





Abstract
Paraquat, MPTP, and rotenone reproduce features of Parkinson's disease (PD) in experimental animals. The exact mechanisms by which these compounds damage the dopamine system are not firmly established, but selective damage to dopamine neurons and inhibition of complex I are thought to be involved. We and others have previously documented that the toxic metabolite of MPTP, MPP+, is transported into dopamine neurons through the dopamine transporter (DAT), while rotenone is not transported by DAT. We have also demonstrated the requirement for complex I inhibition and oxidative damage in the dopaminergic neurodegeneration produced by rotenone. Based on structural similarity to MPP+, it has been proposed that paraquat exerts selective dopaminergic toxicity through transport by the DAT and subsequent inhibition of mitochondrial complex I. In this study we report that paraquat is neither a substrate nor inhibitor of DAT. We also demonstrate that in vivo exposure to MPTP and rotenone, but not paraquat, inhibits binding of 3H-dihydrorotenone to complex I in brain mitochondria. Rotenone and MPP+ were both effective inhibitors of complex I activity in isolated brain mitochondria, while paraquat exhibited weak inhibitory effects only at millimolar concentrations. These data indicate that, despite the apparent structural similarity to MPP+, paraquat exerts its deleterious effects on dopamine neurons in a manner that is unique from rotenone and MPTP.
Parkinson's disease (PD) is a disabling neurodegenerative disorder characterized by the loss of nigrostriatal dopamine neurons. While genetically linked cases of PD exist, these are rare in comparison to late-onset sporadic PD (Sherer et al., 2001; Vila and Przedborski, 2004). Additionally, data from a large twin study found no significant contribution of genetics to sporadic PD (Tanner et al., 1999), suggesting that environmental factors or gene–environment interactions are major contributors to sporadic PD.
The role of environmental factors in PD was first substantiated with the discovery of N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which caused an acute and permanent parkinsonian syndrome in intravenous drug users (Langston et al., 1983). MPTP is selectively toxic to dopamine neurons through conversion to MPP+ and active transport by the dopamine transporter (DAT) (Chiba et al., 1985; Gainetdinov et al., 1997; Javitch et al., 1985). Early studies reported that MPP+ inhibited complex I of the mitochondrial electron transport chain, which was thought to be the mechanism by which MPTP induced parkinsonism (Nicklas et al., 1985). However, the weak inhibitory ability of MPP+ at complex I (Ramsay et al., 1991) has led some to question the role of complex I inhibition in MPTP toxicity (Lotharius and O'Malley, 2000; Nakamura et al., 2000). The recent development of the rotenone model of PD (Betarbet et al., 2000) has now provided evidence that systemic inhibition of complex I may contribute to PD pathogenesis. Additionally, reduction of complex I activity in several tissues of PD patients (Mizuno et al., 1989; Parker et al., 1989; Schapira et al., 1989) has been reported, providing further evidence for a role of complex I inhibition in PD.
Despite their use as animal models of PD, neither rotenone nor MPTP is likely to contribute significantly to the occurrence of PD in the general population. Rotenone has limited uses, poor oral bioavailability, and a short half-life in the environment, while MPTP is not found in the environment. Thus, the identification of environmental agents that may be involved in PD remains a challenge to the research community. Several epidemiological studies have identified pesticides and in particular, the herbicide paraquat as a risk factor for PD (Hertzman et al., 1990; Liou et al., 1997; Semchuk et al., 1992). In animal studies, repeated exposure of mice to paraquat has also been shown to cause a loss of dopamine neurons and aggregation of α-synuclein, two key aspects of the parkinsonian state (Brooks et al., 1999; McCormack et al., 2002). Based upon structural similarity with MPP+, paraquat (1, 1′-dimethyl-4,4′-bipyridinium dichloride) is commonly believed to act through a similar mechanism of action. There has been speculation that paraquat may be actively transported into dopaminergic neurons by DAT (Shimizu et al., 2001, 2003) and inhibit complex I (Fukushima et al., 1994; Tawara et al., 1996). However, the role of the DAT and complex I in paraquat neurotoxicity remains to be established. Given the lack of knowledge as to the mechanism of action of paraquat and its increasing use as an animal model of parkinsonism, the goal of this study was to determine the role of the DAT and complex I in the toxicity of paraquat.
MATERIALS AND METHODS
Construction of inducible iDAT-GFP cell line and general cell culture.
SK-N-MC human neuroblastoma cells (ATCC, Manassas, VA) were maintained at 37°C in minimum essential media and supplemented with 10% fetal bovine serum, 50 U/ml penicillin, 50 μg/ml streptomycin, 2 mM L-glutamine, and 1 mM sodium pyruvate and nonessential amino acids. For generation of the inducible stable cell line, human DAT cDNA (Miller et al., 1997) was inserted into pEGFP-N2 (Clonetech, Palo Alto, CA), in which the C-terminus of DAT was tagged with green fluorescence protein (GFP). The full-length fragment of hDAT-GFP was digested and inserted into the ecdysone-inducible vector (pIND) to generate the plasmid pIND-hDAT-GFP. The ecdysone-inducible mammalian expression system (No et al., 1996) was then used to control expression of DAT in this cell line. Briefly, pIND-hDAT-GFP and the plasmid, pVgRXR (InVitrogen, Carlsbad, CA), which is required for inducible expression, were cotransfected in SK-N-MC cells using Lipofectamine. Forty-eight h post-transfection, cells were plated at 1 × 106 cells per 10-cm culture dish in growth medium containing G418 and Zeocin (InVitrogen, Carlsbad, CA) for selection. Resistant colonies were selected and screened by dopamine uptake (Pifl et al., 1993), Western blotting (Miller et al., 1997), and confocal microscopy. For confocal microscopy, cells were grown to poly-D-lysine coverslips and fixed with 4% paraformaldehyde following 48-h induction with various concentrations of ponasterone A. Images were captured using a Zeiss (Oberkochen, Germany) LSM 510 laser-scanning confocal microscope coupled to a Zeiss 100M axiovert and a 63× Plan-Apochromat oil-immersion lens with detection by the FITC channel (488 excitation, 535 emission). For final output, images were processed simultaneously and identically using Adobe Photoshop 5.5 software (Adobe Systems, San Jose, CA). An individual cell line (iDAT-GFP) with a high level of inducible expression and low background expression was chosen for these studies.
