


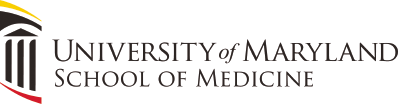



Abstract
The search for liability genes of the world's 2 major psychotic disorders, schizophrenia and bipolar disorder I (BP-I), has been extremely difficult even though evidence suggests that both are highly heritable. This difficulty is due to the complex and multifactorial nature of these disorders. They encompass several intermediate phenotypes, some overlapping across the 2 psychotic disorders that jointly and/or interactively produce the clinical manifestations. Research of the past few decades has identified several neurophysiological deficits in schizophrenia that frequently occur before the onset of psychosis. These include abnormalities in smooth pursuit eye movements, P50 sensory gating, prepulse inhibition, P300, mismatch negativity, and neural synchrony. Evidence suggests that many of these physiological deficits are distinct from each other. They are stable, mostly independent of symptom state and medications (with some exceptions) and are also observed in non-ill relatives. This suggests a familial and perhaps genetic nature. Some deficits are also observed in the BP-I probands and to a lesser extent their relatives. These deficits in physiological measures may represent the intermediate phenotypes that index small effects of genes (and/or environmental factors). The use of these measures in genetic studies may help the hunt for psychosis liability genes and clarify the extent to which the 2 major psychotic disorders share etio-pathophysiology. In spite of the rich body of work describing these neurophysiological measures in psychotic disorders, challenges remain: Many of the neurophysiological phenotypes are still relatively complex and are associated with low heritability estimates. Further refinement of these physiological phenotypes is needed that could identify specific underlying physiological deficits and thereby improve their heritability estimates. The extent to which these neurophysiological deficits are unique or overlap across BP-I and schizophrenia is unclear. And finally, the clinical and functional consequences of the neurophysiological deficits both in the probands and their relatives are not well described.
Introduction
Classification of psychotic illnesses into 3 broad categories of organic, affective, and poor outcome has taken root over the past century, with the French physician Benedict Augustin Morel presenting the concept of premature dementia (demence precoce) describing the concept of a psychotic illness with poor outcome.1 Kraepelin further distinguished dementia praecox from affective psychosis.2 Clinical features such as poor outcome, affective flattening, and autism were stressed in defining schizophrenia (in today's nomenclature would be considered negative symptoms). Morel, Kraepelin, Bleuler, and other investigators of a century ago commented on the familial nature of these disorders. Family studies of the1970s and 1980s used operational criteria to define schizophrenia and bipolar disorders, confirmed these early observations and noted that in general schizophrenia ran true in families of schizophrenia probands, and bipolar in families of bipolar probands, with minimal overlap in familial risks.3 Interestingly, these studies showed that in families where psychotic symptoms were not clear-cut schizophrenia or bipolar affective, there was a blurring of boundaries of familial risk. Twin studies in these 2 disorders are generally consistent with findings observed in the family studies, ie, the 2 disorders ran true in the respective families of bipolar or schizophrenia probands with minimal overlap in the familial risk for the 2 disorders.4,5 However, the diagnostic methods in the twin studies were mostly based on a hierarchal diagnostic system such that an individual is given a schizophrenia diagnosis based on lifetime symptoms of schizophrenia even if in an earlier episodes showed clear manic symptoms. If lifetime manic episodes are considered a separate diagnosis, then there are (1) more than expected rates of comorbidity between manic and schizophrenia syndromes, (2) overlap in familial risks across these 2 syndromes (high twin-pair wise concordance rates), and (3) overlap in genetic risks (significantly higher concordance for monozygotic vs dizygotic twin pairs).6
The subsequent genetic studies in both schizophrenia and bipolar disorder were disappointing, although these studies provided several interesting leads. These include susceptibility loci on chromosome 1q21-44, 5q22-31, 6p24-21, 8p22-21, 13q14-32, 15q13-14, and 22q11-13 that showed a significant linkage and at least one replication. However, consistent replications across several studies are nonexistent. The problem of replication failures is further stressed by recent reports from large sample genome-wide scans, which fail to replicate most of the previous susceptibility loci or the candidate genes for the 2 disorders. Interestingly, several susceptibility loci overlapped across schizophrenia and bipolar disorder. Overall, these findings suggest that both of these major psychotic illnesses are multifactorial, likely caused by various combinations of several genes, all with small effects that jointly and/or interactively (ie, additive and/or multiplicative) lead to the clinical manifestations of the disorders. Thus, an aberrant gene (or a small set of aberrant genes) individually may lead to subtle neurophysiological deficits, but these deficits in various combinations lead to overt clinical disorders such as schizophrenia or bipolar disorder. Within this theoretical framework, an alternative approach to the search for genes is to identify these small effects of genes by using neurophysiological deficits as phenotypes in genetic studies. Gottesman and Shield7 first advocated this approach, arguing that the neurophysiological measure that marks a neuronal deficit nearer to the effects of genes (termed endophenotype) is likely to be a less complex than the clinical syndrome and would therefore simplify the gene hunt. The suggested criteria that define an endophenotype include: the marker is associated with the illness in the