




Abstract
Bathing suit ichthyosis (BSI) is a striking and unique clinical form of autosomal recessive congenital ichthyosis characterized by pronounced scaling on the bathing suit areas but sparing of the extremities and the central face. Here we report on a series of 10 BSI patients. Our genetic, ultrastructural and biochemical investigations show that BSI is caused by transglutaminase-1 (TGase-1) deficiency. Altogether, we identified 13 mutations in TGM1—among them seven novel missense mutations and one novel nonsense mutation. Structural modeling for the Tyr276Asn mutation reveals that the residue is buried in the hydrophobic interior of the enzyme and that the hydroxyl side chain of Tyr276 is exposed to solvent in a cavity of the enzyme. Cryosections of healthy skin areas demonstrated an almost normal TGase activity, in contrast to the affected BSI skin, which only showed a cytoplasmic and clearly reduced TGase-1 activity. The distribution of TGase-1 substrates in the epidermis of affected skin corresponded to the situation in TGase-1 deficiency. Interestingly, the expression of TGase-3 and cathepsin D was reduced. Digital thermography validated a striking correlation between warmer body areas and presence of scaling in patients suggesting a decisive influence of the skin temperature. In situ TGase testing in skin of BSI patients demonstrated a marked decrease of enzyme activity when the temperature was increased from 25 to 37°C. We conclude that BSI is caused by TGase-1 deficiency and suggest that it is a temperature-sensitive phenotype.
INTRODUCTION
Bathing suit ichthyosis (BSI) is described as a unique clinical form of autosomal recessive congenital ichthyosis (ARCI) in South Africa (1,2). Affected individuals are born with collodion membrane encasing the whole body and develop a dark-grey or brownish scaling restricted to the bathing suit areas in the first weeks of life, whereas extremities and central face are almost completely spared. The molecular basis of BSI was unknown so far.
Isolated ARCI encompasses a heterogeneous group of cornification disorders presenting with generalized hyperkeratosis, often accompanied by erythroderma. Associated clinical symptoms are neonatal dehydration, ectropion and hypohidrosis. The more severe phenotype, lamellar ichthyosis (LI) has an estimated prevalence of 1:200 000–300 000. Most patients are born with collodion membrane, which is then gradually replaced by large, dark or plate-like scales (3). In contrast, individuals with non-bullous congenital ichthyosiform erythroderma (NCIE) show a more pronounced erythroderma with fine, white scaling (4). To date, six genes of ARCI have been localized (5–8) and identified (9–13). In about 35–40% ARCI is caused by homozygous or compound heterozygous mutations in TGM1 (14q11.2) resulting in a deficiency of keratinocyte transglutaminase (TGase-1). TGases are Ca2+-dependent enzymes involved in the assembly of the cornified cell envelope (CE). This resilient sheath of ε-(γ-glutamyl) lysine cross-linked proteins is deposited subjacent to the plasma membrane in terminally differentiating keratinocytes. The common reaction catalyzed by each of the eight known active human/rodent TGase isozymes (i.e. Factor XIIIa, TGase-1 through TGase-7) involves attack of a suitable acceptor nucleophile on the γ-carboxamide group of a glutamine residue in a donor protein/peptide. TGase-1 is unique among TGases in which it is anchored to the plasma membrane via specific lipid linkage (14). It has been suggested that TGase-1 can also cross-link ω-hydroxyceramides to the CE (15).
Moreover, ARCI can be associated with missense mutations in ABCA12 on 2q34 (10), the same gene in which large intragenic deletions and frameshift deletions cause Harlequin ichthyosis (16,17). Further mutations were found in the epidermal lipoxygenase genes ALOXE3 and ALOX12B on 17p13 (9), ichthyin on 5q33 (11) and on the new cytochrome P450 gene FLJ39501 on 19p12-q12 (12). In the epidermis, the two lipoxygenases (and probably the gene products of ichthyin and FLJ39501) participate in the same metabolic pathway converting arachidonic acid into specific epoxyalcohol products.
Attempts to refine the classification of LI and NCIE phenotypes by clinical, biochemical and ultrastructural observations have proven difficult, which is illustrated by the fact that the same TGM1 mutation can give rise to either LI or NCIE (18). Moreover, ∼10% of all collodion babies heal completely within the first weeks of life. This distinct phenotype, called self-healing collodion baby (SHCB), can be due to a particular mutation in TGM1 leading to an impaired TGase-1 function at higher intrauterine water pressure (19).
The three main criteria for the diagnosis of BSI are: (a) LI with large dark-grey or brownish scales affecting the trunk and sparing the extremities and central face, (b) a family history consistent with ARCI, and (c) non-syndromic ichthyosis (2). Here, we elucidate the molecular basis of this unique phenotype. We made diagnosis of BSI in a group of 10 individuals from independent families of German, French, Turkish, Moroccan, or Dutch origin. Interestingly, ultrastructural analyses of affected BSI skin revealed the typical morphological signs of ARCI caused by TGase-1 deficiency. Sequencing analyses revealed the presence of unique TGM1 mutations never reported before as well as known mutations. Comparative analysis of affected versus non-affected skin showed a differential expression of TGase-1 in affected and healthy skin. Our investigations suggest that temperature may play a decisive role for the development of the peculiar ichthyosis.
RESULTS
Patients
We ascertained 10 individuals from independent families originating from Germany, France, Turkey, the Netherlands or Morocco, who fulfilled the clinical criteria of BSI (2). All patients were born with collodion membrane. During the first to second month of life they developed a lamellar scaling on the trunk, whereas the four limbs and the face were almost completely spared. However, there was a spectrum concerning the severity of scaling (Table 1). Patient 1 (Fig. 1A and B) displayed large, thick, dark scales with involvement of very specific areas on the extremities and the face, which was also the case in Patients 2, 3, 6 and 10. Patients 4, 5 and 9 only showed mild, brownish scaling that was most pronounced in the axillae and on the neck. Patient 2 and 7 displayed a moderate scaling of the affected areas. Index Patient 8 showed a more variable clinical course. He presented BSI during infancy, but like one of his also affected brothers developed a tendency towards more generalized and rather mild ichthyosis. The family referring to Patient 10 originated from Morocco. Parents were consanguineous and had one boy presenting BSI with moderate scaling on the trunk as well as on particular areas of the limbs (index Patient 10). One brother was also affected and showed a less severe scaling with similar distribution pattern.
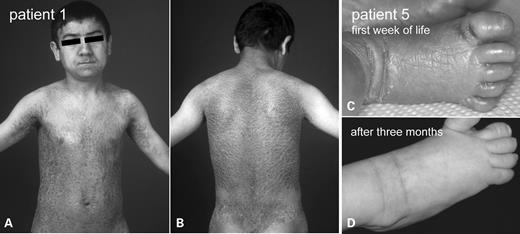
Clinical phenotype of patients with BSI. (A and B) Patient 1 shows lamellar scaling on the trunk, whereas the four limbs and the face were almost completely spared. The scaling was most pronounced in the axillae. Note the symmetric spared area of the suprarenal lumbal region. (C) Patient 5 showed a generalized congenital ichthyosis shortly after birth (right foot). (D) At the age of 3 months the skin lesions had resolved completely on all four extremities.