Dopamine uptake assay.
DAT-mediated dopamine uptake was measured essentially as described by Pifl et al. (1993). Briefly, cells were plated at 1 × 105 cells per well in 24-well trays and treated with various concentrations of ponasterone A. Cells were washed with 1 ml uptake buffer (4 mM Tris, 6.25 mM HEPES, 120 mM NaCl, 5 mM KCl, 1.2 mM CaCl2, 1.2 mM MgSO4, 5.6 mM D-glucose, and 0.5 mM ascorbic acid, pH 7.2) 48 h after treatment and were incubated in 0.5 ml uptake buffer containing 0.25 μCi of 3H-dopamine for 10 min at 37°C. Nonspecific uptake was determined in the presence of 10 μM nomifensine. Uptake was terminated by aspirating the uptake buffer and washing each well twice with 1 ml ice-cold uptake buffer. Cells were lysed by 1% sodium dodecyl sulfate and transferred into vials containing 5 ml of scintillation cocktail, and radioactivity was measured by liquid scintillation counting. For incubations with paraquat or MPP+, cells were incubated with various concentrations of the compounds for 10 min before initiation of dopamine uptake. Uptake values were corrected for the amount of protein to account for variation in plating density, and kinetic values were calculated by nonlinear regression using GraphPad Prism 3.0.
Cell death and viability assays.
Cell viability was determined with the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) reduction assay. Induced cells were incubated with 25 μM MPP+ for 48 h or 10 μM for 72 h. For studies with paraquat, cells were incubated with various concentrations of paraquat for 48 h. Following this incubation, cells were treated with 0.5 mg/ml MTT for 3 h followed by dimethylsulfoxide (DMSO) exposure for 1 h. The optical density of the dissolved formazan grains within the cells was measured spectrophotometrically at 595 nm. The viability of untreated control cells was set to 100%, and the cell loss from each treatment was calculated as a percentage over the nontreated control. Data were normalized to total protein content using the Bio-Rad reagent. Preliminary studies determined that PonA induction was not toxic to the cells and delineated protein concentration and time necessary for determinations in the linear range.
3H-Dihydrorotenone (DHR) binding in isolated mitochondria.
Crude mitochondria were isolated from whole brain of male Lewis rats as described by Hoppel and coworkers (1979). Briefly, whole brain was minced in mitochondrial isolation buffer composed of 70 mM sucrose, 220 mM D-mannitol, and 2 mM HEPES (pH 7.4) and homogenized using a motorized tissue grinder and a Teflon® pestle fitted to a glass mortar. The homogenate was centrifuged at 900 × g for 10 min at 4°C to remove cell debris and nuclei. The resultant supernatant was centrifuged at 10,000 × g for 10 min to isolate the crude mitochondrial fraction. The mitochondrial fraction was resuspended in the same buffer and washed twice by centrifugation. The final pellet was resuspended in 250 mM sucrose and 10 mM HEPES buffer (pH 7.4) and used for the determination of 3H-DHR binding to complex I.
Binding of 3H-DHR to complex I was adapted from a previously described autoradiographic procedure (Higgins and Greenamyre, 1996) to allow for determination of binding to crude mitochondrial preparations in 96-well microtiter plates. Crude mitochondria were isolated as described above and diluted to 1 mg of protein/ml in 50 mM Tris–HCl containing 1% BSA (pH 7.4). BSA was included to reduce high levels of nonspecific binding of 3H-DHR. 3H-DHR binding was conducted in a final volume of 200 μl containing 25 μg of mitochondrial protein and incubated with 5 nM 3H-DHR (50 Ci/mmol, custom synthesized by Amersham Life Sciences, Arlington Heights, IL) for competition binding experiments. In competition experiments the complex I inhibitors, rotenone (0.1–100 nM) and MPP+ (10 μM-10 mM), and the suspected complex I inhibitor, paraquat (10 μM-30 mM), were included in the incubation. Experiments were performed at room temperature and allowed to proceed for 2 h at 25°C to achieve equilibrium. Nonspecific binding was defined by the inclusion of 100 μM unlabeled rotenone dissolved in DMSO. The final concentration of DMSO was 1% or less and did not alter binding. Following the 2-h incubation, samples were rapidly filtered over GF/B 96-well filter plates (Perkin-Elmer, Boston, MA) and washed five times with 50 mM Tris–HCl containing 1% BSA. These washes were followed by an additional five washes with 50 mM Tris–HCl containing no BSA. Filter plates were allowed to dry overnight, sealed, and 50 μl of scintillation fluid added (Microscint-20; Perkin-Elmer, Boston, MA). Plates were vortexed for 1 min, and radioactivity was determined on a Packard TopCount microplate scintillation counter (Perkin-Elmer, Boston, MA). Specific binding was calculated as total binding (incubated without 100 μM unlabeled rotenone) minus nonspecific binding (incubated with 100 μM unlabeled rotenone) and expressed as pmol/mg protein. IC50 values for each compound were determined by nonlinear regression using GraphPad Prism 3.0 (San Diego, CA).