population, heritable, state-independent, co-segregates with the illness within families and is present at a higher rate in unaffected family members of the proband compared with the general population.8 A deficit that is along the causal pathway or at least involved in the biological mechanism of pathogenesis has advantage in genetic studies.9,10 The potential usefulness of this approach is illustrated by the findings of significant linkage in schizophrenia pedigrees of P50 sensory gating deficit to chromosome 15q14 locus and subsequent findings implicating alpha 7 nicotinic receptor gene.11 In spite of the promise of endophenotypes in clarifying etio-pathophysiology of psychotic disorders, several challenges remain. Many neurophysiological measures are still complex exemplifying the final outcome of widely distributed neuronal systems. Further refinement of the different physiological phenotypes is critical in order for the measures to be useful in linkage studies. The review below examines several neurophysiological measures that are known to be abnormal in schizophrenia and to a lesser extent in bipolar disorder. Each section describing the neurophysiological measure attempts to address the following issues. What is the underlying neuronal system subserving the endophenotype, how complex it is, and whether there has been a refinement of the endophenotype to identify a more “elemental” deficit? Is the neurophysiological deficit also observed in bipolar disorder probands and their families? The understanding of the extent of overlap among different psychotic disorders is informative in addressing the question: what is shared and what is unique about the etio-pathophysiologies of different psychotic disorders? What is the clinical and functional significance of the neurophysiological deficit? This point to what clinical and functional outcome measured will be relevant if novel treatments are developed based on the genes associated with the endophenotype. What is known about the genetic underpinning of the endophenotype? What is known about the neurobiology of the endophenotype and what neurotransmitter system affects the measure? With these issues in mind, the review below focuses on physiological measures that are mediated centrally and include smooth pursuit eye movement, sensory gating (P50), sensory-motor gating (prepulse inhibition, PPI), evoked potential measure of P300, mismatch negativity (MMN), and gamma band synchronization (Table 1). These measures were chosen because these endophenotypes have been extensively studied in patients with schizophrenia and their relatives and to some extent in bipolar disorder. Although, the extent of overlap among these different neurophysiological endophenotypes has not been comprehensively studied, recent data suggest that many of these measures are independent of each other likely marking different aspects of psychosis risk.12–16 There are several imaging and cognitive endophenotypes that are not included in this review because they are covered by accompanying papers in this theme issue.
Neurophysiological Markers in Psychotic Disorders
| Biomarker | Schizophrenia/Relatives | Bipolar/Relatives | Heritability Estimates | Linkage Findings | Associated gene | Associated Clinical Phenotype |
| 1. SPEM-predictive pursuit | +++/++ | +/+ | 0.7–0.9 | 6p21 | COMT | ? Positive symptoms |
| Pursuit initiation | +/+ | −/− | Primary negative symptoms | |||
| 2. P50 | ++/+ | +/+ | 0.44 | 15q14 | Alpha-7 nicotine receptor | |
| 3. PPI | ++/+ | +/+ | 0.32–0.50 | 22q11a | Neuregulin 1 | Positive symptoms |
| 4. P300 | ++/+ | +/+ | 0.27–0.69 | 4q22b | ||
| 5. MMN | ++/+ | −/− | Negative symptoms; poor functioning | |||
| 6. Neural synchronization | ++/+ | + | Positive symptoms |
| Biomarker | Schizophrenia/Relatives | Bipolar/Relatives | Heritability Estimates | Linkage Findings | Associated gene | Associated Clinical Phenotype |
| 1. SPEM-predictive pursuit | +++/++ | +/+ | 0.7–0.9 | 6p21 | COMT | ? Positive symptoms |
| Pursuit initiation | +/+ | −/− | Primary negative symptoms | |||
| 2. P50 | ++/+ | +/+ | 0.44 | 15q14 | Alpha-7 nicotine receptor | |
| 3. PPI | ++/+ | +/+ | 0.32–0.50 | 22q11a | Neuregulin 1 | Positive symptoms |
| 4. P300 | ++/+ | +/+ | 0.27–0.69 | 4q22b | ||
| 5. MMN | ++/+ | −/− | Negative symptoms; poor functioning | |||
| 6. Neural synchronization | ++/+ | + | Positive symptoms |
Note: Please see the text for details and appropriate citations. SPEM, smooth pursuit eye movements; COMT, catechol-O-methyltransferase; PPI, prepulse inhibition; MMN, mismatch negativity.
Based on findings of abnormal PPI in 22q11 deletion syndrome.
Based on a linkage findings in alcoholism.
Neurophysiological Markers in Psychotic Disorders
| Biomarker | Schizophrenia/Relatives | Bipolar/Relatives | Heritability Estimates | Linkage Findings | Associated gene | Associated Clinical Phenotype |
| 1. SPEM-predictive pursuit | +++/++ | +/+ | 0.7–0.9 | 6p21 | COMT | ? Positive symptoms |
| Pursuit initiation | +/+ | −/− | Primary negative symptoms | |||
| 2. P50 | ++/+ | +/+ | 0.44 | 15q14 | Alpha-7 nicotine receptor | |
| 3. PPI | ++/+ | +/+ | 0.32–0.50 | 22q11a | Neuregulin 1 | Positive symptoms |
| 4. P300 | ++/+ | +/+ | 0.27–0.69 | 4q22b | ||
| 5. MMN | ++/+ | −/− | Negative symptoms; poor functioning | |||
| 6. Neural synchronization | ++/+ | + | Positive symptoms |
| Biomarker | Schizophrenia/Relatives | Bipolar/Relatives | Heritability Estimates | Linkage Findings | Associated gene | Associated Clinical Phenotype |
| 1. SPEM-predictive pursuit | +++/++ | +/+ | 0.7–0.9 | 6p21 | COMT | ? Positive symptoms |
| Pursuit initiation | +/+ | −/− | Primary negative symptoms | |||
| 2. P50 | ++/+ | +/+ | 0.44 | 15q14 | Alpha-7 nicotine receptor | |
| 3. PPI | ++/+ | +/+ | 0.32–0.50 | 22q11a | Neuregulin 1 | Positive symptoms |
| 4. P300 | ++/+ | +/+ | 0.27–0.69 | 4q22b | ||
| 5. MMN | ++/+ | −/− | Negative symptoms; poor functioning | |||
| 6. Neural synchronization | ++/+ | + | Positive symptoms |
Note: Please see the text for details and appropriate citations. SPEM, smooth pursuit eye movements; COMT, catechol-O-methyltransferase; PPI, prepulse inhibition; MMN, mismatch negativity.