BSI: summary of data in 10 different patients from independent families
| Index patient | Age/Sex | Origin | Mutation | Exon | Effect | In vivo TGase-1 activity | Ultrastructure | Severity of scaling |
|---|---|---|---|---|---|---|---|---|
| 1 | 14 years/male | Turkey | Homozygous c.826T>A | 5 | Tyr265Asn | Reduced in healthy skin areas | No abnormalities in healthy skin areas | Severe (specific involvement of warmer areas of the integument) |
| Abnormal in affected skin areas | Cholesterol clefts in affected skin | |||||||
| 2 | 35 years/female | Netherlands | c.376C>T | 3 | Arg126Cys | Reduced in healthy skin areas | n.d. | Moderate (specific involvement of warmer areas of the integument) |
| c.425G>A | 3 | Arg142His | Abnormal in affected skin areas | |||||
| 3 | 2 years/male | Germany | c.790C>T | 5 | Arg264Trp | n.d. | n.d. | Moderate (specific involvement of warmer areas of the integument) |
| c.877-2A>G | 6 | Splice-site mutation | ||||||
| 4 | 5 years/male | Germany | c.919C>G | 6 | Arg307Gly | Abnormal in affected skin areas | Cholesterol clefts (collodion skin taken during the first week of life) | Mild (most pronounced in the axillae and on the neck) |
| c.1166G>C | 7 | Arg389Pro | ||||||
| 5 | 1 year/male | Germany | c.877-2A>G | 6 | Splice-site mutation | Abnormal in affected skin areas | Cholesterol clefts (collodion skin taken during the first week of life) | Mild (most pronounced in the axillae and on the neck) |
| c.919C>G | 6 | Arg307Gly | ||||||
| 6 | 1 year/female | Morocco | c.791G>A | 5 | Arg264Gln | Abnormal in affected skin areas | Cholesterol clefts (collodion skin taken during the first week of life) | Moderate (specific involvement of warmer areas of the integument) |
| c.1074delC | 7 | Ser358fsX26 | ||||||
| 7 | 3 years/female | Germany | Homozygous | 13 | Arg687His | n.d. | n.d. | Moderate |
| c.2060G>A | ||||||||
| 8 | 16 years/male | Germany | c.877-2A>G | 6 | Splice-site mutation | Reduced in healthy skin areas | n.d. | Moderate (variant expression) |
| c.943C>T | 6 | Arg315Cys | Abnormal in affected skin areas | |||||
| 9 | 5 years/female | France | c.788G>A | 5 | Trp263X | Abnormal in affected skin areas | n.d. | Mild |
| c.919C>G | 6 | Arg307Gly | ||||||
| 10 | 16 years/male | Morocco | Homozygous | 6 | Arg315His | Reduced in healthy skin areas | n.d. | Moderate (specific involvement of warmer areas of the integument) |
| c.944G>A | Abnormal in affected skin areas |
| Index patient | Age/Sex | Origin | Mutation | Exon | Effect | In vivo TGase-1 activity | Ultrastructure | Severity of scaling |
|---|---|---|---|---|---|---|---|---|
| 1 | 14 years/male | Turkey | Homozygous c.826T>A | 5 | Tyr265Asn | Reduced in healthy skin areas | No abnormalities in healthy skin areas | Severe (specific involvement of warmer areas of the integument) |
| Abnormal in affected skin areas | Cholesterol clefts in affected skin | |||||||
| 2 | 35 years/female | Netherlands | c.376C>T | 3 | Arg126Cys | Reduced in healthy skin areas | n.d. | Moderate (specific involvement of warmer areas of the integument) |
| c.425G>A | 3 | Arg142His | Abnormal in affected skin areas | |||||
| 3 | 2 years/male | Germany | c.790C>T | 5 | Arg264Trp | n.d. | n.d. | Moderate (specific involvement of warmer areas of the integument) |
| c.877-2A>G | 6 | Splice-site mutation | ||||||
| 4 | 5 years/male | Germany | c.919C>G | 6 | Arg307Gly | Abnormal in affected skin areas | Cholesterol clefts (collodion skin taken during the first week of life) | Mild (most pronounced in the axillae and on the neck) |
| c.1166G>C | 7 | Arg389Pro | ||||||
| 5 | 1 year/male | Germany | c.877-2A>G | 6 | Splice-site mutation | Abnormal in affected skin areas | Cholesterol clefts (collodion skin taken during the first week of life) | Mild (most pronounced in the axillae and on the neck) |
| c.919C>G | 6 | Arg307Gly | ||||||
| 6 | 1 year/female | Morocco | c.791G>A | 5 | Arg264Gln | Abnormal in affected skin areas | Cholesterol clefts (collodion skin taken during the first week of life) | Moderate (specific involvement of warmer areas of the integument) |
| c.1074delC | 7 | Ser358fsX26 | ||||||
| 7 | 3 years/female | Germany | Homozygous | 13 | Arg687His | n.d. | n.d. | Moderate |
| c.2060G>A | ||||||||
| 8 | 16 years/male | Germany | c.877-2A>G | 6 | Splice-site mutation | Reduced in healthy skin areas | n.d. | Moderate (variant expression) |
| c.943C>T | 6 | Arg315Cys | Abnormal in affected skin areas | |||||
| 9 | 5 years/female | France | c.788G>A | 5 | Trp263X | Abnormal in affected skin areas | n.d. | Mild |
| c.919C>G | 6 | Arg307Gly | ||||||
| 10 | 16 years/male | Morocco | Homozygous | 6 | Arg315His | Reduced in healthy skin areas | n.d. | Moderate (specific involvement of warmer areas of the integument) |
| c.944G>A | Abnormal in affected skin areas |
n.d., not done.
BSI: summary of data in 10 different patients from independent families
| Index patient | Age/Sex | Origin | Mutation | Exon | Effect | In vivo TGase-1 activity | Ultrastructure | Severity of scaling |
|---|---|---|---|---|---|---|---|---|
| 1 | 14 years/male | Turkey | Homozygous c.826T>A | 5 | Tyr265Asn | Reduced in healthy skin areas | No abnormalities in healthy skin areas | Severe (specific involvement of warmer areas of the integument) |
| Abnormal in affected skin areas | Cholesterol clefts in affected skin | |||||||
| 2 | 35 years/female | Netherlands | c.376C>T | 3 | Arg126Cys | Reduced in healthy skin areas | n.d. | Moderate (specific involvement of warmer areas of the integument) |
| c.425G>A | 3 | Arg142His | Abnormal in affected skin areas | |||||
| 3 | 2 years/male | Germany | c.790C>T | 5 | Arg264Trp | n.d. | n.d. | Moderate (specific involvement of warmer areas of the integument) |
| c.877-2A>G | 6 | Splice-site mutation | ||||||
| 4 | 5 years/male | Germany | c.919C>G | 6 | Arg307Gly | Abnormal in affected skin areas | Cholesterol clefts (collodion skin taken during the first week of life) | Mild (most pronounced in the axillae and on the neck) |
| c.1166G>C | 7 | Arg389Pro | ||||||
| 5 | 1 year/male | Germany | c.877-2A>G | 6 | Splice-site mutation | Abnormal in affected skin areas | Cholesterol clefts (collodion skin taken during the first week of life) | Mild (most pronounced in the axillae and on the neck) |
| c.919C>G | 6 | Arg307Gly | ||||||
| 6 | 1 year/female | Morocco | c.791G>A | 5 | Arg264Gln | Abnormal in affected skin areas | Cholesterol clefts (collodion skin taken during the first week of life) | Moderate (specific involvement of warmer areas of the integument) |
| c.1074delC | 7 | Ser358fsX26 | ||||||
| 7 | 3 years/female | Germany | Homozygous | 13 | Arg687His | n.d. | n.d. | Moderate |
| c.2060G>A | ||||||||
| 8 | 16 years/male | Germany | c.877-2A>G | 6 | Splice-site mutation | Reduced in healthy skin areas | n.d. | Moderate (variant expression) |
| c.943C>T | 6 | Arg315Cys | Abnormal in affected skin areas | |||||
| 9 | 5 years/female | France | c.788G>A | 5 | Trp263X | Abnormal in affected skin areas | n.d. | Mild |
| c.919C>G | 6 | Arg307Gly | ||||||
| 10 | 16 years/male | Morocco | Homozygous | 6 | Arg315His | Reduced in healthy skin areas | n.d. | Moderate (specific involvement of warmer areas of the integument) |
| c.944G>A | Abnormal in affected skin areas |
| Index patient | Age/Sex | Origin | Mutation | Exon | Effect | In vivo TGase-1 activity | Ultrastructure | Severity of scaling |
|---|---|---|---|---|---|---|---|---|
| 1 | 14 years/male | Turkey | Homozygous c.826T>A | 5 | Tyr265Asn | Reduced in healthy skin areas | No abnormalities in healthy skin areas | Severe (specific involvement of warmer areas of the integument) |
| Abnormal in affected skin areas | Cholesterol clefts in affected skin | |||||||
| 2 | 35 years/female | Netherlands | c.376C>T | 3 | Arg126Cys | Reduced in healthy skin areas | n.d. | Moderate (specific involvement of warmer areas of the integument) |
| c.425G>A | 3 | Arg142His | Abnormal in affected skin areas | |||||
| 3 | 2 years/male | Germany | c.790C>T | 5 | Arg264Trp | n.d. | n.d. | Moderate (specific involvement of warmer areas of the integument) |
| c.877-2A>G | 6 | Splice-site mutation | ||||||
| 4 | 5 years/male | Germany | c.919C>G | 6 | Arg307Gly | Abnormal in affected skin areas | Cholesterol clefts (collodion skin taken during the first week of life) | Mild (most pronounced in the axillae and on the neck) |
| c.1166G>C | 7 | Arg389Pro | ||||||
| 5 | 1 year/male | Germany | c.877-2A>G | 6 | Splice-site mutation | Abnormal in affected skin areas | Cholesterol clefts (collodion skin taken during the first week of life) | Mild (most pronounced in the axillae and on the neck) |
| c.919C>G | 6 | Arg307Gly | ||||||
| 6 | 1 year/female | Morocco | c.791G>A | 5 | Arg264Gln | Abnormal in affected skin areas | Cholesterol clefts (collodion skin taken during the first week of life) | Moderate (specific involvement of warmer areas of the integument) |
| c.1074delC | 7 | Ser358fsX26 | ||||||
| 7 | 3 years/female | Germany | Homozygous | 13 | Arg687His | n.d. | n.d. | Moderate |
| c.2060G>A | ||||||||
| 8 | 16 years/male | Germany | c.877-2A>G | 6 | Splice-site mutation | Reduced in healthy skin areas | n.d. | Moderate (variant expression) |
| c.943C>T | 6 | Arg315Cys | Abnormal in affected skin areas | |||||
| 9 | 5 years/female | France | c.788G>A | 5 | Trp263X | Abnormal in affected skin areas | n.d. | Mild |
| c.919C>G | 6 | Arg307Gly | ||||||
| 10 | 16 years/male | Morocco | Homozygous | 6 | Arg315His | Reduced in healthy skin areas | n.d. | Moderate (specific involvement of warmer areas of the integument) |
| c.944G>A | Abnormal in affected skin areas |
n.d., not done.
Ultrastructural analyses
Ultrastructural analyses were performed for the morphological classification of congenital ichthyosis. Altogether four BSI patients were examined shortly after birth when presenting as collodion baby. In Patient 1, both affected and healthy skin areas were studied in later life. Affected skin revealed groups of single polygonal clefts within the thickened, massive horny layer, representing remnants of cholesterol crystals (Fig. 2A and C). These so called cholesterol clefts usually associate with TGase-1 deficiency (20). Interestingly, healthy skin of the right arm of BSI Patient 1 showed a normal diameter of the SC and a completely normal ultrastructure (Fig. 2B).