Complex I activity in isolated mitochondria.
Inhibition of complex I activity was determined by monitoring the activity of NADH:quinone reductase essentially as described by Schuler and Casida (2001). Briefly, crude mitochondria were prepared as described above and resuspended in 50 mM sodium phosphate buffer containing 250 mM sucrose, 2 mM potassium cyanide, and 2 μM antimycin A (pH 7.5). Alamethicin (30 μg/ml) was included in the preparation to permeabilize the intact mitochondrial preparation and allow NADH to access the inner membrane. The reaction mixture containing 75 μg of mitochondrial protein was incubated with the inhibitor (10 μl) or vehicle (DMSO for rotenone or buffer for MPP+ and paraquat), 2.5 mg bovine serum albumin (250 μl in substrate buffer), and NADH (200 μM) for 5 min at 30°C in a total volume of 990 μl. The assay was initiated by the addition of decylubiquinone (100 μM final concentration) in ethanol (10 μl) and monitored for 3 min at 340 nm. In some cases, 2 mM glutamic acid and 2 mM malic acid was included in the incubation with freshly isolated mitochondria for assays involving paraquat or MPP+. Rotenone (2 μM in DMSO) was included in parallel samples to define rotenone-sensitive activity and was subtracted from the total activity to yield specific activity expressed as nmol NADH oxidized/min-mg protein. DMSO and buffer vehicles did not affect complex I activity, and the data were collapsed for analysis. IC50 values were determined by nonlinear regression using GraphPad Prism 3.0.
Animals and treatments.
Retired breeder male C57 BL/6J mice (Jackson Laboratories, Bar Harbor, ME; 8–12 months of age) were used for MPTP and paraquat treatments. Mice were administered two injections of 15 mg/kg MPTP subcutaneously 10 h apart (total dosage 30 mg/kg) and sacrificed 1 day after the second injection (Tillerson et al., 2002). Paraquat (10 mg/kg) or saline vehicle was administered intraperitoneally once per week for 3 weeks, and animals were sacrificed 1 day after the last treatment (McCormack et al., 2002). These time points were chosen based upon the appearance of nigral dopamine neuron loss or decreased tyrosine hydroxylase immunoreactivity in these models, which ranges from 25 to 40% for paraquat and MPTP animals at these time points, respectively (Jackson-Lewis et al., 1996; McCormack et al., 2002). The striatum, midbrain containing substantia nigra, and cerebral cortex were rapidly dissected, and crude mitochondria were prepared for 3H-DHR binding as described above. The striatum and midbrain were chosen as regions for analysis based on their high density of the DAT, and the cortex for its low DAT density (Ciliax et al., 1999). For studies with rotenone, male Lewis rats (Charles River Laboratories, Wilmington, MA; 4–5 months of age) were administered rotenone (3 mg/kg/day) or vehicle (DMSO:polyethylene glycol; 1:1) by subcutaneous implantation of osmotic mini-pumps as described previously (Sherer et al., 2003b). The rat model of rotenone administration was used in this study, since administration of rotenone to mice produces less severe dopaminergic toxicity, most likely because of enhanced metabolism of rotenone by mice (Sherer et al., unpublished data). This dosing regimen has been demonstrated to reduce tyrosine hydroxylase immunoreactivity in the substantia nigra and striatum of male Lewis rats by approximately 30–50% following 5–7 days of treatment (Sherer et al., 2003b). Rats were sacrificed 5 days after treatment, and the whole brain was used to prepare crude mitochondria, since we have previously shown that rotenone uniformly inhibits complex I throughout the brain using this dosing paradigm (Betarbet et al., 2000).
Statistical analysis.
All cell culture experiments represent 2–4 independent experiments performed in triplicate. The in vivo studies represent 3–8 independent animals for each treatment. Data were calculated as mean ± SEM and presented as percentage of control in most cases. Statistical analyses were performed on raw data. For the in vivo studies, each treatment was compared to its respective control, and data presented as percentage of control. For illustrative purposes, a single control bar representing 100% is depicted in the graph. Differences between groups were determined by Student's t-test for analyses involving only two groups or by ANOVA followed by mean separation by the Student-Newman-Keuls method for analyses involving more than two groups. Significance is reported at the level of p ≤ 0.05.