Based on findings of abnormal PPI in 22q11 deletion syndrome.
Based on a linkage findings in alcoholism.
Smooth pursuit eye movement abnormality
The eye movement system is a critical physiological function that maintains the image of the object of interest on the fovea, the most sensitive part of the retina. In mammals and other lower animals, this function is served by saccadic eye movements, one of the fastest movements in the body, that serve to capture the image of interest onto the fovea. However, the saccadic system is inefficient and not able to continuously maintain the image on the fovea if the object of interest is moving. Later in the evolutional process, a more sophisticated system emerged, fully developed only in monkey and humans, that is able to generate slow smooth eye movements to match the motion of the object of interest if it starts to move across the visual field. The image motion is processed by specialized neurons at the juncture of occipital and temporal cortex (V5; mediotemporal cortex, MT) that are tuned to the target velocity. In humans, the pursuit system is able to initiate smooth eye movements within 120–150 ms. This smooth pursuit initiation is a unique neurophysiological function in that the response does not occur in the absence of motion information.17,18 Once the eye catches up with the target, matching its speed, the motion of the target image on the retina is near zero; thus, the pursuit system loses the motion signal needed to generate slow pursuit eye movements. To maintain accurate pursuit, the system generates predictive eye velocity using the internal representation of the target velocity, which is derived from the corollary or the efference copy of the motor command processed in the medial superior temporal cortex, and/or a memory trace of the previous retinal motion (processed by posterior parietal cortex17,18). Frontal eye fields drive the smooth pursuit eye movements after integrating the extraretinal motion information and the current retinal error information (ie, differences between eye and target velocities, and positions to a lesser extent).17,18 Thus, one can broadly divide the smooth pursuit system into 2 components: initiation response based on retinal motion and predictive response based on extraretinal motion information (figure 1). Saccadic eye movements are occasionally used to capture the image of the moving object onto the fovea when the eye falls behind. There are other components of smooth pursuit response that are not fully described. These include the role of error monitoring and the effect of learning best noted during initiation that may or may not be different than the processes involved in predictive response.19,20 The interaction between the saccadic system and pursuit system is complex and just beginning to be explored.21,22
![Smooth pursuit eye movements are broadly divided into 2 components: (1) response to the processing of image motion on the retina and (2) the predictive response based on the internal representation of the target motion (ie, memory trace of the previous image motion on the retina and copy of the motor command driving the eye called efference copy). The first component is measured at the time of pursuit initiation when the target image starts moving away from the fovea. The figure illustrates one method of measuring predictive pursuit. In this gaze contingent task, the trial begins with the computer driving the target on the monitor back and forth horizontally and subject's eye following the target (bottom panel; left axis shows the distance traveled in degrees of visual angle from left 15 degrees [−15 degrees] to right 15 degrees and back). During one of the times when the target is changing direction, unbeknownst to the subject the software switches the control such that that now the eye drives the target (for details, see Hong et al.120). Because this happens at the time of change of direction, now the eye based on the internal representation of the target behavior initiates pursuit in the expected direction of the target motion (shown by thick dashed lines in the bottom panel). As can be seen, this subject shows good predictive pursuit with the eye velocity being slightly less than the expected target velocity (slope of the eye trajectory in the bottom panel or the velocity tracing in the top panel shows the magnitude of the eye velocity). Predictive pursuit is abnormal in schizophrenia, highly heritable, and associated with functional variation COMT gene.22,32](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/schizophreniabulletin/34/4/10.1093/schbul/sbn049/2/m_schbulsbn049f01_ht.gif?Expires=1716421711&Signature=ct4XHrD51FaPSAWc-GFgA71uR3-bzY~oUDfvs9f99JvsENMwohH37S1fDHCKNi20LKOER0Z945hsH2C-6MIM9t-IUAVTgTW11tPqyA-YAZVihdw8zrzEYYQQQAQlolcsjvO9woQYFZiaQQWJAhNsgQ8gDqnBsE-mWPg4mnCft9s8JhvZ7TXVPU6J2lv4M0wE3cUirNQM4P56b4icqMFB2eYZ58lVV6vRMgNxMLUif8mTmqP~Mi7cTqL2XaQhkVKFRG0vBhGkC7tqCCpY6YjBzdsmMRL4Rcp7mFboAAoq62M0v456wv~hTuE4atYESAyHfqnvVvSlxKWeTQjheqSR0A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Smooth pursuit eye movements are broadly divided into 2 components: (1) response to the processing of image motion on the retina and (2) the predictive response based on the internal representation of the target motion (ie, memory trace of the previous image motion on the retina and copy of the motor command driving the eye called efference copy). The first component is measured at the time of pursuit initiation when the target image starts moving away from the fovea. The figure illustrates one method of measuring predictive pursuit. In this gaze contingent task, the trial begins with the computer driving the target on the monitor back and forth horizontally and subject's eye following the target (bottom panel; left axis shows the distance traveled in degrees of visual angle from left 15 degrees [−15 degrees] to right 15 degrees and back). During one of the times when the target is changing direction, unbeknownst to the subject the software switches the control such that that now the eye drives the target (for details, see Hong et al.120). Because this happens at the time of change of direction, now the eye based on the internal representation of the target behavior initiates pursuit in the expected direction of the target motion (shown by thick dashed lines in the bottom panel). As can be seen, this subject shows good predictive pursuit with the eye velocity being slightly less than the expected target velocity (slope of the eye trajectory in the bottom panel or the velocity tracing in the top panel shows the magnitude of the eye velocity). Predictive pursuit is abnormal in schizophrenia, highly heritable, and associated with functional variation COMT gene.22,32