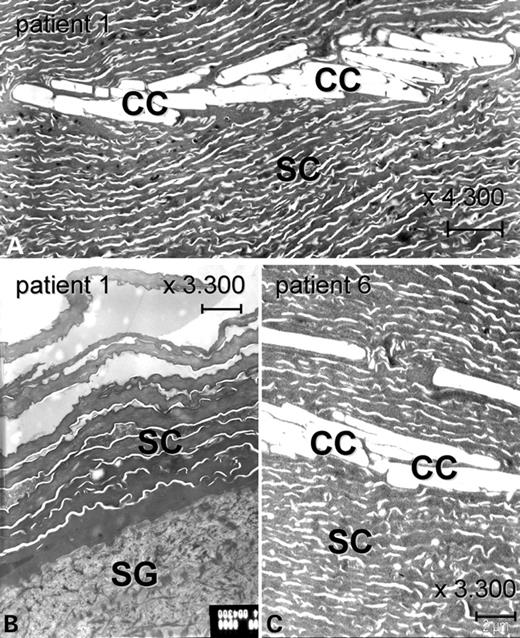
Analysis of ultrastructure in patients with BSI. (A) The massively thickened stratum corneum of the lamellar scaling area of the trunk of Patient 1 displays multiple cholesterol clefts, which are typically observed in ARCI with TGase-1 deficiency. (B) In contrast, the SC of a healthy area (right arm) of Patient 1 has a normal diameter and is not thickened. It does not show any abnormalities. (C) The epidermis of Patient 6 taken at the third day postnatal, when she was still suffering from generalized ichthyosis, also shows a massive SC containing cholesterol clefts. SC: stratum corneum; SG: stratum granulosum; CC: cholesterol clefts. Scale bars: 2 µm.
Mutation analysis of TGM1
The clinical features together with the ultrastructural studies (cholesterol clefts) strongly suggested that BSI is due to TGase-1 deficiency. We therefore performed direct sequencing of TGM1. All BSI individuals analyzed showed homozygous or compound heterozygous mutations (Tables 1 and 2). In a heterozygous status, all mutations could be confirmed in each parental generation.
Observed TGM1 mutations in the BSI group
| Mutation | Domain localization | Suggested effect on TGase-1 function | Reference |
|---|---|---|---|
| Arg126Cys | β-sandwich | Reduced membranous enzyme function | Novel mutation |
| Clinical evidence for the direct or indirect influence of local temperature | |||
| Arg142His | Severe loss of catalytic activity due to altered molecule folding (13) | (13,18,21) | |
| Arg264Trp/Arg264Gln | Reduced membranous enzyme function | Two novel mutations | |
| Clinical evidence for the direct or indirect influence of local skin temperature | |||
| Trp263X | Complete loss of enzyme function | (31,32) | |
| Tyr276Asn | Catalytic core domain | Reduced membranous enzyme function | Novel mutation |
| Clinical and immunochemical evidence for the direct or indirect influence of local skin temperature | |||
| Arg307Gly | Severely diminished cytosolic and membranous activity (of Arg307Try) due to a less stable structure of the catalytic core (28) | Novel mutation, same residue as Arg307Trp (25,27,28) | |
| Arg315Cys | Low specific activity presumably due to protein misfolding or excessively stable protein that can not be processed (30) | (24,29,30) | |
| Arg315His | Similar effect like Arg315Cys | Novel mutation, same residue as Arg315Cys (24,29,30) | |
| Ser358fsX26 | Nonsense mutation leading to a complete loss of enzyme function | Novel mutation | |
| 877-2A>G | Complete loss of enzyme function | (22–24) | |
| Variant transcripts might be possible in different individuals (24) | |||
| Arg389Pro | Within catalytic triad | Probably complete loss of enzyme function because of the localization within the catalytic core (26) | (24–26) |
| Arg687His | Between β-barrel 1 and 2 | Reduced cytosolic and membranous TGase activity (of Arg687Cys) due to a less stable molecule or to an altered substrate binding and specificity (29) | Novel mutation, same residue as Arg687Cys (29) |
| Mutation | Domain localization | Suggested effect on TGase-1 function | Reference |
|---|---|---|---|
| Arg126Cys | β-sandwich | Reduced membranous enzyme function | Novel mutation |
| Clinical evidence for the direct or indirect influence of local temperature | |||
| Arg142His | Severe loss of catalytic activity due to altered molecule folding (13) | (13,18,21) | |
| Arg264Trp/Arg264Gln | Reduced membranous enzyme function | Two novel mutations | |
| Clinical evidence for the direct or indirect influence of local skin temperature | |||
| Trp263X | Complete loss of enzyme function | (31,32) | |
| Tyr276Asn | Catalytic core domain | Reduced membranous enzyme function | Novel mutation |
| Clinical and immunochemical evidence for the direct or indirect influence of local skin temperature | |||
| Arg307Gly | Severely diminished cytosolic and membranous activity (of Arg307Try) due to a less stable structure of the catalytic core (28) | Novel mutation, same residue as Arg307Trp (25,27,28) | |
| Arg315Cys | Low specific activity presumably due to protein misfolding or excessively stable protein that can not be processed (30) | (24,29,30) | |
| Arg315His | Similar effect like Arg315Cys | Novel mutation, same residue as Arg315Cys (24,29,30) | |
| Ser358fsX26 | Nonsense mutation leading to a complete loss of enzyme function | Novel mutation | |
| 877-2A>G | Complete loss of enzyme function | (22–24) | |
| Variant transcripts might be possible in different individuals (24) | |||
| Arg389Pro | Within catalytic triad | Probably complete loss of enzyme function because of the localization within the catalytic core (26) | (24–26) |
| Arg687His | Between β-barrel 1 and 2 | Reduced cytosolic and membranous TGase activity (of Arg687Cys) due to a less stable molecule or to an altered substrate binding and specificity (29) | Novel mutation, same residue as Arg687Cys (29) |
Cursive denotes observation and conclusion of the present study.
Observed TGM1 mutations in the BSI group
| Mutation | Domain localization | Suggested effect on TGase-1 function | Reference |
|---|---|---|---|
| Arg126Cys | β-sandwich | Reduced membranous enzyme function | Novel mutation |
| Clinical evidence for the direct or indirect influence of local temperature | |||
| Arg142His | Severe loss of catalytic activity due to altered molecule folding (13) | (13,18,21) | |
| Arg264Trp/Arg264Gln | Reduced membranous enzyme function | Two novel mutations | |
| Clinical evidence for the direct or indirect influence of local skin temperature | |||
| Trp263X | Complete loss of enzyme function | (31,32) | |
| Tyr276Asn | Catalytic core domain | Reduced membranous enzyme function | Novel mutation |
| Clinical and immunochemical evidence for the direct or indirect influence of local skin temperature | |||
| Arg307Gly | Severely diminished cytosolic and membranous activity (of Arg307Try) due to a less stable structure of the catalytic core (28) | Novel mutation, same residue as Arg307Trp (25,27,28) | |
| Arg315Cys | Low specific activity presumably due to protein misfolding or excessively stable protein that can not be processed (30) | (24,29,30) | |
| Arg315His | Similar effect like Arg315Cys | Novel mutation, same residue as Arg315Cys (24,29,30) | |
| Ser358fsX26 | Nonsense mutation leading to a complete loss of enzyme function | Novel mutation | |
| 877-2A>G | Complete loss of enzyme function | (22–24) | |
| Variant transcripts might be possible in different individuals (24) | |||
| Arg389Pro | Within catalytic triad | Probably complete loss of enzyme function because of the localization within the catalytic core (26) | (24–26) |
| Arg687His | Between β-barrel 1 and 2 | Reduced cytosolic and membranous TGase activity (of Arg687Cys) due to a less stable molecule or to an altered substrate binding and specificity (29) | Novel mutation, same residue as Arg687Cys (29) |
| Mutation | Domain localization | Suggested effect on TGase-1 function | Reference |
|---|---|---|---|
| Arg126Cys | β-sandwich | Reduced membranous enzyme function | Novel mutation |
| Clinical evidence for the direct or indirect influence of local temperature | |||
| Arg142His | Severe loss of catalytic activity due to altered molecule folding (13) | (13,18,21) | |
| Arg264Trp/Arg264Gln | Reduced membranous enzyme function | Two novel mutations | |
| Clinical evidence for the direct or indirect influence of local skin temperature | |||
| Trp263X | Complete loss of enzyme function | (31,32) | |
| Tyr276Asn | Catalytic core domain | Reduced membranous enzyme function | Novel mutation |
| Clinical and immunochemical evidence for the direct or indirect influence of local skin temperature | |||
| Arg307Gly | Severely diminished cytosolic and membranous activity (of Arg307Try) due to a less stable structure of the catalytic core (28) | Novel mutation, same residue as Arg307Trp (25,27,28) | |
| Arg315Cys | Low specific activity presumably due to protein misfolding or excessively stable protein that can not be processed (30) | (24,29,30) | |
| Arg315His | Similar effect like Arg315Cys | Novel mutation, same residue as Arg315Cys (24,29,30) | |
| Ser358fsX26 | Nonsense mutation leading to a complete loss of enzyme function | Novel mutation | |
| 877-2A>G | Complete loss of enzyme function | (22–24) | |
| Variant transcripts might be possible in different individuals (24) | |||
| Arg389Pro | Within catalytic triad | Probably complete loss of enzyme function because of the localization within the catalytic core (26) | (24–26) |
| Arg687His | Between β-barrel 1 and 2 | Reduced cytosolic and membranous TGase activity (of Arg687Cys) due to a less stable molecule or to an altered substrate binding and specificity (29) | Novel mutation, same residue as Arg687Cys (29) |
Cursive denotes observation and conclusion of the present study.
Index patient 1.