RESULTS
Paraquat Toxicity is Independent of DAT Expression
To investigate the role of DAT expression on paraquat toxicity, we developed a neuroblastoma cell line with inducible expression of hDAT tagged with GFP (iDAT-GFP). iDAT-GFP expression was increased in a dose-related manner after addition of ponasterone A, as shown by Western immunoblotting, confocal microscopy, and dopamine uptake (Figs. 1A–1C). Confocal microscopy of induced cells demonstrated a relatively diffuse expression of iDAT-GFP with punctuate expression on the plasma membrane (Fig. 1B). Cell surface expression and function of iDAT-GFP was confirmed by increases in DAT-mediated dopamine uptake following addition of ponasterone A (Fig. 1C). Incubation of 25 μM MPP+ for 48 h with cells induced to varying levels of DAT expression demonstrated that, as DAT expression increased, cell viability decreased (Fig. 2A), and this toxicity could be completely abolished by the DAT inhibitor, nomifensine (data not shown). Conversely, DAT expression had no effect on the toxicity of 100 μM paraquat. We then exposed cells with no induction or maximum induction of DAT expression (10 μM PonA for 48 h) to various concentrations (0.05–1 mM) of paraquat. Paraquat elicited a dose-related decrease in cell viability that was independent of DAT expression (Fig. 2B), suggesting that paraquat is not a substrate for DAT. We then tested the ability of paraquat and MPP+ to act as an inhibitor of DAT-mediated dopamine uptake in cells expressing the highest level of DAT. MPP+ inhibited dopamine uptake in a dose-related manner, with uptake almost completely abolished at 1 mM. In contrast, paraquat had no significant effect on DAT-mediated dopamine except at a concentration of 10 mM, where there was about a 20% decrease (Fig. 2C). Collectively, these data suggest that paraquat is neither a substrate nor inhibitor of DAT.
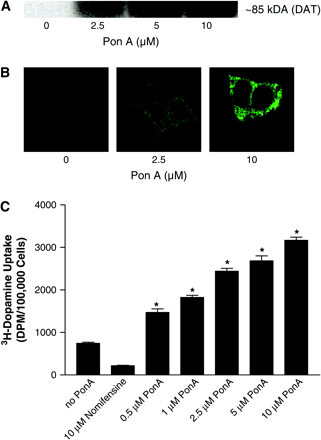
Characterization of the stable cell line with inducible DAT expression (iDAT-GFP). (A) Representative Western blot of DAT after 48 h exposure to 0, 2.5, 5, or 10 μM ponasterone A (Pon A). (B) Representative confocal micrograph of DAT after 48 h exposure to 0, 2.5, or 10 μM ponasterone A (Pon A). (C) DAT-mediated dopamine uptake after 48 h exposure to ponasterone A (n =3). *Represents significantly different (p ≤ 0.05) from no Pon A by Student's t-test.
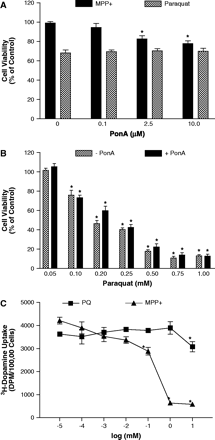
MPP+ but not paraquat toxicity is dependent on DAT expression. (A) Cell viability (percentage of control) of iDAT-GFP cells induced for 48 h with various dosages (0–10 μM) of ponasterone A (Pon A) and then exposed to 25 μM MPP+ or 100 μM paraquat for 48 h. *Represents significantly different (p ≤ 0.05) from no Pon A (0) by Student's t-test (n = 3–4). (B) Cell viability (percentage of control) of iDAT-GFP cells induced for 48 h with 10 μM Pon A and exposed to paraquat (50 μM–1 mM) for 48 h. *Represents significantly different (p ≤ 0.05) from control by Student's t-test (n = 3–4). There were no differences in paraquat toxicity between cells receiving Pon A and those not receiving Pon A. (C) Inhibition of DAT-mediated dopamine uptake by MPP+ and paraquat in iDAT-GFP cells induced with 0 or 10 μM ponasterone A (Pon A) for 48 h. Results represent means ± SEM (n = 3–4). *Represents significantly different (p ≤ 0.05) from control uptake by Student's t-test (n = 3–4).
Paraquat is a Poor Inhibitor of 3H-DHR Binding
To assess the ability of paraquat, MPP+, and rotenone to compete with 3H-DHR for binding to complex I in crude mitochondrial preparations, we used a 96-well microtiter plate assay adapted from a previously described autoradiographic method (Greenamyre et al., 1992). Rotenone was a potent inhibitor of 3H-DHR binding, with an IC50 of 14 nM (Fig. 3), while MPP+ and paraquat were much less potent, with IC50 values of 381 μM and 8.1 mM, respectively. Since MPP+ has been shown to accumulate to a greater extent in freshly isolated mitochondria (Ramsay et al., 1986), we repeated the binding studies in freshly isolated mitochondria with respiration supported by glutamate and malate. The use of freshly isolated mitochondria had no effect on the inhibitor potency of rotenone (data not shown). However, MPP+ was much more potent in competing for 3H-DHR binding, with an IC50 value of 71 μM (Fig. 4A). There was no effect on the weak inhibitory potency of paraquat (Fig. 4B), suggesting paraquat does not accumulate in freshly isolated mitochondria in a similar manner as MPP+.
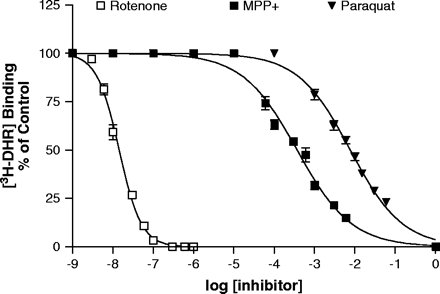
Paraquat competes poorly with 3H-DHR binding to complex I. Competition for 3H-DHR binding in isolated brain mitochondria by rotenone, MPP+ and paraquat. 3H-DHR was used at a single concentration of 5 nM as described in Materials and Methods. Results represent mean ± SEM (n = 3–4). Absence of error bars indicates that the SEM was less than the size of the symbol.