Over the last century, numerous studies have reported abnormality in the smooth pursuit eye movements in schizophrenia (for recent reviews, see Holzman,23 Calkins and Iacono,24 and Levy et al.25). The first observation of the smooth pursuit eye movement abnormality in schizophrenia was made by Diefendorf and Dodge26 in 1908, and the significance of these findings became apparent after its rediscovery by Holzman and colleagues, who also noted a similar abnormality in the non-ill relatives of schizophrenia probands.27,28 Since then about 20 studies have examined smooth pursuit eye movement function in the relatives of schizophrenia patients with vast majority of studies finding abnormality in a proportion of the first-degree relatives of schizophrenia probands.29–48 Recent studies have extensively refined the phenotype in order to identify a more specific physiological deficit(s) associated with schizophrenia. These studies suggest that the pursuit system is highly complex and there may be more than one abnormality within the smooth pursuit system associated with schizophrenia liability. Attentional impairment may contribute to the poor eye tracking, but this does not fully explain poor pursuit in schizophrenia.49 Studies note that schizophrenia subjects make significantly more leading or anticipatory saccades that may indicate abnormality in inhibition of the saccadic system44,49,50. Several studies, but not all, have noted low initiation acceleration in schizophrenia (for review see Hong et al.51). The inconsistency in findings may be due to the fact that the impairment occurs in a subgroup of patients, those with primary and enduring negative symptoms (ie, deficit syndrome probands51). A preliminary report that showed that pursuit initiation abnormality also occurs in relatives of deficit syndrome patients suggests that this specific impairment is marking the liability to develop negative symptoms.51 Consistent with these findings, a recent study observed that early motion processing deficit, which is likely indexed by the initiation impairment, correlates with negative symptoms in schizophrenia.52
It is interesting to note that pursuit initiation acceleration (response to the retinal motion) and the predictive pursuit (response to the extraretinal motion information) are independent of each other and independently contribute to the pursuit maintenance.53 Interestingly, contributions from these 2 components toward maintaining smooth pursuit is different in the first-degree relatives of schizophrenia probands than in the healthy comparison subjects such that normal pursuit maintenance is dependent more on the extraretinal and less on the retinal motion information compared with the relative group.53 The increased dependence on the retinal motion information to maintain pursuit on the part of schizophrenia spectrum subjects may be due to compensation for impaired extraretinal processing. Many patients and their relatives are able to normally pursue the moving target using such a compensatory mechanism.20 Several studies using varied methods now show that schizophrenia probands and their relatives have impaired predictive pursuit, which is thought to be partly due to impaired processing of the efference copy. Impairments in the processing of the efference copy, whether it is in the visual, auditory, or other sensory-motor systems, are thought to contribute to perceptual and reality distortions.54,55. Thus, predictive pursuit abnormality observed in schizophrenia and their relatives may be marking liability to positive psychotic symptoms. This raises the question whether this deficit is also present in patients with other psychotic disorders.
Data in other psychotic disorders are not as rich, and at times conflicting. Some but not all studies note pursuit abnormality in patients with affective disorders even when these patients were in relative remission and not on treatment.44,56–58 More significantly, Rosenberg et al.44 demonstrated that relatives of bipolar patients show pursuit abnormality similar to the relatives of schizophrenia probands in the New York High-Risk sample. These earlier findings were recently replicated.41 Interestingly, a recent study in dyslexia noted that a deficit in detecting coherent motion is associated with low accuracy on reading tests, while ability in discriminating velocities was associated with slow performance on these same tests.59
Two linkage studies in small samples of schizophrenia pedigrees used smooth pursuit eye movement abnormality as a phenotype in the genetic analyses with both reporting a significant linkage to chromosome 6p21-23 region.59,60 Earlier, Kendler et al. also reported a significant linkage to this region using a schizophrenia diagnosis as a phenotype in their Irish sample. Interestingly, the same chromosomal region is implicated in dyslexia, a disorder that is associated with motion processing deficit. Candidate genes of interest in this region are dysbindin, TNF alpha, gamma amino butyric acid (GABA)-B-1 Retinoid X receptor, KIAA0319 and DCDC2 genes. The last 2 have been shown to be associated with dyslexia.61 Our group reported an association of catechol-O-methyltransferase (COMT) gene with the predictive pursuit. Interestingly, the effects of COMT on the pursuit measure interacted with the diagnostic group. In healthy control subjects, low-activity MET/MET COMT genotype, which results in increased synaptic dopamine, was associated with better predictive pursuit than VAL/VAL genotype. The COMT genotype has the opposite effect on the predictive pursuit in the schizophrenia patients than in healthy control subjects.62 A similar finding (ie, improvement with higher dopamine activity in healthy subjects and the opposite effect in schizophrenia patients) was observed in a preliminary study with dopamine transporter (DAT1) gene.63 The opposite effect in the schizophrenia group compared with healthy subjects suggests effects of environmental factors (eg, aberrant stress-related response64) or the presence of another genetic factor that modulates the effects of COMT genotype on the endophenotype.