The most severely affected patient of our cohort showed the homozygous mutation c.826T>A which leads to the missense mutation Tyr276Asn. His Turkish parents were first cousins and both heterozygous for c.826T>A. To our knowledge Tyr276Asn has not yet been reported in patients with ARCI so far.
Index patient 2.
This female patient from the Netherlands showed the well known mutation c.425G>A (Arg142His) (13,18,21) and the new mutation c.376C>T leading to a change from arginine to cysteine at residue 126 (Arg126Cys).
Index patient 3.
This patient of German origin revealed the splice site mutation c.877-2A>G and missense mutation c.790C>T. The latter novel mutation leads to a change from arginine to tryptophan at residue 264 (Arg264Trp). c.877-2A>G has been described earlier (22–24).
Index patient 4.
This patient showed the compound heterozygous genotype c.919C>G/c.1166G>C, leading to missense mutations Arg307Gly and Arg389Pro, respectively. The latter amino acid change has already been reported as well as the mutation Arg389His affecting the neighboring residue (24–26). Arg307Gly is a new mutation; a missense mutation of the same residue (Arg307Trp) has been described before (25,27,28).
Index patient 5.
The analysis in this boy revealed the splice-site mutation c.877-2A>G also found in Patient 3 and the same missense mutation Arg307Gly present in Patients 4 and 9.
Index patient 6.
This young girl showed a disease manifestation similar to Patient 3 and harbored the compound heterozygous genotype c.791G>A/c.1074delC. The mutation leads to Arg264Gln affecting the same residue as, Arg264Trp, seen in Patient 3. The deletion c.1074delC leads to a frameshift from codon 358 resulting in a premature termination codon after 25 residues (Ser358fsX26). This nonsense mutation has not been described so far.
Index patient 7.
As an exception, the mutation found in this family locates outside the TGase-1 core domain. Sequencing of exon 13 revealed the homozygous missense mutation c.2060G>A leading to the amino acid change Arg687His, which alters the same residue as Arg687Cys (29). Consanguinity is not known in this family.
Index patient 8.
The index patient showed the compound heterozygous genotype c.877-2A>G/c.943C>T. The first allele represents the known splice-site mutation also present in Patients 3 and 5. The second exchange leads to the missense mutation Arg315Cys, which has also been described earlier (24,29,30). The brother showing a variant form of BSI had the same genotype.
Index patient 9.
The relatively mildly affected girl showed the compound heterozygous genotype c.788G>A/c.919C>G. The first substitution leads to the known nonsense mutation Trp264X (31,32), the second allele resulting in the missense mutation Arg307Gly was also present in Patients 4 and 5.
Index patient 10.
The consanguineous family from North Africa had two affected brothers (only one is included in our group) who both showed the homozygous mutation c.944G>A, which results in the amino acid change Arg315His. This allele represents a novel missense mutation. Mutation Arg315Cys (24,29,30) present in Patient 8 affects the same residue.
Structural modeling
The likely effect of certain mutations on the resulting protein was modeled in silico based on the atomic structure of TGase-3 enzyme. This modeling predicted which surrounding residues will be affected by the mutation.
Tyr276Asn.
Tyr276 is located on the β-strand consisting of Glu271-Gly278 on the interface between the core domain and β-barrel 2 (Fig. 3). The Tyr276 is buried in the hydrophobic interior of the enzyme and sandwiched from one side by Trp378, Phe435, Gln662, Phe686, His779 and a β-strand encompassing Ile283-Tyr290 from the other side. The hydroxyl side-chain of Tyr276 is exposed to solvent molecules in a cavity.
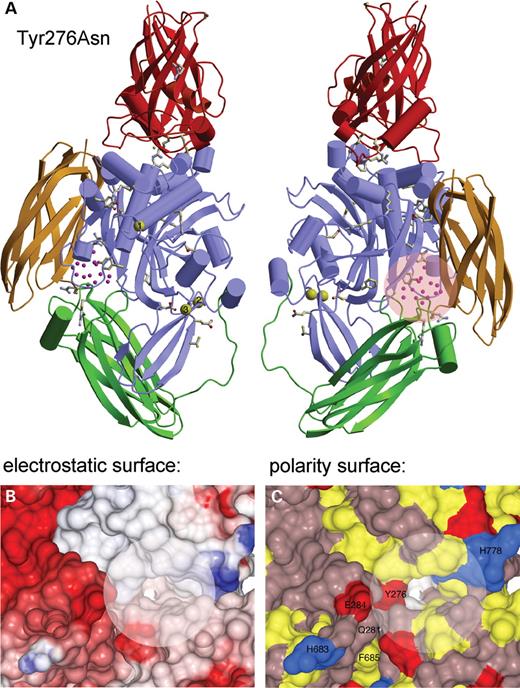
Three-dimensional in silico modeling of the TGase-1 structure concerning the mutation Tyr276Asn. (A) Ribbon image of the model of human TGase-1 structure in the left panel. The four domains are the β-sandwich (red), the catalytic core (light blue), the β-barrel 1 (green) and β-barrel 2 (yellow). The Ca2+ ions are shown in yellow. Residues Asp66–Leu109 from the membrane anchorage region were modelled (orange) and the proteolysis sites are shown. The side-chains of 41 amino acids of TGase-1 shown to be mutated in LI patients, are drawn in ball-and-stick. The Tyr276 residue exposed to a cavity with solvent molecules is shown and is located on the interface between the core domain and β-barrel 2. The rotation of the left image of TGase-1 by 180° is shown in the right panel. (B) View of the electrostatic surface potential of TGase-1 cavity surrounding residue Tyr267. The acidic and basic residues are colored red and blue, respectively. The electrostatic potentials have been mapped onto the surface plan from −15 kT (deep red) to +15 kT (deep blue). (C) View of the polarity surface representation of TGase-1 cavity around residue Tyr267. The hydrophobic residues (Met, Phe, Leu, Ile, Val, Pro and Ala) are colored yellow, positively charged residues (His, Lys and Arg) blue and negatively charged residues (Tyr, Asp and Glu) red. All other polar residues are colored in violet.
Arg264Trp and Arg264Gln.
Arg264 is located in the core domain (Fig. 4), is exposed to the solvent and hydrogen bonded to Tyr257 and Glu261. His130 stacking over Trp250 from the top and His260 stacking over Trp263 from the bottom surround the residue. Other residues surrounding Arg264 from its side are Val258 and Gln265. The change in energy upon mutation is −1.358 to −1.211 kcal/mol (Arg264Trp) and −0.062 to −0.013 kcal/mol (Arg264Gln).
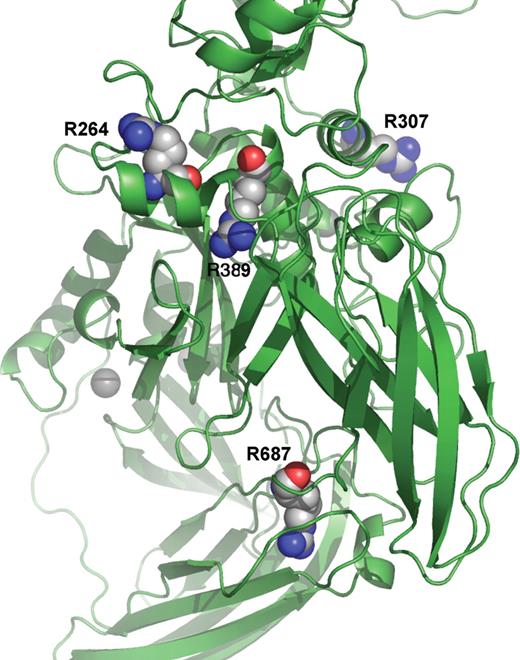
Three-dimensional model of the mutations Arg264Trp/Arg264Gln, Arg307Gly, Arg389Pro and Arg687His. Details of the ribbon image of TGase-1 show the location of the mutations. Arg264 and Arg307 are located in the core domain and both exposed to solvent. Arg389 is also located in the core domain but buried. In contrast, Arg687 is located in the last β-strand of β-barrel 1 leading to β-barrel 2.
Arg307Gly.
Arg307 is located about 9.5 Å apart from the Ca2+ binding site in the core domain. It is hydrogen bonded to Tyr303 and Asn335 and exposed to the solvent. The other surrounding residues are Tyr365 and Met328 from the bottom and Thr368 from the top. The change in energy upon mutation is −1.991 to −2.120 kcal/mol.
Arg389Pro.
Arg389 is located in the core domain, is buried and located within hydrogen distance of Glu266 and Tyr290. The residues Asn270 and Tyr267 surround it. The change in energy upon the mutation is −1.991 to −2.120 kcal/mol.
Arg687His.
Arg687 is located on the last β-strand in β-barrel 1 leading to β-barrel 2. This residue is buried. Arg687 is located within hydrogen distance of Asp587 and Tyr655. The residues Leu594 and Met665 surround it. Change in energy upon the mutation is −1.197 to −1.225 kcal/mol.
TGase-1 antigen and TGase activity
To evaluate the effect of the TGM1 mutations in vivo we performed an immunohistochemical localization of the TGase-1 antigen and a functional in situ test for the TGase-1 enzyme activity. Representative photographs are shown in Figure 5.
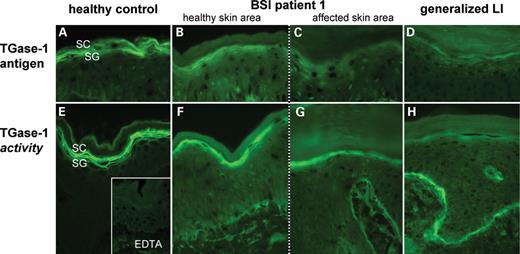
Immunodetection of TGase-1 antigen (A–D) and analysis of TGase(-1) activity (E–H) in healthy controls and BSI patients (only Patient 1 is shown) and generalized LI. (A) Normal skin shows a pericellular TGase-1 antigen of two to three cell layers in the SG, which can also be observed in non-affected BSI skin (B). (C) In contrast, affected BSI skin only shows a faint cytoplasmic staining for TGase-1. (D) For comparison, the example of a patient with generalized ARCI due to TGase-1 deficiency shows an almost complete lack of TGase-1. (E) Using this assay, normal skin shows a pericellular TGase activity of two to three cell layers in the SG, which almost exclusively indicates function of TGase-1. (F) The normal pericellular location of enzyme activity is also present in the healthy BSI skin, but in comparison with normal skin its intensity is reduced. (G) Cryosections of the affected skin only show a cytoplasmic and clearly reduced TGase activity. (F–H) The signal of the basal membrane indicates enzyme activity of TGase-2, which is normally more pronounced in TGase-1 deficient skin. SC: stratum corneum; SG: stratum granulosum; EDTA: negative control for TGase activity.
In healthy BSI skin, the antigen localization revealed a normal TGase-1 staining pattern, which only appeared reduced in its intensity when compared to healthy controls (Fig. 5A and B). In contrast, affected BSI skin displayed a complete lack of the normal pericellular TGase-1 signal in the stratum granulosum (SG). However, a faint diffuse cytoplasmic staining could be observed indicating a reduced and abnormal TGase-1 expression in this skin area (Fig. 5C). For example, the staining pattern of a patient with a generalized form of ARCI with the TGM1 mutation c.877-2A>G/Arg143His likewise showed a faint cytoplasmic signal in the SG (Fig. 5D).
The TGase activity was visualized by the incorporation of biotinyl cadaverine into the CE allowing differentiation between normal, reduced or absent and pericellular (approximately membranous) or cytoplasmatic (approximately cytosolic) TGase activity (33). Enzyme activity in healthy skin corresponds to the TGase-1 antigen mapping and demonstrates a pericellular fluorescence of three to four cell layers in the SG (Fig. 5A and E). Considering TGase-1 as the predominant enzyme isoform in the SG, the assay almost exclusively measures TGase-1 activity, which is reported to be best shown at pH 7.4 (32). The specificity of the assay in the SG is supported by the complete lack of TGase activity in the SG of patients with generalized ARCI due to TGase-1 deficiency. The example shown in Figure 5H refers to the same patient as in Figure 5D. The enhanced fluorescence in the stratum basale (SB) reflects an enhanced signal for TGase-2 activity in the presence of surplus biotinyl cadaverine (33). Healthy BSI skin exhibited a normal zone of pericellular TGase-1 activity although indicating a slightly reduced enzyme function compared to healthy skin. This difference can be semiquantitatively assessed in terms of fluorescence intensity (Fig. 5E and F). In contrast, affected BSI skin appeared intermediate to ARCI with complete loss of TGase-1 activity and showed an abnormal cytoplasmic signal of reduced intensity in the SG and an enhanced signal of TGase-2 in the SB indicating a clear lack of normal membranous TGase-1 activity (Fig. 5G). Experiments have been performed three times each.
Differentiation markers relevant for the CE formation
We wondered about the differential expression and enzyme activity of TGase-1 in affected and healthy BSI skin and therefore evaluated the expression of putative TGase-1 modifier or substrate proteins. An overview of the immunohistologic study performed with healthy and affected BSI skin is given in Table 3.
Staining pattern of transglutaminases and other epidermal markers in three different BSI individuals
| Epidermal markers | BSI individuals (n=3) | |
|---|---|---|
| Healthy skin areas | Affected skin areas | |
| Transglutaminase-1 | Pericellular signal of two to three cell layers in the SG | Very weak and only diffuse signal in the SG |
| Reduced intensity when compared with normal controls | ||
| Transglutaminase-3 | Strong lamellar staining of the overall SC | Reduced signal concentrated in the lower third of the SC |
| More pronounced when compared to normal control | ||
| Transglutaminase-5 | Pericellular signal of nearly all epidermal cell layers (except SC) | No difference of healthy and affected BSI skin |
| Most pronounced in the SG | ||
| Cathepsin D | Strong cytoplasmic signal of one to two cell layers in the upper SG and lower SC | Marked reduction and partial absence of the normal signal |
| Involucrin | Granular signal of approximately two cell layers in the SG like normal skin | More diffuse signal ‘shifted to the SC’ |
| Loricrin | Linear pericellular signal of approximately two cell layers in the upper SG | Slightly broadened signal also present in the SC |
| Annexin 1 | Pericellular signal throughout all layers except SC and most pronounced in the SB | No difference of healthy and affected BSI skin |
| Reduced intensity when compared with normal skin | ||
| Annexin 2 | Pericellular signal throughout all layers except SC | No difference of healthy and affected BSI skin |
| Less regular when compared with normal skin | ||
| Elafin | Very slight or negative signal in the SC | Strong positive lamellar signal in the SC |
| Like normal skin | ||
| Human β-Defensin-2 | Negative | Granular signal in the SC |
| Like normal skin | ||
| PAI-2 | Pericellular signal of two to three cell layers in the SG | Positive but only diffuse signal in the SG |
| Reduced when compared to the controls | ||
| Epidermal markers | BSI individuals (n=3) | |
|---|---|---|
| Healthy skin areas | Affected skin areas | |
| Transglutaminase-1 | Pericellular signal of two to three cell layers in the SG | Very weak and only diffuse signal in the SG |
| Reduced intensity when compared with normal controls | ||
| Transglutaminase-3 | Strong lamellar staining of the overall SC | Reduced signal concentrated in the lower third of the SC |
| More pronounced when compared to normal control | ||
| Transglutaminase-5 | Pericellular signal of nearly all epidermal cell layers (except SC) | No difference of healthy and affected BSI skin |
| Most pronounced in the SG | ||
| Cathepsin D | Strong cytoplasmic signal of one to two cell layers in the upper SG and lower SC | Marked reduction and partial absence of the normal signal |
| Involucrin | Granular signal of approximately two cell layers in the SG like normal skin | More diffuse signal ‘shifted to the SC’ |
| Loricrin | Linear pericellular signal of approximately two cell layers in the upper SG | Slightly broadened signal also present in the SC |
| Annexin 1 | Pericellular signal throughout all layers except SC and most pronounced in the SB | No difference of healthy and affected BSI skin |
| Reduced intensity when compared with normal skin | ||
| Annexin 2 | Pericellular signal throughout all layers except SC | No difference of healthy and affected BSI skin |
| Less regular when compared with normal skin | ||
| Elafin | Very slight or negative signal in the SC | Strong positive lamellar signal in the SC |
| Like normal skin | ||
| Human β-Defensin-2 | Negative | Granular signal in the SC |
| Like normal skin | ||
| PAI-2 | Pericellular signal of two to three cell layers in the SG | Positive but only diffuse signal in the SG |
| Reduced when compared to the controls | ||
SB, stratum basale; SG, stratum granulosum; SC, stratum corneum.
Staining pattern of transglutaminases and other epidermal markers in three different BSI individuals
| Epidermal markers | BSI individuals (n=3) | |
|---|---|---|
| Healthy skin areas | Affected skin areas | |
| Transglutaminase-1 | Pericellular signal of two to three cell layers in the SG | Very weak and only diffuse signal in the SG |
| Reduced intensity when compared with normal controls | ||
| Transglutaminase-3 | Strong lamellar staining of the overall SC | Reduced signal concentrated in the lower third of the SC |
| More pronounced when compared to normal control | ||
| Transglutaminase-5 | Pericellular signal of nearly all epidermal cell layers (except SC) | No difference of healthy and affected BSI skin |
| Most pronounced in the SG | ||
| Cathepsin D | Strong cytoplasmic signal of one to two cell layers in the upper SG and lower SC | Marked reduction and partial absence of the normal signal |
| Involucrin | Granular signal of approximately two cell layers in the SG like normal skin | More diffuse signal ‘shifted to the SC’ |
| Loricrin | Linear pericellular signal of approximately two cell layers in the upper SG | Slightly broadened signal also present in the SC |
| Annexin 1 | Pericellular signal throughout all layers except SC and most pronounced in the SB | No difference of healthy and affected BSI skin |
| Reduced intensity when compared with normal skin | ||
| Annexin 2 | Pericellular signal throughout all layers except SC | No difference of healthy and affected BSI skin |
| Less regular when compared with normal skin | ||
| Elafin | Very slight or negative signal in the SC | Strong positive lamellar signal in the SC |
| Like normal skin | ||
| Human β-Defensin-2 | Negative | Granular signal in the SC |
| Like normal skin | ||
| PAI-2 | Pericellular signal of two to three cell layers in the SG | Positive but only diffuse signal in the SG |
| Reduced when compared to the controls | ||
| Epidermal markers | BSI individuals (n=3) | |
|---|---|---|
| Healthy skin areas | Affected skin areas | |
| Transglutaminase-1 | Pericellular signal of two to three cell layers in the SG | Very weak and only diffuse signal in the SG |
| Reduced intensity when compared with normal controls | ||
| Transglutaminase-3 | Strong lamellar staining of the overall SC | Reduced signal concentrated in the lower third of the SC |
| More pronounced when compared to normal control | ||
| Transglutaminase-5 | Pericellular signal of nearly all epidermal cell layers (except SC) | No difference of healthy and affected BSI skin |
| Most pronounced in the SG | ||
| Cathepsin D | Strong cytoplasmic signal of one to two cell layers in the upper SG and lower SC | Marked reduction and partial absence of the normal signal |
| Involucrin | Granular signal of approximately two cell layers in the SG like normal skin | More diffuse signal ‘shifted to the SC’ |
| Loricrin | Linear pericellular signal of approximately two cell layers in the upper SG | Slightly broadened signal also present in the SC |
| Annexin 1 | Pericellular signal throughout all layers except SC and most pronounced in the SB | No difference of healthy and affected BSI skin |
| Reduced intensity when compared with normal skin | ||
| Annexin 2 | Pericellular signal throughout all layers except SC | No difference of healthy and affected BSI skin |
| Less regular when compared with normal skin | ||
| Elafin | Very slight or negative signal in the SC | Strong positive lamellar signal in the SC |
| Like normal skin | ||
| Human β-Defensin-2 | Negative | Granular signal in the SC |
| Like normal skin | ||
| PAI-2 | Pericellular signal of two to three cell layers in the SG | Positive but only diffuse signal in the SG |
| Reduced when compared to the controls | ||
SB, stratum basale; SG, stratum granulosum; SC, stratum corneum.
The staining of TGase-3 was similar to normal skin, but appeared more pronounced in healthy BSI skin and reduced in the affected one suggesting a slightly different TGase-3 expression in BSI (Fig. 6A–E). The TGase-5 signal was present in all skin sections showing a normal pericellular signal in the overall epidermis except the SC (Fig. 6F–J). In contrast, cathepsin D only displayed a weak signal in the SG of affected BSI skin, when compared to normal and healthy BSI skin (Fig. 6K–O). In affected BSI skin, components of the CE such as involucrin, loricrin and PAI-2 (34,35) revealed a localization typically observed in TGase-1 deficient epidermis, e.g. the CE precursor proteins appeared to be ‘shifted to the SC’. No differences to normal controls were observed for annexin 1 and 2. Interestingly, elafin, a CE precursor protein with low expression in healthy skin (36) showed a strong lamellar signal in the SC of affected BSI skin, whereas healthy BSI skin was similar to normal controls almost negative. We also looked for the presence of human β-defensin-2, which is normally—like elafin—only associated with inflammatory skin lesions such as psoriasis (37). No immunostaining was achieved in healthy BSI skin, but affected BSI skin demonstrated a granular signal in the SC (not shown).
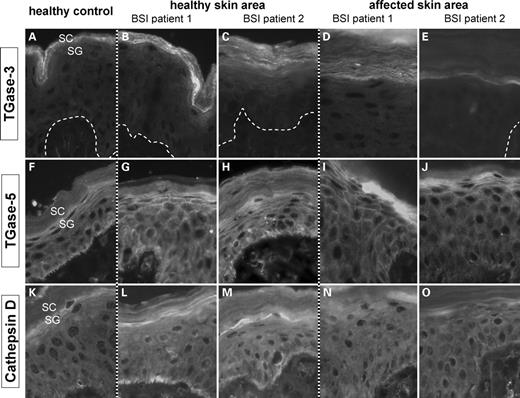
Immunodetection of TGase-3 (A–E), TGase-5 (F–J) and cathepsin D (K–O) in healthy and affected skin areas of BSI patients compared to normal controls. (A) In healthy controls, TGase-3 shows a lamellar signal predominantly in the lower third of the stratum corneum. (B,C) Healthy skin of BSI Patient 1 and 2 displays a pronounced and clearly broadened signal of the overall SC. (D,E) Interestingly, the TGase-3 expression is diminished in the affected BSI area, in particular visible in Patient 2. (F–J) Staining for TGase-5 antigen resulted in a linear pericellular staining of the overall SG and of upper parts of the SP. No obvious differences were observed between healthy controls and BSI skin sections. (K–O) Cathepsin D shows a cytoplasmic signal in the SG and at the basal site of basal keratinocytes in healthy controls as well as in healthy BSI skin. It is of note, that affected BSI skin exhibits a marked reduction of Cathepsin D in the SG. SC: stratum corneum; SP: stratum spinosum; SG: stratum granulosum.
Digital thermal imaging
The study of TGase-1 activity as well as the immunohistologic characterization of affected and unaffected BSI skin highlighted a complex alteration of the normal terminal differentiation. However, this observation does not answer the question why both clinical disease and biochemical defect manifest only on the trunk, although the mutant protein must be present in the entire skin. We suspected an environmental influence, e.g. moisture content, pH value or temperature, and therefore decided to analyze more profoundly the peculiar clinical features in BSI patients. Patients 1, 2, 3, 6 and 10 showed a distinct scaling of the elbow fossae and popliteal space, a fine linear scaling on the inner forearm and a very pronounced scaling in the axillae, on the neck and on medial parts of the trunk sparing the suprarenal lumbal areas (Figs 1 and 7C). To us, this clinical re-analysis strongly suggested a possible temperature effect on the phenotype.
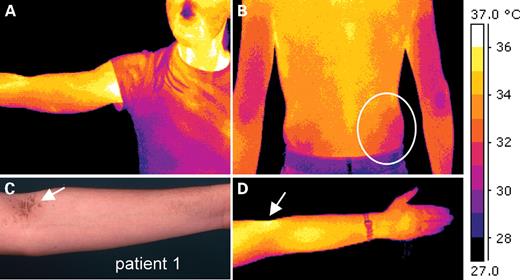
Physiological skin temperature pattern as demonstrated by digital thermal imaging of healthy individuals (n=4). (A) The pseudocolor image of a healthy female volunteer demonstrates well-known temperature zones of arms, elbows, axillae and on the face. (B and D) These temperature images of healthy volunteers were taken after 10 min of acclimatization (RT=22.4°C). (C) The symmetric temperature differences exhibit a striking correlation to the distribution of lamellar scaling as seen, for example, in Patient 1 (compare with Fig. 1) 3, 6 or 10. Regarding the skin temperature zones, one has to conclude that skin areas of more than 33–34°C obviously predispose to the local development of ichthyotic skin.
To evaluate the ‘normal temperature’ of the skin and especially its local differences we performed digital infrared thermal imaging (38) with four different healthy volunteers (Fig. 7). Body areas were imaged at fixed time points after disrobing and revealed a striking and well known temperature pattern. Comparison of the particular distribution of lamellar scaling and skin temperature revealed a striking correlation. The mean temperature of warmer body sites, which show a clear-cut scaling in BSI, was greater than 33–34°C. In contrast, colder areas appeared normal. Lamellar scaling, most probably leads to a different absorption index of the skin (ε), which would cause a confounding effect. Therefore, we did not directly take infrared photographs of BSI individuals.
Functional in situ TGase-1 testing under different temperature conditions
The healthy BSI skin of Patient 1 with the homozygous mutation Tyr276Asn offered the possibility for an in vivo study of its enzyme activity under different temperatures. The functional TGase test was therefore performed at 25°C and 37°C at pH 7.4 (Fig. 8) and at pH 8.4 (not shown). First of all, normal controls (n=5) did not show a reduction of TGase activity at higher temperature, neither at pH 7.4 nor at pH 8.4. In contrast, healthy BSI skin assayed at 37°C demonstrated a reduced TGase activity in the SG at pH 7.4 (Fig. 8B versus D). When the TGase test was performed at pH 8.4 and 25°C, an additional activity signal is observed in the SC, which in BSI skin disappears at 37°C. It is of note that changes relating to the assay temperature were not observed in skin sections of four different TGase-1 deficient skin sections of generalized ARCI, which in two cases also showed a residual TGase signal in the SC at pH 8.4 (not shown). The experiments performed with healthy BSI skin of Patient 2 and 10 likewise showed a reduction of TGase-1 activity under higher temperature, even though this difference appeared less pronounced (not shown). For Patient 1 other experiments were performed at 33°C. Interestingly, the most relevant reduction of the pericellular fluorescence appeared between 33 and 37°C (not shown).
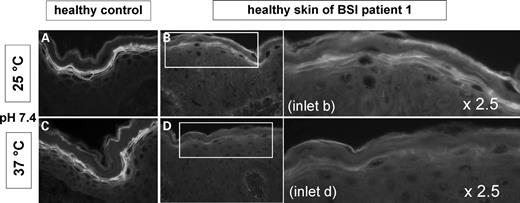
TGase activity test at pH 7.4: 25°C (A, B) versus 37°C (C, D). For control, images on the left side show skin sections of normal skin. Images on the right side including their 2.5× magnification demonstrate TGase activity of the healthy skin of BSI Patient 1. (A, C) Normal skin shows a strong pericellular TGase activity zone in the SG at 25°C as well as at 37°C. (B, D) However, BSI skin exhibits a marked reduction of the fluorescence at 37°C when compared with 25°C indicating a loss of TGase-1 activity under higher temperature in BSI.
DISCUSSION
BSI is a striking and unique clinical phenotype that has been observed in black people from South Africa. It is characterized by a collodion membrane at birth encasing the entire skin. The clinical healing of the ichthyosis on arms and legs occurs during the first weeks of life. In the present article, we show that this remarkable type of ARCI can also be found in patients from Europe and the Mediterranean area, and by ultrastructural, enzymatic and molecular genetic studies, we show that BSI is caused by TGase-1 deficiency.
Sequencing analyses confirmed TGM1 mutations in BSI individuals: We have ascertained a group of 10 BSI individuals from independent families. Altogether, we identified 13 mutations in TGM1—10 different missense, two nonsense mutations and one splice-site mutation (Table 1). The mutations Arg126Cys, Arg264Trp, Arg264Gln, Tyr276Asn, Arg307Gly, Arg315His, Ser358fsX26 and Arg687His are here reported for the first time (Table 2). Three index patients showed a homozygous genotype, the others were compound heterozygous. Regarding the genotypes of the patients in more detail, it is of note that there are particular residues affected more than once within this BSI group: Arg307Gly is present in three, Arg264Trp or Arg264Gln in two, and Arg315Cys or Arg315His also in two independent patients. Tyr276Asn is only observed in Patient 1 but located close to the mutations at Arg264. The residues affected by a missense mutation are all located within the first part of the TGase-1 core domain (Fig. 9). As an exception, Arg126Cys reported in Patient 2 is located in the β-sandwich domain, and Arg687His only reported in Patient 7 affects a residue exactly between β-barrel 1 and β-barrel 2. The A–G mutation in the splice acceptor of intron 5 (c.877-2A>G) has been reported to result in two different mRNA transcripts—one retaining intron 5 and the other one only leading to a G nucleotide insertion between exon 5 and 6. Both transcripts lead to a premature termination codon (22–24). Their translation products as well as the translation products of the mutations Ser358fsX26 and Trp263X are unlikely to be functional (22–26,31). We suggest that these nonsense mutations predispose for the generalized (and severe) type of ARCI due to TGase-1 deficiency. This association may also account for missense mutations such as Arg142His and Arg389Pro (Fig. 9). Regarding the almost normal TGase activity in healthy BSI skin, we conclude that the missense mutations—being present on at least one allele in all BSI patients—produce a functional active protein (Table 2). For instance, Patient 6 shows the nonsense mutation Ser358fsX26 coding for a non-functional TGase-1 protein and the missense mutation Arg264Gln, which apparently codes an enzyme with sufficient residual activity as far as the healthy skin areas are concerned (Table 1).
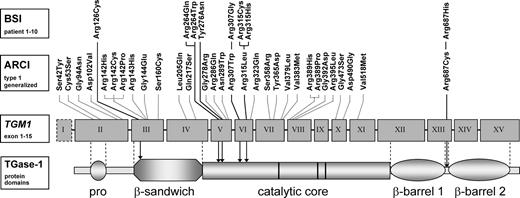
Overview of all missense mutations so far described in TGase-1 deficiency. The first line (top) shows all missense mutations associated with BSI. The other missense mutations (second line) have been described in patients with generalized ARCI due to TGase-1 deficiency. Most BSI mutations are located in exon 5 and 6 leading to an amino acid change in the first part of the catalytic core domain. As an exception, Arg126Cys (exon 3) affects a residue at the beginning of the β-sandwich domain and Arg687His (exon 13) a residue exactly between β-barrel 1 and 2. Nonsense and splice-site mutations are not included in this figure. BSI: bathing suit ichthyosis; ARCI: autosomal recessive congenital ichthyosis; TGase-1: transglutaminase-1.
Regarding the particular skin phenotype of BSI it is important to note that a genetic mosaicism caused by TGM1 mutations of post-zygotic origin can be clearly ruled out. Generalized ichthyosis at birth together with an autosomal recessive trait and the present distribution pattern of affected and healthy skin do not fit to the concept of cutaneous mosaicism (39). From the clinical point of view, BSI is reminiscent of the self-healing collodion baby (SHCB), in which, likewise, a specific TGM1 mutation has been identified in one family (19). In the past, single cases of LI have been reported, which in retrospect have to be categorized as having mild BSI. The first report concerns a patient with the heterozygous mutation Val382Met (40), two other patients with LI ‘mainly on the trunk’ were described for the heterozygous mutation Arg307Trp (27,28), whereas the heterozygous mutation Arg307Gly was found in Patients 4, 5 and 9 reported here.
Three-dimensional modeling suggested that particular BSI mutations do not result in a major error of TGase-1 structure: The crystal structure of TGase-3 has been used as a homology model to generate a structure for the TGase-1 enzyme (41). Modeling of various mutations showed that the first homozygous mutation in Patient 1 affects a residue (Tyr276) on the β-strand on the interface between the core domain and β-barrel 2, where it is buried in the hydrophobic interior of the enzyme. This mutation can have the following consequences (1): Tyr276Asn does not appear to introduce any structural modifications or errors in protein folding. A lack of activity in vivo could possibly involve protein–protein interactions, solvent molecules and the pH of the environment of Tyr276. Because of the high pKa of the tyrosine side chain (pKa=∼10) it usually does not ionize at physiological pH. But the local environment can lower its pKa, and then it could participate in acid–base reactions and phosphorylation (2): The propensities for the Tyr residue are much greater than for the Asn residue, and it is suggested that His, Tyr, Phe residues make a particularly good ‘glue’ for sticking protein subunits together. Hence, Tyr276Asn could possibly diminish the interface between TGase-1 protein subunits such as the core domain and β-barrel 2. Furthermore, when Tyr276 is mutated, complementarities (electrostatic interactions and shape) are perturbed. Generally, electrostatic interactions are highly dependent on the pH, salt, concentration, and dielectric constant of the medium (3): Moreover, it is important to emphasize that among all the known crystal structures of TGases, only TGase-2 and TGase-3 have been shown to bind and hydrolyze GTP (42). Homology modeling suggested that TGase-1 as well as TGase-5 and TGase-7 may also be able to bind to GTP/GDP (43). In fact, Tyr276 is located five residues away from the nucleotide-binding pocket, which could possibly affect the hydrogen bonding of GTP.
The mutations Arg307Gly, Arg264Trp and Arg264Gln—similar to Tyr276Asn—are not likely to introduce any structural modifications or errors in protein folding, and they are also exposed to the solvent. Very surprisingly, there is at least one tyrosine close to each of the mutated residues. One might speculate that these tyrosine residues are susceptible to phosphorylation. Hence, a different pH of the environment of the residues would have an influence on this reaction. Consequently, the mutation Tyr276Asn or mutations near a putative phosphorylation site would affect the TGase-1 activity. However, hitherto phosphorylation sites of TGase-1 have not been experimentally shown.
There is a strong correlation of phenotype, ultrastructure and TGase activity: A major finding of our investigations is that TGase-1 is differentially expressed in healthy and affected BSI skin. The in situ monitoring of the TGase activity in the healthy skin area revealed an almost normal membranous TGase activity. Affected skin only showed a residual cytoplasmic enzyme activity. Very remarkably, the ultrastructure of the epidermis including the SC in the healthy skin area did not show any abnormalities, in contrast to the affected skin, which revealed typical morphological signs of TGase-1 deficiency suggesting that BSI can be regarded a particular subtype of ARCI.
Possible modifier proteins and TGase-1 substrates show a differential expression pattern in affected and healthy BSI skin: The observation of an almost normal TGase-1 antigen and activity in the healthy skin area (Fig. 5) supported the in silico result. Interestingly, we observed a difference of TGase-3 expression between healthy and affected BSI skin, where the TGase-3 signal was still present, but not as strong as in the healthy BSI skin. TGase-3 might be upregulated in a situation of mild TGase-1 deficiency, but surely it cannot replace the essential cross-linking performed by the absent or strongly reduced TGase-1 enzyme. Somewhat surprising elafin and human β-Defensin 2, which are normally only expressed in the context of inflammation (36,37), showed a clear-cut signal in the SC of the affected skin. This upregulation might reflect a compensative reaction of the innate immune response taking into account the impaired epidermal barrier of ichthyotic skin (15). Upon the terminal differentiation of keratinocytes, TGase-1 is proteolytically cleaved at two sites, which leads to an up to 10-fold increase in specific activity (44). Cathepsin D, an aspartatic protease involved in the degradation of intracellular proteins, has an important regulatory effect on the TGase-1 activation as has been demonstrated in mouse epidermis (45). Interestingly, this protease showed a reduced expression in the SG of TGase-1-deficient BSI when compared with healthy skin. Other differentiation markers and TGase-1 substrates such as involucrin, loricrin and PAI-2 revealed a staining pattern in accordance with a reduced TGase-1 function (34). Summarizing the results of the immunohistologic investigations (Table 3), we favor the idea that most of the observed changes are due to an altered proliferation and differentiation program secondary to TGase-1 deficiency, but it cannot be ruled out that interactions with cathepsin D have a primary influence on the differential loss of TGase-1 function in BSI.
Bathing suit ichthyosis: a temperature-sensitive phenotype. Looking at the differential TGase activity and altered differentiation of healthy and affected BSI skin, we assumed that an external factor in the presence of the observed TGM1 mutations predisposes to the particular ichthyosis. Therefore, we performed a thorough re-analysis of the clinical phenotype. To validate the hypothesis that warmer skin temperature has a primary influence on the development of local scaling, digital thermography was performed in healthy individuals, all of whom showed the typical symmetric distribution of the physiologic skin temperature pattern. Our infrared photographs (Fig. 7) are in line with those of similar studies (46). Comparing the distribution pattern of the ichthyosis it became obvious that there is a striking correlation between warmer body sites and scaling—especially in Patient 1, 2, 3, 6 and 10. This was even true for small areas with a physiological temperature of >33°C such as the skin above the superficially localized popliteal artery and vein (not shown). Another characteristic clinical sign was the circumscribed suprarenal healthy skin area, which is normally ∼2–3° colder than the surrounding area. We concluded that skin temperatures above ∼33°C predispose to the development of ichthyosis thus causing a lamellar scaling on the bathing suit areas.