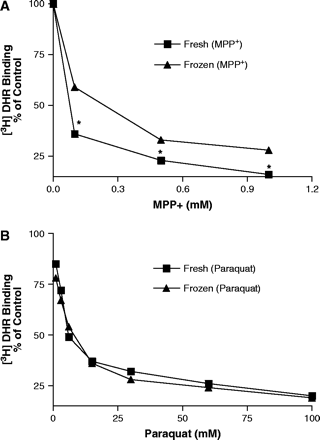
MPP+ but not paraquat potency is enhanced in freshly isolated mitochondria. Competition for 3H-DHR binding by MPP+ and paraquat in fresh and frozen mitochondria. Results represent mean ± SEM (n = 3). Absence of error bars indicates that the SEM was less than the size of the symbol. *Represents significantly different (p ≤ 0.05) from frozen mitochondria by Student's t-test.
Paraquat is a Poor Inhibitor of NADH:Quinone Reductase (Complex I) Activity
Inhibition of the binding of 3H-DHR has been shown to correlate well with the inhibition of complex I activity (Higgins and Greenamyre, 1996). Therefore, we determined the inhibitory potency of rotenone, MPP+ and paraquat for NADH:quinone reductase activity using the same mitochondrial preparation as used in the binding studies. Rotenone exhibited a dose-related reduction in complex I activity, with an IC50 of 7.75 nM, while paraquat was without significant effect at concentrations up to 10 mM (Fig. 5). MPP+ (5 mM) did not inhibit complex I activity in frozen preparations, in agreement with that found by others (Ramsay et al., 1986). However, MPP+ inhibited complex I activity, with an IC50 of 142 μM, in freshly isolated intact mitochondria, supporting the observation of active accumulation of MPP+ (Fig. 5). This effect is also similar to that observed with freshly isolated mitochondria in the 3H-DHR assays, as MPP+ potency was increased and paraquat potency was unaffected. Rotenone potency was similar in both fresh and frozen preparations, similar to that observed in the binding assays (data not shown). In support of previous studies (Higgins and Greenamyre, 1996), there was excellent agreement between the IC50 values obtained for rotenone and MPP+ in the 3H-DHR binding assays and the IC50 values obtained with the activity assays.
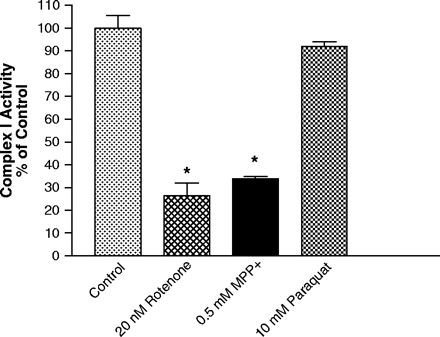
Rotenone and MPP+, but not paraquat, inhibit complex I activity. Inhibition of NADH:quinone reductase (complex I) activity by rotenone, MPP+, and paraquat in freshly isolated mitochondria. Results represent mean ± SEM (n = 3–4). *Indicates significantly from control (p ≤ 0.05) by ANOVA followed by Student-Newman Keuls test.
In Vivo Administration of Rotenone and MPTP, but Not Paraquat Reduces 3H-DHR Binding
Systemic in vivo administration of the complex I inhibitor rotenone has been shown to reduce 3H-DHR to complex I (Betarbet et al., 2000). Likewise, systemic administration of MPTP to Swiss Albino mice has been shown to reduce NADH:quinone reductase activity in the striatum (Sriram et al., 1997). High-level acute exposure of rats to paraquat (100 mg/kg iv) has been shown to reduce NADH:quinone reductase activity in the brain and lung (Tawara et al., 1996). However, there are no data on the effects of paraquat on complex I in mice treated with lower dosages of paraquat shown to kill dopamine neurons. Therefore, we sought to determine the effects of in vivo administration of rotenone, MPTP, and paraquat on 3H-DHR binding using dosage paradigms previously shown to result in nigrostriatal degeneration or reduced 3H-DHR binding. Systemic administration of 3 mg/kg rotenone to rats for 5 days caused a 60% reduction in 3H-DHR in mitochondria prepared from whole brain (Fig. 6). Consistent with the selective uptake of MPP+ by DAT, administration of MPTP (2 × 15 mg/kg sc) to retired breeder male C57 BL/6J mice resulted in a 27% decrease of 3H-DHR in mitochondria prepared from the striatum and a 30% decrease in midbrain, with no corresponding decrease in mitochondria prepared from the frontal cortex, 24 h after administration of the second dose. Paraquat administration (10 mg/kg ip; 1× per week for 3 weeks) did not reduce 3H-DHR binding in mitochondria prepared from the frontal cortex, striatum, or midbrain 24 h after the last dose.
