal surface for a prolonged period of time to allow (1) an increase in the time interval available for absorption, and (2) a localized increase in the concentration gradient between luminal and serosal sides of the membrane.7 Sustained release of drugs upon administration of nebulized WGA-coated PLG-NPs indicates the binding of WGA on the alveolar epithelium. WGA-functionalized liposomes have been reported to bind to and to be internalized by alveolar cells.18 The bioavailability of drugs encapsulated in WGA-functionalized PLG-NPs was increased significantly (P < 0.001), as depicted from the pharmacokinetic evaluation, with a prolonged Tmax, and increased AUC0–∞ and MRT through the oral/aerosol route, as compared with oral free drugs.
The presence of isoniazid, rifampicin and pyrazinamide in tissues, i.e. lungs, liver and spleen, for 15 days on administration of oral/nebulized WGA-functionalized PLG-NPs favours its application against tuberculosis where infection is largely localized in tissues. Because of the fact that the drug levels were present at above MIC in the organs, the chances of emergence of drug resistance are also minimal. Accordingly, a fortnightly schedule of chemotherapy was designed for WGA-coated PLG-NPs. In terms of achieving undetectable cfu, it was observed that only three oral/nebulized doses of this formulation, every 15 days, were equiefficient to 45 doses of oral free drugs administered daily. Histopathologically, a significant improvement was seen in lung tissue of animals subjected to chemotherapy with oral/aerosolized WGA-coated PLG-NPs, as compared with controls. The formulation was found to elicit neither any immune responses nor signs of hepatotoxicity.
Recent studies from our laboratory demonstrated that when PLG-NPs containing antitubercular drugs were administered through the oral9/aerosol19 route in animals, mycobacterial cfu were undetectable with the formulation, following five doses of chemotherapy every 10 days. However, in the present communication, a fortnightly schedule (three doses) has been shown to be of equal chemotherapeutic benefit, clearly indicating the advantage of lectin conjugation in PLG-NPs. Although the aerosol route should be the route of choice for administration of antitubercular drugs as TB infection is localized mainly in lungs, the oral route is more patient friendly. WGA-coated PLG-NPs bear equivalent chemotherapeutic potential through the oral and aerosol routes. This study reports for the first time the application of WGA-coated PLG-NPs as a carrier for antitubercular drugs, in order to improve their therapeutic benefit and, in turn, patient compliance in TB control, which is cost-effective ($1 versus $17 mg/kg) for a full course of chemotherapy in guinea pigs.
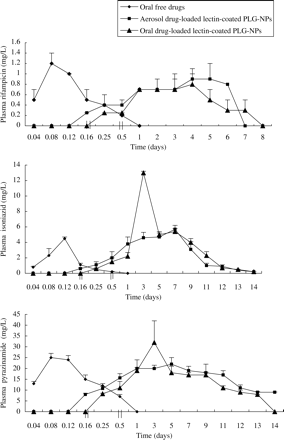
Plasma drug levels following single administration of oral/aerosol drug-loaded WGA-conjugated PLG-NPs and free drugs to guinea pigs.
Pharmacokinetic profile following oral/aerosol administration of WGA lectin-functionalized PLG-NPs as compared with free drugs