In conclusion, abnormality(ies) in smooth pursuit eye movements is one of the consistently reported findings in schizophrenia and their relatives. There are only a few studies in probands with bipolar disorder and their relatives, and the findings are mixed. There are 2 positive linkage studies using smooth pursuit eye movements as a phenotype in schizophrenia, both noting a significant linkage to chromosome 6p21-23 locus.
Sensory gating (P50) deficit
The nervous system is constantly bombarded by incoming sensory information and is faced with a task of screening out unwanted and redundant stimuli while selecting sensory information for further processing. This is accomplished by several mechanisms during preattentive and attentive stages of information processing. The initial screening during the preattentive stages of information processing occurs by gating (or screening out) of the redundant stimuli. This process is studied in the laboratory by examining the scalp electro-encephalography (EEG). In the laboratory, the scalp EEG registers a small positive change in the electrical potential around 50 ms following a presentation of a stimulus (P50), which is usually hidden in the noise but can be uncovered by averaging across a large number of trials that cancels out the noise and retains the evoked response. The inhibition of this positive wave occurring at 50 ms (P50 gating) after a redundant (second) stimulus indexes the sensory gating (figure 2). Schizophrenia patients show none or muted inhibition (see figure 2 as an illustration of lack of P50 gating), and this deficit is not consistently affected by medication status or clinical state.65–68 de Wilde et al.69 recently carried out a meta-analysis of published P50 studies in schizophrenia and estimated the average effect size to be 1.28 (SD = 0.72) when comparing schizophrenia proband with healthy control subjects. The meta-analysis was based on 28 studies that included a total of 891 schizophrenia subjects and 686 control subjects. They also examined 6 studies carried out in the relatives of schizophrenia probands and noted an estimated effect size of 0.85 (SD = 0.42). Based on their calculations of the number of additional studies necessary to reverse the overall probability obtained in the meta-analysis, the authors concluded that these findings were not due to publication bias. However, they noted a high variability in the effects sizes across studies that could at least in part be explained by variability in subject sample and testing procedures.69 Diminished suppression of P50 auditory stimulus response is also demonstrated in patients with bipolar disorder with a lifetime history of psychosis, whereas subjects with bipolar illness and no history of psychosis did not exhibit abnormal P50 suppression.70,71 Recent studies have noted P50 suppression abnormality in the relatives of bipolar disorder I probands with psychotic features.72–74
The figure illustrates P50 gating measure. Evoked potential responses to S1 (solid line) and S2 (dashed line) stimuli are overlaid in the figure for illustrative purposes. Subject was presented with 150 trials of paired clicks (S1 and S2) 500 ms apart; the intertrial interval was 10 s. The figure gives averaged data across all trials that were not rejected because of artifacts. P50 amplitudes were measured by the absolute differences between the positive peak and the preceding negative trough occurring near 50 (operationally, S1 amplitude is within 35–70 ms of stimulus, and S2 within a window of the S1 latency ± 10 ms for the subject). As one can see, the S2 amplitude was not reduced in this schizophrenia subject with S2/S1 ratio being about 1 suggesting failure to gate.
Although P50 gating is thought to be a deficit in preattentive phase of information processing, studies find a more pervasive sensory gating abnormality throughout the mid-latency auditory-evoked responses, both in preattentive and attentive phases of the information processing.75 The underlying mechanism of the gating remains unclear, but in order for the recognition that the second stimulus is redundant, there has to be appropriate processing of the first stimulus beyond perception. Studies note that the initial response to S1 is observed as a high gamma band oscillatory activity. However after about 200 ms, the response switches to beta frequency oscillations which may reflect an encoding of the sensory perception.76–79 This post–S1 beta frequency response is inversely correlated to the S2 response in patients with schizophrenia.80–83 Examination of evoked responses to S1 and S2 in frequency domain allows one to evaluate the gating responses in single trials.84 This refined method may yield a more “elemental” phenotype than the measure based on averaging across trials. This is because rhythmic activity of neural assemblies codes information processing within and across cortical circuits and modulates neuronal excitability.78,85–87 Hong et al.84 used a discrete wavelet transform technique to identify single-trial oscillatory components in response to S1 and S2 stimuli and identified a finite range of time-frequency specific oscillatory components. In this study, oscillatory components in beta and alpha frequency range best indexed the strength of sensory gating in healthy controls.
P50 gating is served by a widely distributed neuronal circuitry that includes temporoparietal and prefrontal cortical regions, particularly during the early phases of processing and hippocampus during the later phase of gating. The CA3-CA4 area of the hippocampus is thought to play an important role involving the cholinergic input from the septal nucleus that is mediated by low-affinity nicotinic receptors88 and affects CA3-CA4 interneurons. In turn, these GABAergic interneurons transiently inhibit pyramidal neurons and thus mediate the gating of the second stimulus. The role of low-affinity nicotinic receptors in the P50 gating abnormality in schizophrenia is supported by genetic,89 physiological,89 and pharmacological90,91 studies. Noradrenergic system is also thought to play a role suggested by disruption of gating by alpha-2–selective noradrenergic antagonist yohimbine and a significant correlation of 3-methoxy-4-hydroxyphenylglycol levels with P50 gating.92 Clozapine, which affects cholinergic as well as noradrenergic activity in the brain, normalizes P50 gating.