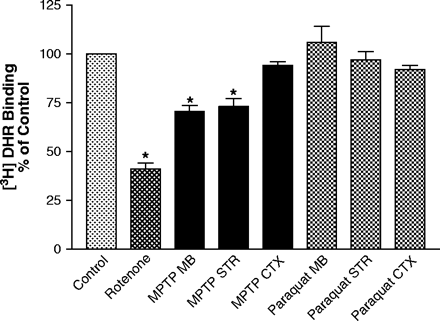
In vivo exposure to rotenone and MPTP, but not paraquat, reduces 3H-DHR binding. Rats were exposed to vehicle (DMSO:PEG; n = 5) or 3 mg/kg rotenone (n = 7), and mice to vehicle (saline; n = 4–5) or 10 mg/kg paraquat (n = 4) or 15 mg/kg MPTP (n = 5) as described in the materials and methods. Animals were sacrificed 1 day after the last exposure, and mitochondria were isolated from whole brain for rotenone treated animals, the striatum (STR), midbrain (MB), and cortex (CTX) for MPTP-treated animals, and STR, CTX, and MB for paraquat-treated animals and assayed for 3H-DHR binding. Results represent mean ± SEM. *Indicates significantly different from control (DMSO:PEG for rotenone and saline for MPTP and paraquat) for each tissue and treatment group (p ≤ 0.05) by ANOVA followed by Student-Newman Keuls test. A control bar without SEM representing 100% of control activity is included for illustrative purposes.
DISCUSSION
While rotenone, MPTP, and paraquat are used to produce animal models of PD, the mechanism(s) by which these compounds target and kill dopaminergic neurons has remained elusive. Recently, we demonstrated that complex I inhibition is required for rotenone toxicity (Sherer et al., 2003a), and it is well established that MPP+ targets dopamine neurons through selective uptake by the DAT (Javitch et al., 1985; Miller et al., 1999; Uhl, 1998). However, the mechanism by which paraquat kills dopamine neurons is not clear. The present study demonstrates that the neurotoxic actions of paraquat are not the result of transport by the DAT or complex I inhibition.
Systemic administration of paraquat to mice has been shown to result in a specific loss of tyrosine hydroxylase–positive neurons of the substantia nigra pars compacta (Brooks et al., 1999; McCormack et al., 2002). However, the mechanism by which paraquat specifically targets dopamine neurons is unknown. Based upon structural similarity with MPP+, it has been thought that paraquat may gain access to dopaminergic neurons through the dopamine transporter (DAT). Indeed, it has been suggested that the DAT transports paraquat, since GBR 12909 protected against paraquat toxicity in midbrain organotypic cultures (Shimizu et al., 2003). However, DAT function was not measured in this study and GBR 12909 has been shown to protect against rotenone and MPP+ toxicity in cerebellar granule cells, which do not contain the DAT (Shang et al., 2003). Here, we developed a neuroblastoma cell line in which the expression of DAT is controlled by the ecdysone system (No et al., 1996) to directly test the role of DAT in the toxicity of paraquat. Using this system, we determined that paraquat toxicity was independent of DAT expression, suggesting that paraquat is not a substrate for the DAT. Likewise, paraquat did not inhibit DAT-mediated dopamine uptake, similar to that observed in mouse brain synaptosomes by Barlow and coworkers (2003). Therefore, our data directly refute a role for DAT in paraquat transport and damage to dopamine neurons.
Once transported into the cell by the DAT, MPP+ is concentrated in the mitochondria and inhibits complex I by binding to the same site as rotenone (Ramsay et al., 1991). Here, we adapted a previously utilized autoradiographic assay of 3H-DHR binding for use with isolated mitochondria along with enzymatic assay of complex I activity to determine whether paraquat inhibits DHR binding to complex I in a similar manner as MPP+ and rotenone. We found the IC50 for rotenone competition with DHR and complex I activity to be 14 and 7.75 nM, similar to that previously reported in brain slices (Higgins and Greenamyre, 1996). The IC50 for MPP+ competition for DHR was 351 μM, which is much more potent than the IC50 (3.96 mM) found with the previous autoradiographic assay, but similar to that (300 μM) observed to decrease State 3 mitochondrial respiration in isolated mitochondria (Nicklas et al., 1985). The discrepancy between the current results and the previous autoradiographic data is most likely the result of using an isolated mitochondrial preparation, as compared to the brain slices used for autoradiography. By using brain slices, MPP+ is free to bind to or be sequestered by the DAT, the vesicular monoamine transporter 2, and possibly other sites (Del Zompo et al., 1990), which may reduce the amount available for binding to complex I. MPP+ was without effect on complex I activity at concentrations up to 10 mM in assays using previously frozen mitochondrial preparations, similar to that observed by Ramsay and coworkers (1986). However, in the freshly isolated mitochondria, MPP+ inhibited complex I activity and competed for DHR binding with IC50s of 142 μM and 71 μM. These results are in good agreement with those of Ramsay and coworkers (1986), who reported that 0.5 mM MPP+ totally abolished State 3 mitochondrial respiration in freshly isolated mitochondria. Paraquat was a much less potent inhibitor of DHR binding (IC50 of 7 mM) and was without significant effect on complex I activity at concentrations of up to 10 mM, supporting Palmeira and coworkers (1995) who found that paraquat had no significant effect on State 3 mitochondrial respiration in isolated liver mitochondria. Likewise, paraquat had no significant effect on complex I in freshly isolated mitochondria, suggesting that paraquat is not actively accumulated by the mitochondria. Taken together, these data suggest that there is no direct interaction of paraquat with complex I.