| Isoniazid | Rifampicin | Pyrazinamide | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| oral NP-lectin | aerosol NP-lectin | free drug (oral) | oral NP-lectin | aerosol NP-lectin | free drug | oral NP-lectin | aerosol NP-lectin | free drug | |||||||
| Cmax (mg/L) | 13.54 ± 1.64 | 5.79 ± 0.52 | 2.3 ± 1.33 | 0.79 ± 0.23 | 1.07 ± 0.41 | 1.22 ± 0.23 | 50.2 ± 3.52 | 22.52 ± 3.45 | 25.46 ± 1.86 | ||||||
| Tmax (h) | 80 ± 3.85 | 120 ± 0 | 2 ± 0 | 40 ± 27.71 | 72 ± 2.4 | 2 ± 0 | 72 ± 0 | 96 ± 0 | 2 ± 0 | ||||||
| kel | −0.005 ± 0.00 | −0.02 ± 0 | −0.21 ± 0 | −0.008 ± 0.002 | −0.01 ± 0.005 | −0.15 ± 0 | −0.008 ± 0 | −0.007 ± 0 | −0.10 ± 0.01 | ||||||
| t½ (h) | 129.25 ± 38.98 | 34.65 ± 0 | 3.11 ± 0.53 | 92.5 ± 26.84 | 53.31 ± 20.01 | 4.5 ± 0.53 | 80.3 ± 9.75 | 99 ± 0 | 6.5 ± 1.43 | ||||||
| AUC0–∞(mg·h/L) | 1525 ± 98.3*** | 584 ± 25*** | 10.99 ± 3 | 110.04 ± 42*** | 117.2 ± 26.28*** | 9.7 ± 1.11 | 6273 ± 145*** | 4670 ± 101*** | 6.213 ± 1.15*** | ||||||
| Relative bioavailability | 138.76*** | 53.1*** | 1 | 10.19*** | 13.4*** | 1 | 29.45*** | 22.1*** | 1 | ||||||
| Isoniazid | Rifampicin | Pyrazinamide | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| oral NP-lectin | aerosol NP-lectin | free drug (oral) | oral NP-lectin | aerosol NP-lectin | free drug | oral NP-lectin | aerosol NP-lectin | free drug | |||||||
| Cmax (mg/L) | 13.54 ± 1.64 | 5.79 ± 0.52 | 2.3 ± 1.33 | 0.79 ± 0.23 | 1.07 ± 0.41 | 1.22 ± 0.23 | 50.2 ± 3.52 | 22.52 ± 3.45 | 25.46 ± 1.86 | ||||||
| Tmax (h) | 80 ± 3.85 | 120 ± 0 | 2 ± 0 | 40 ± 27.71 | 72 ± 2.4 | 2 ± 0 | 72 ± 0 | 96 ± 0 | 2 ± 0 | ||||||
| kel | −0.005 ± 0.00 | −0.02 ± 0 | −0.21 ± 0 | −0.008 ± 0.002 | −0.01 ± 0.005 | −0.15 ± 0 | −0.008 ± 0 | −0.007 ± 0 | −0.10 ± 0.01 | ||||||
| t½ (h) | 129.25 ± 38.98 | 34.65 ± 0 | 3.11 ± 0.53 | 92.5 ± 26.84 | 53.31 ± 20.01 | 4.5 ± 0.53 | 80.3 ± 9.75 | 99 ± 0 | 6.5 ± 1.43 | ||||||
| AUC0–∞(mg·h/L) | 1525 ± 98.3*** | 584 ± 25*** | 10.99 ± 3 | 110.04 ± 42*** | 117.2 ± 26.28*** | 9.7 ± 1.11 | 6273 ± 145*** | 4670 ± 101*** | 6.213 ± 1.15*** | ||||||
| Relative bioavailability | 138.76*** | 53.1*** | 1 | 10.19*** | 13.4*** | 1 | 29.45*** | 22.1*** | 1 | ||||||
P < 0.001 as compared with free drug group.
Pharmacokinetic profile following oral/aerosol administration of WGA lectin-functionalized PLG-NPs as compared with free drugs
| Isoniazid | Rifampicin | Pyrazinamide | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| oral NP-lectin | aerosol NP-lectin | free drug (oral) | oral NP-lectin | aerosol NP-lectin | free drug | oral NP-lectin | aerosol NP-lectin | free drug | |||||||
| Cmax (mg/L) | 13.54 ± 1.64 | 5.79 ± 0.52 | 2.3 ± 1.33 | 0.79 ± 0.23 | 1.07 ± 0.41 | 1.22 ± 0.23 | 50.2 ± 3.52 | 22.52 ± 3.45 | 25.46 ± 1.86 | ||||||
| Tmax (h) | 80 ± 3.85 | 120 ± 0 | 2 ± 0 | 40 ± 27.71 | 72 ± 2.4 | 2 ± 0 | 72 ± 0 | 96 ± 0 | 2 ± 0 | ||||||
| kel | −0.005 ± 0.00 | −0.02 ± 0 | −0.21 ± 0 | −0.008 ± 0.002 | −0.01 ± 0.005 | −0.15 ± 0 | −0.008 ± 0 | −0.007 ± 0 | −0.10 ± 0.01 | ||||||
| t½ (h) | 129.25 ± 38.98 | 34.65 ± 0 | 3.11 ± 0.53 | 92.5 ± 26.84 | 53.31 ± 20.01 | 4.5 ± 0.53 | 80.3 ± 9.75 | 99 ± 0 | 6.5 ± 1.43 | ||||||
| AUC0–∞(mg·h/L) | 1525 ± 98.3*** | 584 ± 25*** | 10.99 ± 3 | 110.04 ± 42*** | 117.2 ± 26.28*** | 9.7 ± 1.11 | 6273 ± 145*** | 4670 ± 101*** | 6.213 ± 1.15*** | ||||||