Biochemical evidence that the enzyme function of the mutant Tyr276Asn is inhibited by increased temperature: As Patient 1 harbored a homozygous mutation, analysis of his healthy skin, where a normal TGase-1 antigen and activity was present, allowed a direct test of the mutant Tyr264Asn. Indeed, the TGase activity test performed with an increased temperature of 37°C at pH 7.4 or 8.4 revealed a striking reduction of enzyme activity. The reduced pericellular activity in the SG strongly suggests a temperature sensitivity of the mutant TGase-1 (Fig. 8). Same experiments were performed with 33 versus 37°C, likewise demonstrating clear differences of TGase activity. In contrast, healthy skin or skin sections of ARCI patients with generalized TGase-1 deficiency did not show any influence of the temperature on TGase activity, neither at pH 7.4 nor at pH 8.4.
The Tyr276Asn modeling and in vivo testing provide first biochemical evidence for the notion that particular BSI mutations affect the TGase-1 function depending on temperature. The results are strongly supported by the clinical observation that local skin temperature predisposes to the ichthyosis explaining the particular BSI phenotype. Of course, temperature need not necessarily have a direct effect on the TGase-1 protein, e.g. through a protein destabilization. The influence of temperature on the epidermal homeostasis in vivo is complex (47) and most probably causes a considerable change of the micromilieu subjacent to the cell membrane, e.g. a change of the pH or activity of proteolytic enzymes. Recent experiments demonstrated a loss of the proteolytic processing of the mutant TGase-1 Arg315Leu (30). A similar effect with temperature sensitivity could explain the BSI phenotype caused by the mutations Arg315Cys and Arg315His found in our patients.
We conclude that BSI also occurs in European and Mediterranean populations, is caused by particular mutations in TGM1, and from a clinical point of view is a temperature-sensitive phenotype. Our biochemical in situ investigations exemplarily done for the mutant Tyr276Asn assert the notion that the mutations found in BSI may directly or indirectly render the enzyme sensitive to differences in body temperature.
MATERIALS AND METHODS
Subjects and samples
The study was approved by the institutional review board of the University Hospital of Münster, all patients enrolled gave their informed consent. Clinical data were recorded by five dermatologists (V.O., H.T., J.M.H., P.M.S. and W.K.). A detailed medical and dermatological history was obtained from all affected persons. EDTA blood was collected of all patients and their parents. Punch biopsies (4 mm) stored at −80°C were taken from eight BSI patients (from the affected skin), 10 patients with generalized ARCI caused by TGM1 mutations and 10 healthy control individuals. Healthy skin areas of BSI patients were additionally examined in four of them.
Ultrastructural analysis
All specimens were fixed for at least 2 h at room temperature in 3% glutaraldehyde solution in 0.1 cacodylate buffer pH 7.4, cut into pieces of ca. 1 mm3, washed in buffer, post-fixed for 1 h at 4°C in 1% osmium tetroxide, rinsed in water, dehydrated through graded ethanol solutions, transferred in propylene oxide, and embedded in epoxy resin (glycidether 100). Semi-thin and ultra-thin sections were cut with an ultramicrotome (Reichert Ultracut E). Semi-thin sections were stained with methylene blue. Ultra-thin sections were treated with uranyl acetate and lead citrate, and examined with an electron microscope (Philips EM 400).
Genetic analysis
DNA was extracted from peripheral blood leukocytes using standard procedures. The translated exons 2–15 of TGM1 including the exon–intron boundaries were amplified by polymerase chain reaction (PCR) using intronic primers designed according to the genomic sequence of TGM1 (NC_000014.7). Reactions were performed with 0.5 µm of each primer, 0.1 mm dNTPs and 0.4 U Taq DNA polymerase in 30 cycles of 10 s at 94°C, 10 s at annealing temperature depending on the respective primer pair and 10 s at 72°C as described earlier (32). All mutations found in the index patients were confirmed in their parents. Mutations were not seen in at least 100 chromosomes from control persons.
Structural modeling
Crystal structures from various sources having sequence identity of more than 30% [i.e. TGase 2 (PDB: 1kv3) and TGase 3 (PDB: 1L9 N)], and the model of TGase 5 (data not shown) were chosen to build the TGase-1 model structure. Comparative protein modeling was performed with the Composer module of Sybyl 6.9 (Tripos Assoc. Inc., St Louis, MO, USA). Energy minimizations and molecular dynamics were accomplished in the Discover module of InsightII 2000 (Accelrys Inc., San Diego, CA, USA). The geometrical and local environmental consistencies of the models were evaluated with the ProStat and Profiles-3D (48) modules of InsightII 2000 and the MatchMaker module of Sybyl 6.9. Coordinates for the structurally conserved regions were assigned from the TGase-3 template molecules. The coordinates of structurally variable regions SVRs were obtained using the conformational search program GeneFold and MatchMaker modules. The SVR loop regions were built using the protein loop search protocol (49) as available in Composer. The model obtained was then refined by minimization using a similar protocol as described earlier (50). The TGase-1 model has been subjected to energy minimization refinement to eliminate any steric conflicts. The Ramachandran plot revealed a normal distribution the residues occupying the allowed region indicating that no major distortion was introduced to eliminate steric conflicts.
Histochemical TGase-1 assay
The in situ TGase activity test was performed as described previously (33,34). In brief, unfixed cryosections of 3–4 µm were blocked with 1% BSA in PBS for 30 min and directly incubated with 0.1 mm TGase substrate biotinyl cadaverine (Molecular Probes, Leiden, The Netherlands), which is incorporated into the CE in the presence of calcium ions (5 mm). At physiological pH 7.4 (100 mm Tris–HCl) this reaction is almost exclusively performed by TGase-1 (reaction time 90 min). Incubation with 5 mm EDTA served as negative control. The incorporated biotinylated substrate was incubated with fluorochrome-coupled streptavidin 1:100 (Jackson ImmunoResearch Laboratories Inc., West Grove, PA, USA). Slides were viewed with an Axioscope 2 and digital images taken using an Axiocam HR video camera and Axiovision 3.0 software (all Carl Zeiss, Jena, Germany).
To test for temperature-resistance, TGase activity test was performed under different temperatures (25, 33 and 37°C) and the pH was set to 7.4 or 8.4. The experiment was repeated three times under the same conditions. The respective slides were evaluated by different observers.
Immunofluorescence study
Indirect immunofluorescence staining was performed on acetone fixed (−20°C for 10 min) or unfixed cryosections. The following primary antibodies and dilutions were used: Mouse MAb BT-621 (clone B.C1) against human keratinocyte TGase-1, 1:40 (Biomedical Technologies, Stoughton, MA, USA), polyclonal rabbit anti-TGase-3, 1:3200 (Immundiagnostik, Bensheim, Germany), polyclonal rabbit anti-TGase-5, 1:200 (N-Zyme Biotec GmbH, Darmstadt, Germany), rabbit anti-involucrin, 1:200 (CellSystems Biotechnologie Vertrieb GmbH, St Katharinen, Germany), mouse monoclonal anti-cathepsin D, 1:100 (ab6313, Abcam, Cambridge, UK), rabbit anti- loricrin, 1:2500 (PRB-145P, BabCo, Berkeley, CA, USA), rabbit anti-elafin, 1:2000, and rabbit anti-human β-Defensin-2, 1:2000 (both: Peptide Institute Inc., Minoh-shi Osaka, Japan), rabbit anti-annexin I, 1:200, and mouse-anti annexin II, 1:400 (both: Zymed, San Francisco, USA), and monoclonal mouse anti-PAI-2, 1:200 (#3750, American Diagnostica, Greenwich, CT, USA). After 30 min blocking with 10% normal goat serum, primary antibodies were incubated for 16–20 h at RT. Rabbit-antibodies were detected with FITC–goat anti-rabbit IgG, 1:200, and mouse-antibodies with FITC–goat anti-mouse IgG, 1:100 (both: Jackson ImmunoResearch Laboratories).
Digital infrared imaging
Infrared images were taken by an experienced technician (Thomas Maue, Handwerkskammer Bildungszentrum Münster, Germany) using a Thermovision 550 system (Agema, Sweden). Subjects (n=4) were asked to disrobe the body areas to be imaged and stand up for 5 min to allow equilibration with an ambient temperature (22.4°C). Photographs were taken at 10 and 15 min with a camera distance of 3 m in a daylight-free room at a humidity of 30%. The absorption index of the skin (ε) taken for calibration of the pseudocolor temperature scale was 0.97.
Online Mendelian inheritance in man
Lamellar ichthyosis (LI; MIM 242300); lamellar ichthyosis 2 (LI2; MIM 601277); lamellar ichthyosis 3 (LI3; MIM 604777); lamellar ichthyosis 5 (LI5; MIM 606545); lamellar ichthyosis type 6 (LI6; 609383); Harlequin ichthyosis (HI; MIM 607800); transglutaminase-1 (TGM1; MIM 190195) (http://www.ncbi.nlm.nih.gov/Omim).
Other databases
http://www.genome.ucsc.edu/, http://www.expasy.uniprot.org/index.shtml.
ACKNOWLEDGEMENTS
We are grateful to all the patients and other probands who participated in the study. We thank Mrs Bückmann, Mr Wissel and Mr Thomas for the help with the photographs. Our work is supported by the Selbsthilfe Ichthysose e.V., Deutsche Forschungsgemeinschaft (Tr 228/6-2), GENESKIN European coordination action and by the Bundesministerium für Bildung und Forschung as part of the Network for rare diseases NIRK (GFGM01143901). The expert technical assistance of Dr T. Maue, Cordula Focke, Andrea Wissel and Marc Nätebus is gratefully acknowledged as well as the fruitful discussion with Dr Markus Höwel. Special thanks to Dr Eva-Maria Schnäker and Christian Gorzelanny, Brigitte Willis and to all other persons including our families who have very much supported this work.
Conflict of Interest statement. None declared.
REFERENCES
Author notes
Deceased July, 24 2006.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}