We next sought to determine whether complex I inhibition, as measured by DHR binding, is observed following in vivo exposure to these compounds. Rotenone administration resulted in a 59% decrease in DHR binding, similar to that observed by the autoradiographic method (Betarbet et al., 2000). MPTP decreased DHR binding by 27% in the striatum and 30% in midbrain with no corresponding change in the cortex, consistent with the low amount of DAT in the cortex (Miller et al., 1997; Ciliax et al., 1999). This finding is similar to the decrease in complex I activity found in the striatum after a single 30-mg/kg dose of MPTP (Sriram et al., 1997), but different than that found by Gerlach and coworkers (1996), who observed no change in complex I activity 1 week after exposure of marmosets to MPTP. There are a number of possible explanations for these differences. First, MPP+ binds to complex I with lower affinity than rotenone and can be dissociated during the centrifugation procedure used to obtain isolated mitochondria (Krueger et al., 1990), which would reduce the amount of MPP+ bound to mitochondria during assay. Second, MPP+ is rapidly cleared from the body (Yang et al., 1988). Therefore, the inhibition we observed may be reversible and dependent upon time and experimental conditions. In spite of this, even transient inhibition of complex I activity by MPTP may contribute to nigrostriatal damage by generation of reactive oxygen species (Lotharius and O'Malley, 2000) and/or decreases in ATP production (Bates et al., 1994). Paraquat administration caused no reduction in DHR binding, confirming the lack of effect on complex I observed in our in vitro studies. In contrast, Tawara and coworkers (1996) found that administration of 20 mg/kg paraquat iv every other day for 5 days caused a 50% reduction in complex I activity in whole-brain homogenates. However, extensive lipid peroxidation of mitochondrial membranes was observed, which may have contributed to the reduction in complex I activity (Picklo et al., 1999). This suggests that the actions of paraquat on complex I are nonspecific and do not involve direct inhibition of complex I.
Taken in concert, our data suggest paraquat does not specifically target dopamine neurons through the DAT as is the case with MPTP. However, the question of how paraquat, a charged hydrophilic compound, enters the brain remains to be answered. Recently Shimizu et al. (2001) suggested that paraquat might be specifically transported into the brain by a neutral amino acid transporter, most likely the system L carrier (LAT-1). Likewise, McCormack and Di Monte (2003) reported that pretreatment of mice with L-valine, a LAT-1 substrate, prevented the dopaminergic neurotoxicity of paraquat. These results suggest that the LAT-1 may be a candidate for paraquat translocation into the brain. However, these studies provided no direct evidence for the involvement of the LAT-1 and, more specifically, no evidence for why dopamine neurons are specifically affected by paraquat exposure. While it is known that LAT-1 resides near the blood–brain barrier (Kanai and Endou, 2001), there are currently no data demonstrating uptake of paraquat by this transporter. Thus, further study is required to determine the mechanism by which paraquat gains access to the brain.
Our data also suggest that paraquat differs from rotenone and MPTP by its lack of effect on complex I. In the case of rotenone, we have previously demonstrated that uniform inhibition of brain complex I results in oxidative damage and specific damage to dopamine neurons (Betarbet et al., 2000; Sherer et al., 2003a). Thus, it appears that dopamine neurons are uniquely susceptible to oxidative damage. The exact reason for this susceptibility is not known, but dopamine neurons are believed to exist in a constant state of oxidative stress. During the normal process of synthesis and metabolism of dopamine several enzymes in these pathways promote the formation of hydrogen peroxide as a normal byproduct. Additionally, the substantia nigra may be particularly vulnerable to oxidative damage, because it contains neuromelanin and has intrinsically low levels of antioxidant molecules such as glutathione (reviewed by Bharath et al., 2002). Likewise, paraquat is able to cause oxidative damage through a free radical mechanism (Bus et al., 1976) and cause oxidative damage and lipid peroxidation in brain (McCormack et al., 2005). Therefore, we hypothesize that the reason for the selective dopaminergic neurodegeneration in paraquat-treated animals may involve general oxidative damage affecting the more vulnerable dopamine neurons rather than specific uptake into dopamine neurons and subsequent mitochondrial inhibition and dopamine neuron death. A better understanding of the mechanisms by which these compounds damage dopaminergic neurons may provide insight into the pathogenic process of PD and provide targets for therapeutic intervention aimed at slowing the progression of PD.
The authors certify that all research involving human subjects was done under full compliance with all government policies and the Helsinki Declaration.
Current address: Department of Environmental and Occupational Medicine, University of Medicine and Dentistry-New Jersey/Robert Wood Johnson Medical School and Environmental and Occupational Health Sciences Institute, Piscataway, NJ 08854.
Current address: The Michael J. Fox Foundation for Parkinson's Disease Research, New York, NY 10163.
Current address: Pittsburgh Institute for Neurodegenerative Diseases Pittsburgh, PA 15261.
This work was supported by NIH Grants U54 ES012068 as part of the Collaborative Centers for Parkinson's Disease Environmental Research (JTG & GWM), RO1 NS037031 (GWM), RO1 ES009248 (GWM), and F32ES013457 and T32NS07480 (JRR). T.B.S. was supported by a Michael J. Fox Foundation Fellowship. Conflict of interest: none declared.