| Relative bioavailability | 138.76*** | 53.1*** | 1 | 10.19*** | 13.4*** | 1 | 29.45*** | 22.1*** | 1 | ||||||
| Isoniazid | Rifampicin | Pyrazinamide | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| oral NP-lectin | aerosol NP-lectin | free drug (oral) | oral NP-lectin | aerosol NP-lectin | free drug | oral NP-lectin | aerosol NP-lectin | free drug | |||||||
| Cmax (mg/L) | 13.54 ± 1.64 | 5.79 ± 0.52 | 2.3 ± 1.33 | 0.79 ± 0.23 | 1.07 ± 0.41 | 1.22 ± 0.23 | 50.2 ± 3.52 | 22.52 ± 3.45 | 25.46 ± 1.86 | ||||||
| Tmax (h) | 80 ± 3.85 | 120 ± 0 | 2 ± 0 | 40 ± 27.71 | 72 ± 2.4 | 2 ± 0 | 72 ± 0 | 96 ± 0 | 2 ± 0 | ||||||
| kel | −0.005 ± 0.00 | −0.02 ± 0 | −0.21 ± 0 | −0.008 ± 0.002 | −0.01 ± 0.005 | −0.15 ± 0 | −0.008 ± 0 | −0.007 ± 0 | −0.10 ± 0.01 | ||||||
| t½ (h) | 129.25 ± 38.98 | 34.65 ± 0 | 3.11 ± 0.53 | 92.5 ± 26.84 | 53.31 ± 20.01 | 4.5 ± 0.53 | 80.3 ± 9.75 | 99 ± 0 | 6.5 ± 1.43 | ||||||
| AUC0–∞(mg·h/L) | 1525 ± 98.3*** | 584 ± 25*** | 10.99 ± 3 | 110.04 ± 42*** | 117.2 ± 26.28*** | 9.7 ± 1.11 | 6273 ± 145*** | 4670 ± 101*** | 6.213 ± 1.15*** | ||||||
| Relative bioavailability | 138.76*** | 53.1*** | 1 | 10.19*** | 13.4*** | 1 | 29.45*** | 22.1*** | 1 | ||||||
P < 0.001 as compared with free drug group.
Chemotherapeutic efficacy of drug-loaded PLG-NPs coated with WGA administered orally/aerosolized to guinea pigs
| Group | Organ | Log cfu/mL (mean ± S.D.) |
|---|---|---|
| Untreated controls | lung spleen | 5.6 ± 0.4 5.2 ± 0.41 |
| Empty WGA nanoparticles (oral) | lung spleen | 6 ± 0.38* 5.3 ± 0.24* |
| Free antitubercular drugs (oral) | lung spleen | <1 <1 |
| Drug-loaded WGA nanoparticles (oral) | lung spleen | <1 <1 |
| Drug-loaded WGA nanoparticles (aerosolized) | lung spleen | <1 <1 |
| Group | Organ | Log cfu/mL (mean ± S.D.) |
|---|---|---|
| Untreated controls | lung spleen | 5.6 ± 0.4 5.2 ± 0.41 |
| Empty WGA nanoparticles (oral) | lung spleen | 6 ± 0.38* 5.3 ± 0.24* |
| Free antitubercular drugs (oral) | lung spleen | <1 <1 |
| Drug-loaded WGA nanoparticles (oral) | lung spleen | <1 <1 |
| Drug-loaded WGA nanoparticles (aerosolized) | lung spleen | <1 <1 |
Values are means ± S.D. of 6–8 animals.
P > 0.05, as compared with untreated controls.
Chemotherapeutic efficacy of drug-loaded PLG-NPs coated with WGA administered orally/aerosolized to guinea pigs
| Group | Organ | Log cfu/mL (mean ± S.D.) |
|---|---|---|
| Untreated controls | lung spleen | 5.6 ± 0.4 5.2 ± 0.41 |
| Empty WGA nanoparticles (oral) | lung spleen | 6 ± 0.38* 5.3 ± 0.24* |
| Free antitubercular drugs (oral) | lung spleen | <1 <1 |
| Drug-loaded WGA nanoparticles (oral) | lung spleen | <1 <1 |
| Drug-loaded WGA nanoparticles (aerosolized) | lung spleen | <1 <1 |
| Group | Organ | Log cfu/mL (mean ± S.D.) |
|---|---|---|
| Untreated controls | lung spleen | 5.6 ± 0.4 5.2 ± 0.41 |
| Empty WGA nanoparticles (oral) | lung spleen | 6 ± 0.38* 5.3 ± 0.24* |
| Free antitubercular drugs (oral) | lung spleen | <1 <1 |
| Drug-loaded WGA nanoparticles (oral) | lung spleen | <1 <1 |
| Drug-loaded WGA nanoparticles (aerosolized) | lung spleen | <1 <1 |
Values are means ± S.D. of 6–8 animals.
P > 0.05, as compared with untreated controls.
Biochemical hepatotoxicity upon administration (oral/nebulized) of WGA-coated PLG-NPs every 15 days to infected guinea pigs as compared with free drugs
| Control | Empty WGA-coated PLG-NPs | Oral WGA-coated PLG-NPs | Nebulized WGA-coated PLG-NPs | |
|---|---|---|---|---|
| ALP activity (U/L) | 1.63–2.44 | 1.41–3.8 | 4–4.5 | 4.1–5.2 |
| ALT activity (U/L) | 2.01–3.5 | 2–3.2 | 3.8–4.3 | 2.7–3.8 |
| Bilirubin (U/L) | 1.19–3.39 | 3.41–3.43 | 2.8–4.1 | 3.9–4.2 |
| Control | Empty WGA-coated PLG-NPs | Oral WGA-coated PLG-NPs | Nebulized WGA-coated PLG-NPs | |
|---|---|---|---|---|
| ALP activity (U/L) | 1.63–2.44 | 1.41–3.8 | 4–4.5 | 4.1–5.2 |
| ALT activity (U/L) | 2.01–3.5 | 2–3.2 | 3.8–4.3 | 2.7–3.8 |
| Bilirubin (U/L) | 1.19–3.39 | 3.41–3.43 | 2.8–4.1 | 3.9–4.2 |
Biochemical hepatotoxicity upon administration (oral/nebulized) of WGA-coated PLG-NPs every 15 days to infected guinea pigs as compared with free drugs
| Control | Empty WGA-coated PLG-NPs | Oral WGA-coated PLG-NPs | Nebulized WGA-coated PLG-NPs | |
|---|---|---|---|---|
| ALP activity (U/L) | 1.63–2.44 | 1.41–3.8 | 4–4.5 | 4.1–5.2 |
| ALT activity (U/L) | 2.01–3.5 | 2–3.2 | 3.8–4.3 | 2.7–3.8 |
| Bilirubin (U/L) | 1.19–3.39 | 3.41–3.43 | 2.8–4.1 | 3.9–4.2 |
| Control | Empty WGA-coated PLG-NPs | Oral WGA-coated PLG-NPs | Nebulized WGA-coated PLG-NPs | |
|---|---|---|---|---|
| ALP activity (U/L) | 1.63–2.44 | 1.41–3.8 | 4–4.5 | 4.1–5.2 |
| ALT activity (U/L) | 2.01–3.5 | 2–3.2 | 3.8–4.3 | 2.7–3.8 |
| Bilirubin (U/L) | 1.19–3.39 | 3.41–3.43 | 2.8–4.1 | 3.9–4.2 |
We thank Dr Ranjana Minz (Department of Immunopathology, PGIMER, Chandigarh) for her assistance in carrying out histopathological studies. Anjali Sharma gratefully acknowledges CSIR for providing a Junior Research Fellowship. This research was financially supported by a grant from the Indian Council of Medical Research.
References
Mo, Y. & Lim, L. Y. (
Lehr, C. M. (
Ezpeleta, I., Arangoa, M. A., Irache, J. M. et al. (
Lis, H. & Sharon, N. (
Kilpatrick, D. C., Pusztai, A., Grant, G. et al. (
Clark, M. A., Hirst, B. H. & Jepson, M. A. (
Montisci, M. J., Dembri, A., Giovannuci, G. et al. (
Abu-Dahab, R., Schafer, U. F. & Lehr, C. M. (
Pandey, R., Zahoor, A., Sharma, S. et al. (
Ertl, B., Franziska, H., Wirth, M. et al. (
Pandey, R. & Khuller, G. K. (
Scott, E. H. & Wright, R. C. (
Saito, H. & Tomioka, H. (
Gurumurthy, P., Nair, N. G. & Sarma, G. R. (
Andersen, P., Askgaard, D., Ljungqvist, L. et al. (
Tao, S. L., Lubeley, M. W. & Desai, T. A. (
Montisci, M. J., Giovannuci, G., Duchene, D. et al. (
Bruck, A., Abu-Dahab, R., Borchard, G. et al. (

{kind=link}