Arguably, sensory gating deficit may lead to sensory overload, and perhaps positive psychotic symptoms. Empirical data supporting this theory are inconclusive in patients. However, this correlation is better examined in individuals with schizophrenia spectrum personality traits where positive symptoms are trait-like; a recent such study reports a positive correlation between poor P50 suppression and perceptual anomalies and magical thinking. Schizophrenic patients with predominantly negative symptoms are not different on the P50 measure compared with other patients.93 Potter et al. recently reviewed the relationship between P50 gating deficit and cognitive impairments in schizophrenia. They noted that most studies have not found significant correlations of P50 gating deficits with cognitive impairments in schizophrenia.94 However, modest correlations are observed between measures of sustained attention and p50 gating.94
Freedman et al.95 provided evidence for linkage to chromosome 15q14. An association study has implicated the promoter region of the alpha-7 nicotinic cholinergic receptor subunit gene.89 Interestingly, the presence of promoter polymorphisms in control subjects as well as in bipolar subjects was associated with failure to inhibit P50.96
In conclusion, numerous studies report P50 sensory gating abnormality in schizophrenia. The abnormality is also observed in their relatives. Although, not as extensively studied, similar findings are also noted in families of bipolar disorder probands with psychotic features. A linkage study implicate chromosome 15q14 locus in the P50 abnormality. Several studies suggest an association of the gating deficit with variations in alpha-7 nicotinic receptor gene.
Sensory-motor gating: PPI
Conceptually “sensory-motor gating” is similar to sensory gating in that it helps filter out excessive stimuli so that organism can focus on the salient features of the environment.97 This sensory-motor gating is traditionally examined in the context of the startle reflex, which is a defensive motor response consisting of sudden contractions of skeletal and facial muscle groups in response to a sudden, loud, or excessive auditory (can be tactile or visual) stimulus. When a muted stimulus precedes the loud sound (or other strong stimulus), the response is inhibited (figure 3).98 Usually, this is observed when the muted stimulus (prepulse) precedes the strong stimulus (pulse) by 30–300 ms.98 The prepulse inhibition occurs even when the prepulse and pulse use different sensory modalities.98 There are about 20%–30% of individuals in whom startle reflex does not occur consistently, making it difficult to evaluate prepulse inhibition. In individuals where one is reliably able to measure startle response, the PPI is a trait-like phenomenon with high test-retest reliability.
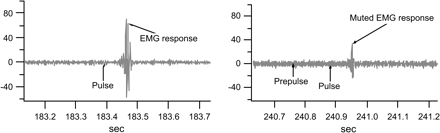
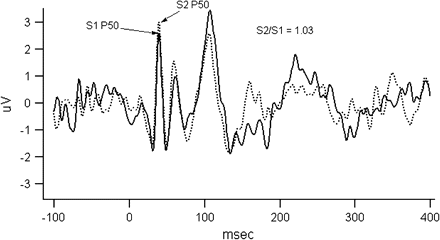
The figure shows examples of recording of the orbicularis oculi electromyographic (EMG) activity in response to pulse alone (left panel) and prepulse-pulse stimulus (right panel). In this experiment, subjects were acclimatized to a 3-min period with 70-dB white noise; startling pulse stimulus was 116 dB white noise lasting 40 ms, and the prepulse was a 20-ms, 80 dB white noise. The EMG recording was processed offline with a 100-Hz high pass filter and baseline correction using 100 ms prestimulus baseline. Response onset was defined by the first crossing from the baseline activity within a 20–120 ms window after stimulus onset. Peak response amplitude was calculated by the difference of the most positive peak and most negative trough in a 20–150 ms window after pulse onset.
PPI is typically examined in humans by measuring the magnitude of contraction of orbicularis oculi muscle using electromyography. Data suggest that when the prepulse precedes the pulse by 120 ms, the maximum inhibition is observed. Several studies carried out over the past 30 years have shown that individuals with schizophrenia show a reduced inhibition associated with the prepulse, even when the startle reflex is generally within the normal range (for review, see Braff et al.97). Although many drugs, including drugs that affect dopaminergic system, modulate PPI, the sensory-motor impairment observed in schizophrenia is not due to their drug treatment or any other disease related secondary factors such acute psychotic symptoms or medications. PPI impairment is seen in relatively nonpsychotic patients, patients not on medications, schizotypal personality disorder patients, and in first-degree relatives who are clinically unaffected.97 The observed impairment in schizophrenia is stable and thought to mark schizophrenia liability.97 The heritability estimates associated with the PPI range from 0.32 to 0.50.99
If PPI impairment marks vulnerability to psychosis as suggested by the data, then the deficit may also be observed in other psychotic disorders. Indeed, Perry et al.100 reported abnormal response by the preceding prepulse in acutely manic patients. Subsequent studies also noted similar findings in remitted bipolar disorder patients,101–103; however, there were 2 negative studies in remitted adult and pediatric bipolar disorder patients. 104,105 It is possible that differences in the medications across different studies may explain these inconsistencies. Consistent with this argument are the findings of abnormal PPI in the relatives of bipolar patients.101,106,107 The latter findings strongly suggest that PPI deficit, as in schizophrenia, may mark the genetic predisposition to bipolar disorder (perhaps marking the psychosis in this disorder). In addition, PPI impairment is observed in several other neuropsychiatric disorders including obsessive-compulsive disorder, comorbid attention-deficit hyperactivity disorder and tic disorder, and Huntington's Disease (for review, see Braff et al.97). These disorders may share the impairment in the limbic cortico-striatopallido-pontine circuitry that is thought to mediate the inhibition of the startle response.
The PPI is easily modeled in laboratory animals, and the measure is one of the most extensively studied endophenotypes across species with extensive data on pharmacological response. The neurobiology of PPI has been extensively investigated both in animal and human studies. Data suggest that several cortical and subcortical regions are involved in mediating the inhibition of the startle response. The limbic cortico-striato-pallido-thalamic circuitry mediating the inhibition of pulse response include hippocampus, amygdala, and mesial temporal and medial prefrontal cortical regions as well as several subcortical regions including striatum, pallidum, and nucleus accumbens.108,109 Modulation of dopaminergic system within this circuitry reliably affects the PPI response. Dopamine agonist impairs PPI both in animal models and humans. Interestingly, dopamine antagonist haloperidol has a similar but milder effect in some studies; haloperidol is shown to reliably block the effect of dopamine agonist. Depletion of tryptophan impairs PPI.110 In humans, the effects of ketamine, an NMDA antagonist, on PPI are complex with some studies showing no effects or increased inhibition, while others decreased inhibition if ketamine is combined with haloperidol.111–114 Nicotine also plays a role in modulating the PPI response; acute nasal administration of nicotine reverses the PPI deficits in schizophrenia patients.115–117
Following the initial observations of impaired gating in schizophrenia, Braff and others speculated that failure to gate overwhelming sensory information may lead to a breakdown in the sensory processing system resulting in perceptual distortions and other psychotic symptoms.97,118,119 However, examination of the relationship between positive or negative symptoms and PPI has yielded mixed results; this may be due to the problem of examining correlations between state and trait-related measures.
Although several studies have examined the genetics of PPI in laboratory animals,120,121 few studies have done so in humans. The original report on the association of neuregulin 1 with schizophrenia had noted that neuregulin knockout mice have PPI deficits. Based on these findings, our group examined the effects of variation in the neuregulin 1 gene on PPI in schizophrenia and healthy control subjects. The study found that a missense mutation in the gene significantly affected PPI in both subject groups.122 Based on animal studies suggesting a significant role of dopamine and related genes on PPI,123 several investigators have examined this relationship in human genetic studies and note significant effects of variations in COMT and dopamine receptor 3 (DRD3) genes on the PPI in humans.124,125 Preliminary findings from our laboratory suggest that another dopamine-related gene, dopamine transporter gene (DAT1), affects PPI in Caucasian healthy control subjects but not schizophrenia patients. The negative finding in patients may be related to the modulating effects of their antipsychotic treatment on the association between PPI and DAT1.126
In conclusion, PPI is found to be abnormal in schizophrenia probands and their relatives. Similar findings are also observed in probands with bipolar disorder (although there are a few negative studies in euthymic patients) and their first-degree relatives. Several genetic studies suggest associations of PPI deficit with dopamine-related genes. Neuregulin 1 knockout mice show abnormal PPI; a recent study shows that missense mutation in this gene results in deceased PPI in schizophrenia and healthy control subjects.
P300 component of the evoked potential
In response to an infrequent, task relevant or novel stimulus embedded within a train of repeated stimuli, there is a change in brain activity reflected by a positive deflection on the scalp EEG occurring about 300 ms after the oddball stimulus. This positive deflection at around 300 ms (P300) is thought to index cognitive processes such as updating working memory (ie, “context updating”) and directed attention. In the laboratory, P300 component of the event-related potential (called P300 henceforth) is extracted from the background EEG and noise associated with the measurement by presenting many trials and then averaging across trials to reduce noise. The main 2 outcome measures are the latency of the response and the amplitude of the response. Recent studies have examined this response under different target conditions that recruit different cognitive processes and elicit different subcomponents of the P300, P3a and P3b. P3a indexes frontal attention engagement mediated by precentral areas and insula. It is observed in response to a novel or unexpected stimuli within a train of repeated stimuli.127 P3a occurs little early and is observed to be most robust in the frontal and central electrodes. In contrast, P3b is observed most robustly in the posterior regions (parietal and inferior temporal areas) and is thought to index context updating of the working memory. Other studies have noted topographic anteriorization of the P300 signal associated with NoGo condition in a Go-NoGo task that elicits response inhibition; the robust response in the anterior electrodes likely indexes anterior cingulate and other prefrontal cortical activity under the NoGo condition.128
In general, schizophrenia patients do not significantly differ from healthy subjects in their ability to recognize the novel or the unexpected stimulus randomly presented among a series of repeated stimuli. In spite of the comparable performance, the P300 response is significantly different in the patient group showing reduced P300 response amplitude and increased latency.129 Although the measure is affected to some extent by state-related factors,130 findings of similar impairment in individuals with schizophrenia spectrum disorders,131 and in relatives of schizophrenia probands132–134 suggest that the deficit is heritable and related to the etiology of schizophrenia. Several studies have calculated heritability estimates based on twin pair samples and noted that both the latency and the amplitude measures are highly heritable.135–138 van Beijsterveldt and colleagues carried out a meta-analysis of these data and report an aggregate heritability estimate of 60% for P300 amplitude and 51% for P300 latency.138
Several studies examined P300-evoked potential in probands with bipolar disorder and noted reduced amplitude and prolonged latency in this patient group.139–141 Salisbury et al.141 noted that topography differed in bipolar patients compared with schizophrenia subjects group. Bipolar patients showed anterior reduction while schizophrenia showing posterior reduction compared with the healthy control subjects. P300-evoked potential has not been extensively examined in the relatives of bipolar disorder patients. A recent study noted delayed P300 latency (but normal amplitude) in bipolar probands and their relatives.142 These data suggest that P300 component of the event-related potential may mark illness liability in bipolar disorder.
Several dopamine-related genes including DRD2, DRD3, and COMT are known to affect P300 measure.143–145 Blackwood and colleagues noted that (1;11) translocation, that disrupts DISC1 and DISC2 genes located on chromosome 1q42, was significantly linked to clinical syndromes defined by both schizophrenia and affective disorders (including bipolar disorder). The translocation carriers, regardless of whether they had clinical phenotype, showed decreased P300 amplitude and increased latency similar to the findings in schizophrenia patients. These findings suggest a role of DISC1 and DISC2 genes in P300 both in individuals with vulnerability to schizophrenia or affective disorders.146 Several studies also note similar findings in patients with bipolar disorder147 and their first-degree relatives.148 Abnormality is also noted in patients with alcoholism149 and in patients with depression; in the latter group, the P300 normalizes after antidepressant treatment.
Other electrophysiological measures
There are several other electrophysiological measures that have not been as extensively examined in schizophrenia as those described above but still show promise as schizophrenia/psychosis endophenotypes. These include MMN, which is a negative ERP that occurs between 100 and 250 ms of a “deviant” stimulus most consistently observed in response to an auditory stimulus. As the name suggests, the response is elicited by the deviancy from an expected event, eg, presentation of a different tone in a series of auditory stimuli or a repetition of a tone in a sequence of descending tones. In the latter instance, the stimulus eliciting the MMN is the same as the preceding tone; however, the fact that it is deviant from what was expected is the critical feature eliciting the response.150 The magnitude of deviancy from the expected event correlates with the amplitude of the MMN. Several studies now show impaired MMN in schizophrenia (for review, see Michie151); a similar impairment is also seen in the relatives of schizophrenia probands suggesting that the deficit marks schizophrenia liability.152,153 Although the measure has not been extensively studied in bipolar disorder, the available literature suggests that the MMN is normal in bipolar patients.154 Studies in schizophrenia suggest that the MMN impairment correlates with their negative symptoms,155 and with functioning (impaired individuals more likely to live in highly structured environment, while less MMN-impaired patients tend to live in independent settings.156
In contrast to the measures discussed above, which are based on time-domain analyses and average EEG signals across different frequencies, recently there has been interest in examining brain's electrical activities in frequency domain. There are several advantages to frequency domain analyses. These measures may better represent the underlying neuronal activities than the time-domain–based measures. These analyses have particularly helped with understanding the mechanism for integrating sensory information across different modalities and cortical areas and are less vulnerable to variability in measurement introduced by the scalp-related factors. Recent studies in medicated schizophrenia have found reduced 40 Hz steady-state auditory-evoked potential (SSAEP) that is generated by applying repetitive auditory stimuli in at 40 Hz to entrain the EEG to the same frequency.157 Reduced power observed in gamma band in schizophrenia patients has been replicated by several subsequent studies.158–160 A study by Hong et al. suggested that these impairments may be heritable occurring in first-degree relatives of schizophrenia patients. This study also suggested that certain antipsychotic medications may affect the measure.161 Spencer et al.162 showed that a measure of gamma oscillation was positively correlated with positive psychotic symptoms such as visual hallucinations, thought disorder, conceptual disorganization, and attention. In other disorders with psychosis, such as bipolar disorder, similar reductions of the 40-Hz SSAEP are also noted.163 Several neurotransmitters including glutamate, acetylcholine, and GABA are to thought mediate gamma band synchronization.164–170
In conclusion, there are several physiological deficits observed in schizophrenia that are stable, independent of state-related factors, and are also observed in a proportion of non-ill first-degree relatives. The latter suggests that these impairments are familial, and likely genetic, in nature. Several studies have examined the heritability estimates and find that most of these neurophysiological measures are associated with moderate to high heritability. Recent data suggest that many of these physiological deficits are independent of each other. In the context of schizophrenia being a complex and multifactorial syndrome, it is possible that these independent physiological deficits represent effects of different genetic factors. Physiological deficits in various combinations lead to clinical phenotypes that are part of the schizophrenia syndrome. Thus, some of the physiological deficits may mark the liability to develop a negative symptom phenotype, while others mark a psychosis phenotype. The latter impairments are also observed in other psychotic illnesses particularly bipolar disorder. Although these measures have not been extensively studied in the families of bipolar disorder probands, data suggest that some but not all of these impairments also run true in bipolar families. As suggested by Gottesman and several other investigators, these core, heritable physiological deficits provide important tools to potentially identify liability genes of major psychotic disorders, clarify the etio-pathophysiologic pathways in these disorders, understand the extent of overlap among the psychotic disorders, and identify novel treatment approaches.
Funding
NIMH grants MH077852-02, MH-67014, and MH-49826; General Clinical Research Center grant M01-RR16500; and the VA Capitol Health Care Network (VISN 5) Mental Illness Research, Education, and Clinical Center (MIREC).

{kind=link}
{kind=link}
{kind=link}