References
Barlow, B. K., Thiruchelvam, M. J., Bennice, L., Cory-Slechta, D. A., Ballatori, N., and Richfield, E. K. (
Bates, T. E., Heales, S. J., Davies, S. E., Boakye, P., and Clark, J. B. (
Betarbet, R., Sherer, T. B., MacKenzie, G., Garcia-Osuna, M., Panov, A. V., and Greenamyre, J. T. (
Bharath, S., Hsu, M., Kaur, D., Rajagopalan, S., and Andersen, J. K. (
Brooks, A. I., Chadwick, C. A., Gelbard, H. A., Cory-Slechta, D. A., and Federoff, H. J. (
Bus, J. S., Cagen, S. Z., Olgaard, M., and Gibson, J. E. (
Chiba, K., Trevor, A. J., and Castagnoli, N., Jr. (
Ciliax, B. J., Drash, G. W., Staley, J. K., Haber, S., Mobley, C. J., Miller, G. W., Mufson, E. J., Mash, D. C., and Levey, A. I. (
Del Zompo, M., Ruiu, S., Maggio, R., Piccardi, M. P., and Corsini, G. U. (
Fukushima, T., Yamada, K., Hojo, N., Isobe, A., Shiwaku, K., and Yamane, M. (
Gainetdinov, R. R., Fumagalli, F., Jones, S. R., and Caron, M. G. (
Gerlach, M., Gotz, M., Dirr, A., Kupsch, A., Janetzky, B., Oertel, W., Sautter, J., Schwarz, J., Reichmann, H., and Riederer, P. (
Greenamyre, J. T., Higgins, D. S., and Eller, R. V. (
Heikkila, R. E., Hess, A., and Duvoisin, R. C. (
Hertzman, C., Wiens, M., Bowering, D., Snow, B., and Calne, D. (
Higgins, D. S., Jr., and Greenamyre, J. T. (
Hoppel, C., DiMarco, J., and Tandler, B. (
Jackson-Lewis, V., Jakowek, M., Burke, R. E., and Przedborski, S. (
Javitch, J. A., D'Amato, R. J., Strittmatter, S. M., and Snyder, S. H. (
Kanai, Y., and Endou, H. (
Krueger, M. J., Singer, T. P., Casida, J. E., and Ramsay, R. R. (
Langston, J. W., Ballard, P., Tetrud, J. W., and Irwin, I. (
Liou, H. H., Tsai, M. C., Chen, C. J., Jeng, J. S., Chang, Y. C., Chen, S. Y., and Chen, R. C. (
Lotharius, J., and O'Malley, K. L. (
McCormack, A. L., Thiruchelvam, M., Manning-Bog, A. B., Thiffault, C., Langston, J. W., Cory-Slechta, D. A., and Di Monte, D. A. (
McCormack, A. L., and Di Monte, D. A. (
McCormack, A. L., Atienza, J. G., Johnston, L. C., Andersen, J. K., Vu, S., and Di Monte, DA. (
Miller, G. W., Gainetdinov, R. R., Levey, A. I., and Caron, M. G. (
Miller, G. W., Staley, J. K., Heilman, C. J., Perez, J. T., Mash, D. C., Rye, D. B., and Levey, A. I. (
Mizuno, Y., Ohta, S., Tanaka, M., Takamiya, S., Suzuki, K., Sato, T., Oya, H., Ozawa, T., and Kagawa, Y. (
Nakamura, K., Bindokas, V. P., Marks, J. D., Wright, D. A., Frim, D. M., Miller, R. J., and Kang, U. J. (
Nicklas, W. J., Vyas, I., and Heikkila, R. E. (
No, D., Yao, T. P., and Evans, R. M. (
Palmeira, C. M., Moreno, A. J., and Madeira, V. M. (
Parker, W. D., Jr., Boyson, S. J., and Parks, J. K. (
Picklo, M. J., Amarnath, V., McIntyre, J. O., Graham, D. G., and Montine, T. J. (
Pifl, C., Giros, B., and Caron, M. G. (
Ramsay, R. R., Dadgar, J., Trevor, A., and Singer, T. P. (
Ramsay, R. R., Krueger, M. J., Youngster, S. K., Gluck, M. R., Casida, J. E., and Singer, T. P. (
Schapira, A. H., Cooper, J. M., Dexter, D., Jenner, P., Clark, J. B., and Marsden, C. D. (
Schuler, F., and Casida, J. E. (
Semchuk, K. M., Love, E. J., and Lee, R. G. (
Shang, T., Uihlein, A. V., Van Asten, J., Kalyanaraman, B., and Hillard, C. J. (
Sherer, T. B., Betarbet, R., and Greenamyre, J. T. (
Sherer, T. B., Betarbet, R., Testa, C. M., Seo, B. B., Richardson, J. R., Kim, J. H., Miller, G. W., Yagi, T., Matsuno-Yagi, A., and Greenamyre, J. T. (
Sherer, T. B., Kim, J. H., Betarbet, R., and Greenamyre, J. T. (
Shimizu, K., Matsubara, K., Ohtaki, K., and Shiono, H. (
Shimizu, K., Ohtaki, K., Matsubara, K., Aoyama, K., Uezono, T., Saito, O., Suno, M., Ogawa, K., Hayase, N., Kimura, K., et al. (
Sriram, K., Pai, K. S., Boyd, M. R., and Ravindranath, V. (
Tanner, C. M., Ottman, R., Goldman, S. M., Ellenberg, J., Chan, P., Mayeux, R., and Langston, J. W. (
Tawara, T., Fukushima, T., Hojo, N., Isobe, A., Shiwaku, K., Setogawa, T., and Yamane, Y. (
Tillerson, J. L., Caudle, W. M., Reveron, M. E., and Miller, G. W. (
Uhl, G. R. (
Vila, M., and Przedborski, S. (
Author notes
*Center for Neurodegenerative Disease, School of Medicine and †Department of Environmental and Occupational Health, Rollins School of Public Health, Emory University, Atlanta, Georgia 30322, and ‡Division of Pharmacology and Toxicology, College of Pharmacy, University of Texas at Austin, Austin, Texas 78712

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments