




Abstract
Selection technologies such as phage and ribosome display, which provide a physical linkage between genetic information and encoded polypeptide, are important tools for the engineering of proteins for diagnostic and therapeutic applications. We have recently described a selection strategy called covalent DNA display, in which individual proteins are covalently linked to the cognate encoding DNA template in separate droplets of a water-in-oil emulsion. We here report on the optimization of several experimental steps in covalent DNA display technology, such as the elution conditions and the PCR strategy used for the amplification of selected DNA templates. A PCR assembly strategy was developed, which allows the amplification of the DNA templates over repeated rounds of selection. In addition, we could demonstrate that ∼50% of the DNA templates form a covalent adduct with the corresponding proteins in the compartments of a water-in-oil emulsion. In model selection experiments, differences in recovery efficiency <100 000 per round of selection could be observed when comparing a specific binding polypeptide with a binder of irrelevant specificity. Furthermore, the optimized protocol was successfully applied for the selection of single domain proteins, capable of specific binding to mouse serum albumin (MSA). A mutant derived from the SH3 domain of the Fyn kinase, with millimolar affinity to MSA, was affinity matured using covalent DNA display and yielded several MSA binding FynSH3 variants with dissociation constants in the 100 nM range.
Introduction
The development of next-generation protein drugs with optimal biophysical properties is dependent on the availability of powerful selection technologies and high-throughput screening methods. In order to obtain a potent biopharmaceutical with a good safety profile, protein characteristics such as target binding (affinity and specificity), stability, solubility and the pharmacokinetic profile have to be optimized.
The engineering of the biophysical properties of a protein can be performed by generating a large pool of protein mutants, followed by the identification of those polypeptides, which have the desired biological activity (e.g. binding to a target molecule of interest). The generation of protein repertoires comprising billions of different protein variants is relatively straightforward, whereas the identification of those few protein mutants in the repertoire having a desired phenotype remains a daunting challenge. Screening each of the mutants one by one is very time consuming and, therefore, selection technologies have been developed that allow the rapid identification of protein variants with desired characteristics.
A variety of different selection technologies are described in the literature, and all of them require a physical linkage between the genotype (genetic information encoded by DNA or RNA) and the corresponding phenotype (typically a binding or catalytic activity of the encoded protein or RNA). A first class of selection techniques, such as phage display ( Winter et al. , 1994 ), yeast surface display ( Feldhaus et al. , 2003 ) and bacterial surface display ( Olsen et al. , 2000 ), need living cells in at least one step of the selection procedure, whereas alternative methods such as ribosome display ( Mattheakis et al. , 1994 ; Schaffitzel et al. , 1999 ), mRNA display ( Roberts and Szostak, 1997 ), CIS display ( Odegrip et al. , 2004 ) and DNA display ( Tabuchi et al. , 2001 ) take place fully in vitro . Some in vitro selection strategies make use of artificial, cell-like compartments [ in vitro compartmentalization ( Tawfik and Griffiths, 1998 ; Sepp et al. , 2002 ; Griffiths and Tawfik, 2003 )] for the linkage of phenotype and genotype. In vitro compartmentalization has been used to directly link DNA templates with the encoded polypeptide ( Doi and Yanagawa, 1999 ; Yonezawa et al. , 2003 ).
The first class of selection methodologies involving live organisms is well established and relatively robust. However, all these methodologies require the transformation of living cells with suitable plasmid DNA, a process that can be laborious and limit library size.
In order to circumvent the transformation of living cells, research has been focussed on the development of selection strategies such as ribosome display, which take place fully in vitro . However, the ternary complex of mRNA, ribosome and displayed polypeptide is stable only under certain conditions (high salt concentrations, low temperatures), thus restraining the experimental conditions in which selections can be performed. In addition, RNA is very sensitive to enzymatic contaminations, which may lead to a rapid degradation of the RNA.
We reasoned that an ideal in vitro system for the directed evolution of proteins would comprise the following features: (i) rapid generation of genetic diversity (library), (ii) facile and irreversible linkage between phenotype (protein) and genotype (genetic material), (iii) high expression levels of protein mutants in the format used for the selection process, (iv) a carrier of genetic information, which is stable under a wide range of conditions (e.g. high and low salt concentrations, extremes of temperature and pH, denaturing agents) and (v) control of the number of proteins to be linked to the genetic material (valence of display).
Recently, we have proposed a selection technology ( Bertschinger and Neri, 2004 ) called covalent DNA display, where a library of linear DNA molecules is co-packaged with an in vitro transcription/translation mixture in the compartments of a water-in-oil emulsion ( Tawfik and Griffiths, 1998 ) (Fig. 1 A, B). Ideally, one DNA molecule is present in each water compartment and is used as a coding template for the synthesis of a fusion protein that consists of a constant domain and a variable domain. The constant domain M.Hae III (Hae III DNA-methyltransferase of Haemophilus aegypticus ) of the fusion protein can react with a modified methylation target DNA sequence (5′-GGFC-3′; F = 5-fluoro-2′-deoxycytidine) ( Chen et al. , 1991 ) site within the DNA molecule that is present in the same water compartment, and thereby a covalent DNA-protein fusion is formed. M.Hae III fusion proteins linked to their encoding DNA template in each compartment of the water-in-oil emulsion are extracted from the emulsion. The DNA molecules, which display a protein with the desired binding specificity, are selected from the pool of DNA-protein fusions by affinity panning. The genetic information of the selected DNA-protein fusions is amplified by PCR and can be used either for a further round of selection or for cloning and characterization of the selected mutants (Fig. 1 A).
![(A) Concept of covalent DNA display: A library of linear DNA molecules is co-packaged with an in vitro transcription/translation mix into a water-in-oil emulsion (1). Ideally, one compartment contains one DNA molecule. The DNA molecules each code for a Hae III DNA-methyltransferase fusion protein and contain a mechanism-based inhibitor for the covalent cross-linking of the DNA-methyltransferase fusion proteins (green star) [see also (B)]. After in vitro expression and formation of the DNA-protein complexes, the water phase is extracted from the emulsion (2) and DNA molecules displaying a protein with desired binding properties are selected from the pool of DNA-protein fusions by affinity selection (3). The genetic information of selected DNA-protein fusions is amplified by PCR (4) and can either be used for a further round of selection (5) or for cloning and characterization of the selected mutants (6). (I–III) Improvements in the selection procedure presented in this work: (I) visualization of covalent DNA-protein adducts formed in the water compartment of a water-in-oil emulsion; (II) general elution procedure compatible with subsequent PCR amplification of eluted DNA templates; (III) PCR strategy allowing for repeated rounds of affinity selection and amplification of functional DNA molecules. (B) Enlarged view of a water compartment of the water-in-oil emulsion: the DNA molecule is transcribed into mRNA, which is translated into a fusion protein consisting of two domains: the N-terminal Hae III DNA-methyltransferase (yellow) and a C-terminal potential binding domain (red). Due to the catalytic activity of the Hae III DNA-methyltransferase, the fusion proteins form a covalent bond via the modified methylation target sequence 5′-GGFC-3′ (F = 5-fluoro-2′-deoxycytidine) with their encoding DNA molecule, which is present in the same compartment of the water-in-oil emulsion.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/peds/20/2/10.1093_protein_gzl055/2/m_gzl05501.jpeg?Expires=1716628985&Signature=UwEctE2V9JRcmqqT2r4qHoSc6IrON1Kvtw9LdOyDg3gy1qGsaVIE0PwHKmhslRfIMbh8GA5~m-D4mdUfBD6GisTswZyvku8POkYCg6ugcqkubw32hJNNs4T7vcmRKEmCaJvQLXOSrJdUyJXOlIWK3MdeiQ-WJVwQZupDexAO5pp9cTgXs10UCHTt6zGWAIQs2tE~fe9XeQTaHsxITPkuOSOoR4Ikhtwh5sZH6S6INSDGDOk7X-PArrBan3aM8Cd3jB6APh9PF~x46RKU0E5Lv-7L4mZU~DHjxhB5q0uoy6XPARP742oH06WhNACBdCWnfeoA58x0L22gI6ep9xSH2w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
(A) Concept of covalent DNA display: A library of linear DNA molecules is co-packaged with an in vitro transcription/translation mix into a water-in-oil emulsion (1). Ideally, one compartment contains one DNA molecule. The DNA molecules each code for a Hae III DNA-methyltransferase fusion protein and contain a mechanism-based inhibitor for the covalent cross-linking of the DNA-methyltransferase fusion proteins (green star) [see also (B)]. After in vitro expression and formation of the DNA-protein complexes, the water phase is extracted from the emulsion (2) and DNA molecules displaying a protein with desired binding properties are selected from the pool of DNA-protein fusions by affinity selection (3). The genetic information of selected DNA-protein fusions is amplified by PCR (4) and can either be used for a further round of selection (5) or for cloning and characterization of the selected mutants (6). (I–III) Improvements in the selection procedure presented in this work: (I) visualization of covalent DNA-protein adducts formed in the water compartment of a water-in-oil emulsion; (II) general elution procedure compatible with subsequent PCR amplification of eluted DNA templates; (III) PCR strategy allowing for repeated rounds of affinity selection and amplification of functional DNA molecules. (B) Enlarged view of a water compartment of the water-in-oil emulsion: the DNA molecule is transcribed into mRNA, which is translated into a fusion protein consisting of two domains: the N-terminal Hae III DNA-methyltransferase (yellow) and a C-terminal potential binding domain (red). Due to the catalytic activity of the Hae III DNA-methyltransferase, the fusion proteins form a covalent bond via the modified methylation target sequence 5′-GGFC-3′ (F = 5-fluoro-2′-deoxycytidine) with their encoding DNA molecule, which is present in the same compartment of the water-in-oil emulsion.
In our previous work ( Bertschinger and Neri, 2004 ), we could enrich DNA molecules in model selection experiments on the basis of the protein they encode from an excess of DNA molecules coding for a protein with irrelevant binding specificity. In this article, we show that M.Hae III fusion proteins can be expressed and covalently cross-linked with their encoding DNA molecule within the water compartments of a water-in-oil emulsion, and that the covalent bond is necessary for the efficient recovery of DNA molecules coding for binding proteins in model selection experiments. In addition, we have optimized several steps of the selection protocol, such as emulsion preparation, elution of the selected DNA molecules and their subsequent PCR amplification (Fig. 1 A).
Finally, we have applied the optimized experimental protocol for the isolation of a single domain protein binding to mouse serum albumin (MSA). Over the last few years, several research groups and biotech companies have focussed their research on the development of small globular proteins as substitutes for antibody-based drugs with improved biophysical and, eventually, therapeutic properties (reviewed in Binz et al. , 2005 ; Hey et al. , 2005 ). Albumin represents a particularly attractive target, since it can modulate pharmacokinetic properties in vivo ( Dennis et al. , 2002 ; Nguyen et al. , 2006 )
We have used a src homology 3 (SH3) domain called FynSH3 (amino acid residues 83–145 in the sequence reported by Kawakami et al. , 1986 ; Semba et al. , 1986 ) as a scaffold for the construction of a protein repertoire, from which we selected FynSH3 mutants that specifically bind to MSA. FynSH3 is a small domain of 63 amino acid residues of the proto-oncogene tyrosine-protein kinase Fyn, and its amino acid sequence is fully conserved between man, mouse, rat and monkey (gibbon). FynSH3 domains from chicken and from Xenopus laevis differ from their human counterpart at one and two amino acid positions, respectively. FynSH3 is composed of two antiparallel β-sheets and contains two flexible loops (called RT-Src and n-Src-loop) to interact with other proteins. We were able to isolate FynSH3 derivatives binding to MSA using phage display and covalent DNA display.
Materials and methods
Preparation of DNA templates for in vitro transcription/translation
The gene coding for Hae III DNA-methyltransferase was ordered at ATCC (strain 11116). The DNA templates for the expression of the various M.Hae III constructs were assembled following the instructions of the RTS E. coli Linear Template Generation Set (Roche Applied Sciences) with the exception that the oligonucleotides ext ba 2 [5′-GAT GCC GGC CAC GAT GCG TCC GGC GTA GAG-3′ (Qiagen)] and ext 2 fo [5′-GCT AAT TA G GFC ACC ACA CCC GTC CT-3′, modified substrate sequence in bold letters, (Microsynth, Switzerland)] were used for the final PCR assembly instead of the primers supplied with the Linear Template Generation Set. For the generation of DNA templates without the modification 5′-GGFC-3′ the primer ext 1 fo [GCT AAT TA G GCC ACC ACA CCC GTC CT-3′ (Microsynth, Switzerland)] instead of ext 2 fo was used. DNA was purified using the QIAquick Gel extraction kit or the QIAquick PCR purification kit (Qiagen). If DNA was used directly for in vitro transcription/translation reactions, DNA samples never were purified over an agarose gel because agarose inhibits in vitro protein expression (Manual RTS E.coli HY kit, Roche Applied Science).
Further details concerning the assembly of the different M.Hae III fusions are as follows; M.Hae III-Flag tag: the amino acid residues GGSG-DYKDDDK followed by two stop codons were appended to the DNA-methyltransferase gene in order to avoid the expression of the 6xHis tag, which is included in the Linear Template Generation Set.
M.Hae III-EDB/CaM: the sequences coding for calmodulin (CaM) and the extra-domain B of fibronectin (EDB) were available in our laboratory. The amino acid residues GGSGAS were used as linker for the C-terminal fusion of EDB/CaM to M.Hae III.
Preparation of water-in-oil emulsions
Water-in-oil emulsions were essentially prepared as described in Tawfik and Griffiths (1998) . A 10-fold concentrated solution of oil phase was prepared by mixing 50% (v/v) of mineral oil (Sigma-Aldrich) with 45% (v/v) Span 80 (Fluka, Switzerland) and 5% (v/v) Tween 80 (Fluka, Switzerland). For the preparation of the emulsions, this concentrated stock solution was diluted 10-fold in mineral oil and 950 µl were used for the encapsulation of 50 µl water phase. The mixing of the water and the organic phase was done by the addition of 50 µl of ice-cooled water phase to ice-cooled oil phase in a 2 ml micro-centrifuge tube. Stirring was done at 11 000 r.p.m. (speed 3) using a Ultra-Turrax T8 homogenizer (IKA, Germany) with a 5 mm dispersing tool. After addition of water phase, stirring was continued for 5 min.
Extraction of DNA-protein fusions from emulsions
After protein expression and formation of the covalent DNA-protein adducts, emulsions were broken by centrifugation for 10 min at 7000 r.p.m. in a table-top centrifuge (Eppendorf). Water droplets formed a pellet and the organic supernatant was removed. One hundred and fifty microliter of room temperature breaking buffer was added [PBS (or TBS with 1 mM CaCl 2 for selections with calmodulin (TBSC)], pH = 7.4, 5 µM biotinylated ds DNA fragments [5′-biotin-GGA GCT TCT GCA TTC TGT GTG CTG-3′ (Qiagen)], followed by 1 ml room temperature diethyl ether (water saturated). The tube was vortexed at full speed for 2× 10 s. After separation of the water phase and the organic phase, the water phase was removed from the bottom of the tube and transferred to a 24-well plate (NUNCLON Surface, Nunc). The extracted water phase was air-dried at room temperature for 10 min until residual Et 2 O was completely evaporated.
Purification and visualization of DNA-protein fusions
One hundred nanogram biotinylated DNA templates were used for expression in emulsion. After expression at 30°C for 3.5 h, the water phases were extracted from each emulsion. Instead of PBS, 100 µl binding buffer (5 mM Tris, 0.5 mM EDTA, 1 M NaCl, 0.1% Tween 20, pH = 7.5) was used as breaking buffer. DNA molecules present in each water phase were captured on streptavidin coated magnetic Dynabeads M-280 (Dynal, Norway). Twenty microliter beads had been previously prepared by washing them two times with 40 µl binding buffer. One hundred microliter extracted water phase was then mixed with 40 µl beads in binding buffer and incubated for 15 min at RT. The beads were subsequently washed three times with 80 µl TE-buffer (10 mM Tris, 1 mM EDTA, pH = 7.5) and suspended in 20 µl sterile, deionized water. After addition of 5 µl biotinylated ds DNA fragments [5′-biotin-GGA GCT TCT GCA TTC TGT GTG CTG-3′ (Qiagen); final concentration 125 µM], the samples were heated for 2 min to 70°C. After cooling, the supernatants were removed from the beads in a magnetic separator and analyzed on a 6% TBE gel (200 V, 15 mA, 75 min). DNA was visualized with SYBR green I (Molecular Probes, USA), and Smart Ladder (Eurogentec) was used as DNA marker (1:10 diluted).
Model selection experiments with M.Hae III-CaM and M.Hae III-EDB fusion proteins
Each 100 ng DNA coding either for M.Hae III-CaM-6xHis tag (with or without 5-fluoro-2′-deoxycytidine) or M.Hae III-EBD-6xHis tag (with 5-fluoro-2′-deoxycytidine) was used for expression in emulsion (RTS 100 E.coli HY kit, Roche Applied Science). During protein expression, 3 × 50 µl magnetic streptavidin coated Dynabeads (M-280, Dynal, Norway) were incubated for 15 min with the capture agent at room temperature [biotinylated calmodulin binding peptide (biotin-CAAARWKKAFIAVSAANRFKKIS ( Montigiani et al. , 1996 ), which was kindly provided by L. Lozzi, Dipartimento di Biologia Molecolare, Universita' di Siena, Italy) (4/50 µl beads, 400 nM)]. The beads were washed once with TBSC 0.1% Tween 20 (TBSCT) and blocked with 5 µM biotinylated ds DNA fragments [5′-biotin-GGA GCT TCT GCA TTC TGT GTG CTG-3′ (Qiagen)] for 15 min at room temperature. After expression for 3 h at 30°C and extraction of the DNA-protein fusions from the emulsions, the magnetic beads were incubated with the extracted water phase for 45 min at room temperature. Samples were shaken gently from time to time. The magnetic beads were washed six times with 100 µl TBSCT, followed by one wash with 100 µl TBSC using a magnetic separator (Dynal, Norway). The beads were resuspended in 100 µl water and used directly for PCR amplification of the DNA molecules remaining on the beads using the primers Ampl ba [5′-CCC GCG AAA TTA ATA CGA CTC A-3′ (Qiagen)] and Ampl fo [5′-AAA ACC CCT CAA GAC CCG TT-3′ (Qiagen)]. Smart Ladder (Eurogentec) was used as DNA marker on the agarose gel.
Affinity selections with M.Hae III-Flag tag and elution with KOH
Each 120 ng M.Hae III-Flag tag and M.Hae III-CaM-6x His tag DNA were used for non-emulsified expression in vitro in separate tubes. During protein expression, 2 × 50 µl magnetic streptavidin coated Dynabeads (M-280, Dynal, Norway) were incubated for 15 min with biotinylated α-Flag antibody (M2, 4 µl or 400 µg/50 µl beads) (Sigma-Aldrich). The beads were washed once with PBS 0.1% Tween 20 (PBST) and blocked with 5 µM biotinylated ds DNA fragments [5′-biotin-GGA GCT TCT GCA TTC TGT GTG CTG-3′ (Qiagen)] for 15 min at room temperature. After protein expression for 2.5 h at 30°C, 150 µl PBS containing 5 µM biotinylated ds DNA fragments [5′-biotin-GGA GCT TCT GCA TTC TGT GTG CTG-3′ (Qiagen)] was added to each tube. Subsequently, the magnetic beads were incubated with the bacterial lysates/PBS mixtures for 45 min at room temperature at 120 r.p.m. on a rotary shaker. The magnetic beads were washed six times with 100 µl PBST, followed by one wash with 100 µl PBS using a magnetic separator (Dynal, Norway). The beads were resuspended in 100 µl water and each 1 µl of the two bead suspensions was removed for real-time PCR analysis. Then, the two samples were each split into 4 × 25 µl and the supernatants were removed. For elution, the beads were suspended in 25 µl 1, 3, 6 or 10 mM KOH solution. After an incubation time of 2 min, the supernatants were removed and 1 µl of each sample was used for real-time PCR analysis. The ABI PRISM 7900 Fast Real-Time PCR System instrument (Applied Biosystems), ABI PRISM 96-well Optical Reaction Plates and TaqMan Universal PCR Master Mix (Applied Biosystems) were used for real-time PCR measurements. All reactions were done in triplicate for each sample. Primers and TaqMan probes specific for M.Hae III were designed with the Primer Express 1.5 software (Applied Biosystems). Primers were used at a concentration of 200 nM and the TaqMan probe at 100 nM.
FynSH3 library construction
The gene encoding the FynSH3 domain was amplified from a human cDNA library [human fetal MTC panel, brain (#K1425-1) (AMS Biotechnology (Europe) Ltd., Switzerland)] using the primers hFynSH3 domain ba [5′-AT CGC GGA TCC GGA GTG ACA CTC TTT GTG GCC CTT TAT-3′ (Operon)] and hFynSH3 domain fo [5′-GA AGA TCT CTG GAT AGA GTC AAC TGG AGC CAC ATA-3′ (Operon)]. The resulting DNA fragment was cloned in the vector pQE-12 (Qiagen) using the restriction sites BamHI and BglII. Starting from the clone G4, an affinity maturation library was constructed with six randomized amino acids in the n-Src-loop. Two independent PCRs were performed with G4 DNA as template. The first was done with the primer LMB3 long ba [5′-CAG GAA ACA GCT ATG ACC ATG ATT AC-3′ (Qiagen)] and the degenerate primer FynSH3 loop 2 fo [5′-GGA GCG GGC TTC CCA CCA ATC TCC MNN MNN MNN MNN MNN MNN CAA TAT TTG AAA TTT TTC TCC TTT-3′ (Operon)], whereas the second PCR was done with the primers FynSH3 loop 2 ba [5′-GGA GAT TGG TGG GAA GCC CGC T-3′ (Operon)] and FD seq long fo [5′-GAC GTT AGT AAA TGA ATT TTC TGT ATG AGG-3′ (Operon)]. Full-length FynSH3 mutants were assembled by PCR and the DNA library was further amplified using Fyn vitro ba [5′-GGT AAT GGC GGA TCA GGT GCA AGC GGA GTG ACA CTC TTT GTG GCC CTT-3′, (Operon)] and Fyn Xho fo long [5′-GTG GCT CCA GTT GAC TCT ATC CAG CTC GAG CGA GCT CCC GGG GGG GGT TCT CAT CAT CA-3′, (Operon)] as primers. The resulting DNA fragment was purified from an agarose gel using the QIAquick Gel extraction kit (Qiagen).
In order to construct DNA templates coding for M.Hae III-Fyn library fusion proteins, M.Hae III DNA alone was first cloned into the vector pIVEX2.3d (Roche Applied Science) using the restriction enzymes Nco I and Xho I. From this vector pJB25/3, two DNA fragments [(i) T7 promotor-M.Hae III and (ii) T7 terminator)] were amplified using the primers (i) ext 5′ ba (5′-TCG GTG CGG GCC TCT T-3′) and M.Hae lin fo (5′-GCA TTG GAA ATT TGT AAA GGT AAT GGC GGA TCA GGT GCA AGC-3′) and (ii) Ass His ba (5′-GGG GGG GGT TCT CAT CAT CAT CAT CAT CAT TAA TAA AAG-3′) and ext 3′ fo (5′-CGA AAC AGC TAT GGF CTG ATT ACG AAT TC-3′). Full-length M.Hae III-Fyn library DNA templates for in vitro transcription/translation were then assembled by PCR in two steps. The first PCR assembly of T7 promotor-M.Hae III DNA with Fyn library DNA was performed using the primers ext 5′ ba and Fyn Xho fo long (linker between M.Hae III and FynSH3 = GGSGASG). The resulting DNA fragment was purified from an agarose gel using the QIAquick Gel extraction kit (Qiagen). In a second step, T7 promotor-M.Hae III-Fyn library DNA was assembled with T7 terminator DNA using the primers ext 2 ba and ext 2 fo, and the full-length DNA template was purified with the QIAquick PCR purification kit (Qiagen).
Affinity maturation with covalent DNA display
Fifty nanogram DNA template was used as input for the first round of panning with covalent DNA display against MSA as target molecule. For further rounds of selection 10 ng DNA was used as input. After emulsification, protein expression was performed for 2.5–3 h at 30°C. Fifty microliter streptavidin coated magnetic Dynabeads M-280 (Dynal, Norway) were washed once with PBS Tween 0.1% and then loaded with 2.8 µg biotinylated MSA. After incubation at RT for 15 min, the beads were washed once with PBS Tween 0.1% and then blocked with 5 µM biotinylated ds DNA fragments [5′-GGA GCT TCT GCA TTC TGT GTG CTG-3′ (Qiagen)] for 10 min at RT. After extraction of the DNA-protein fusions from the emulsion, they were mixed with the streptavidin coated magnetic Dynabeads M-280 and incubated on a rotary shaker (120 r.p.m. for 60–90 min). Then, the magnetic beads were washed in a magnetic separator (Dynal, Norway) six times with 100 µl PBS Tween 0.1%, once with 100 µl PBS and once with 100 µl sterile water. After the last washing step, the beads were suspended in 30 µl 6 mM KOH (pH = 11.7) for elution. To neutralize the KOH, 27 µl of the eluate were added to 3 µl of 10 × PCR buffer (FastStart High Fidelity System, Roche Applied Science).
PCR amplification
Selected FynSH3 variants were amplified by PCR with the primers Fyn vitro ba and Fyn Xho fo long (FastStart High Fidelity System, Roche Applied Science) using the PCR program 95°C(2′)—[95°C(30″)—58°C(30″)—72°C(10′)] 35 —72°C(10′). PCR products were purified from a 1.6% agarose gel (QIAquick Gel extraction kit, Qiagen). The DNA fragment was then assembled with T7 promotor-M.Hae III DNA (discussed earlier) using the primers ext 5′ ba and Fyn Xho fo long [PCR program: 94°C(3′)—[94°C(1′)—52°C (1′)—72°C(2′)] 25 —72(10′)] and the HighFidelity Expand PCR System (Roche Applied Science). The product of the PCR assembly was purified from a 1.2% agarose gel. The product of the first DNA assembly was then assembled with T7 terminator DNA in further step using the primers ext 2 ba and ext 2 fo [PCR program: 94°C(3′)—[94°C(1′)—56°C(1′)—72°C(2′15 ″ )] 25 —72°C(10′)] and the HighFidelity Expand System (Roche Applied Science). The product of the assembly was purified with the QIAquick PCR purification kit (Qiagen).
Screening of selected FynSH3 mutants by ELISA
For ELISA screening, selected FynSH3 variants were ligated into the vector pQE-12 (Qiagen) using the restriction sites BamHI and HindIII and electroporated into TG1 E. coli bacteria. The transformed bacteria were plated on agar plates containing 100 µg/ml ampicillin and 1% (w/v) glucose and grown overnight in an incubator at 37°C. The next day, colonies were picked from the agar plate and grown in a round bottom 96-well plate (Nunc, cat. no. 163320) in 200 µl 2xYT medium containing 100 µg/ml ampicillin and 0.1% (w/v) glucose. Protein expression was induced after growth for 3 h at 37°C and 200 r.p.m. by adding 1 mM IPTG (Applichem, Germany). Proteins were expressed overnight in a rotary shaker (200 r.p.m., 30°C). Subsequently, the 96-well plate was centrifuged at 1800 g for 10 min and the supernatant was discarded. The bacterial pellets were resuspended in 60 µl lysis buffer (50 mM NaH 2 PO 4 , 300 mM NaCl, 10 mM imidazole, pH = 8.0) containing 1 mg/ml lysozyme and left for 30 min on ice. Afterwards, the bacterial cells were lysed by sonication in a water bath (six bursts for 10 s) and then centrifuged at 1800 g for 10 min. Fifty microliter of the supernatant was added together with 50 µl 4% MPBS to the wells of a 96-well MaxiSorp plate (Nunc, cat. no. 442404) that had been previously coated with 100 µl MSA (100 µg/ml). After 2 h at RT, the plate was washed three times each with PBS, which was followed by the addition of 100 µl α-6xHis-HRP immunoconjugate (Sigma-Aldrich) diluted 1:1000 in 2% MPBS. The 96-well plate was left for 1 h at RT and then washed three times with PBS 0.1% Tween and PBS. Colorimetric detection was done by the addition of 100 µl of BM blue POD substrate (Roche Applied Science, cat. no. 1484281) and the reaction was stopped with 60 µl 1 M H 2 SO 4 .
The specificity ELISA was performed as described earlier but with purified proteins dialyzed against PBS. To coat the MaxiSorp plate (Nunc, cat. no. 442404, 100 µl of HSA, RSA, MSA, ovalbumin (OVA), and BSA (all 100 µg/ml) or 2% MPBS were used.
Sequencing of isolated MSA binding proteins
Positive clones from the ELISA were used as templates in a PCR reaction using the primers EDB His ba [5′-GAA AAG TGC CAC CTG ACG TCT AA-3′ (Operon)] and EDB His fo [5′-CGG TCT GGT TAT AGG TAC ATT GAG C-3′ (Operon)] and the PCR products were purified from an agarose gel (QIAquick Gel extraction kit, Qiagen). Sequencing was done according to the instructions of the sequencing kit's manufacturer (BigDye ® Terminator v.3.1 Cycle sequencing kit, Applied Biosystems), and the sequencing reactions were analyzed on an Applied Biosystems 3130 Genetic Analyzer.
Expression of MSA binding FynSH3 mutants
Selected FynSH3 variants were inoculated in 100 ml 2xYT medium containing 100 µg/ml ampicillin and 0.1% (w/v) glucose and grown at 37°C in a rotary shaker at 200 r.p.m. At an OD (600 nm) of 0.6, protein expression was induced by the addition of 1 mM IPTG (Applichem, Germany). After 16 h at 30°C in a rotary shaker (200 r.p.m.), the bacterial cells were harvested by centrifugation and resuspended in 4 ml lysis buffer (50 mM NaH 2 PO 4 , 300 mM NaCl, 10 mM imidazole, pH = 8.0). One microgram per microliter lysozyme was added and the cells were incubated on ice for 30 min. After cell lysis by sonication, the lysates were centrifuged for 25 min at 11 000 r.p.m. One milliliter of Ni 2+ -NTA slurry (Qiagen) was added to the cleared lysate to capture M.Hae III-6xHis tag proteins (incubation at 4°C for 1 h while shaking on a rotary shaker). The resin was washed two times with 5 ml wash buffer (50 mM NaH 2 PO 4 , 300 mM NaCl, 20 mM imidazole, pH = 8.0) and the protein was eluted with 2 ml elution buffer (50 mM NaH 2 PO 4 , 300 mM NaCl, 250 mM imidazole, pH = 8.0).
Determination of apparent dissociation constants by ELISA
Dilution series of the purified FynSH3 mutants were prepared in elution buffer (50 mM NaH 2 PO 4 , 300 mM NaCl, 250 mM imidazole, pH = 8.0). Fifty microliter of each protein dilution was added together with 50 µl 4% MPBS to the well of a 96-well MaxiSorp plate (Nunc, cat. no. 442404) that had been previously coated with 100 µl MSA (100 µg/ml). Detection of bound FynSH3 variants was performed as described earlier.
Surface plasmon resonance experiments
Affinity measurements were performed using a BIAcore3000 instrument (Biacore). For the interaction analysis between MSA and the FynSH3 mutants, a CM5 chip (Biacore) was used with 18680 RU MSA immobilized. The running buffer was PBS, 0.1% NaN 3 , Surfactant P20 (Biacore). The interactions were measured at a flow of 20 µl/min and injections of different concentrations of FynSH3 mutants.
Results
Formation of covalent DNA-protein adducts
In a previous article ( Bertschinger and Neri, 2004 ), we had shown that DNA molecules containing the modified methylation target sequence 5′-GGFC-3′ (F = 5-fluoro-2′-deoxycytidine) can be covalently cross-linked with purified recombinant M.Hae III at low concentrations of both the DNA (2 nM) and the enzyme (38 nM). However, it remained unclear whether expression of M.Hae III followed by in situ cross-linking with the corresponding encoding DNA template would be equally efficient in emulsion, since we had observed degradation of DNA templates caused by the presence of nucleases in in vitro transcription/translation mixtures ( data not shown ). Nucleolytic removal of the cross-linking sites (5′-GGFC-3′) from the extremities of the DNA templates would impede the formation of covalent DNA-protein fusions. For this reason, we analyzed the efficiency of DNA-protein adduct formation within the compartments of a water-in-oil emulsion.
Biotinylated DNA templates coding for M.Hae III-6xHis tag or M.Hae III-Flag tag (either with or without the modified DNA sequence needed for the formation of DNA-protein fusions) were prepared. The four different DNA templates were packaged in four separate emulsions (at a final concentration of 2 nM) together with in vitro transcription/translation mixture, and the emulsions were incubated for 3.5 h at 30°C to allow for protein expression and formation of covalent DNA-protein adducts. Subsequently, the water phase was extracted from the emulsion and the biotinylated DNA templates were captured on the surface of streptavidin coated magnetic beads. After washing, bound DNA molecules were released from the beads by heating the samples in sterile water for 2 min to 70°C ( Holmberg et al. , 2005 ) in the presence of an excess of short biotinylated, double-stranded DNA fragments. The eluted DNA molecules were loaded on a TBE gel to visualize the relative amounts of DNA-protein fusions and unmodified DNA molecules (Fig. 2 ).
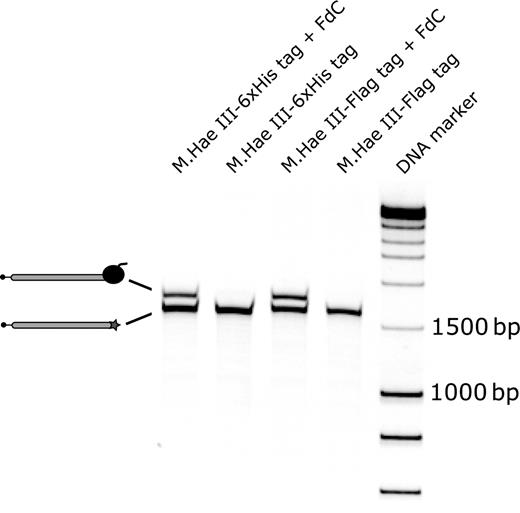
Formation of covalent DNA-protein fusions in the compartments of a water-in-oil emulsion. Biotinylated M.Hae III-6xHis tag and M.Hae III-Flag tag DNA templates with or without the covalent attachment site 5′-GGFC-3′ were prepared. The four different DNA templates were used for protein expression in emulsion. Subsequently, the biotinylated DNA fragments present in the extracted water phases were immobilized on the surface of streptavidin coated magnetic beads. After washing, the beads were suspended in sterile, ion-free water and then heated to 70°C in the presence of an excess of short biotinylated DNA fragments to elute the captured biotinylated DNA templates. The eluted fractions were analyzed on a 6% TBE gel on which the DNA was stained with SYBR green I. A second, upper DNA band representing the DNA-protein fusions was detected in the presence of the cross-linking site 5′-GGFC-3′. FdC : 5-Fluoro-2′-deoxycytidine. DNA marker : Smart ladder DNA marker.
Formation of covalent DNA-protein fusions was observed only in the presence of the modified methylation target sequence 5′-GGFC-3′, whereas DNA molecules without the cross-linking site were not associated with M.Hae III proteins (Fig. 2 ). From the relative intensities of the bands on the gel, we estimate that ∼50% of the DNA molecules were covalently bound to a M.Hae III protein, which correlates well with the results obtained in cross-linking experiments with purified recombinant M.Hae III protein ( Bertschinger and Neri, 2004 ).
Affinity selection experiments with covalent DNA-protein fusions
Hae III DNA-methyltransferase not only binds covalently to the modified target site 5′-GGFC-3′ but has also been shown to interact non-covalently with DNA in different ways. Using various double-stranded substrate DNAs, the dissociation constants of M.Hae III for its substrates ( KdS ) were measured in presence of 80 µM S-adenosylhomocysteine by electrophoretic mobility shift assays ( Chen et al. , 1993 ). M.Hae III bound hemimethylated DNA substrate with a dissociation constant of 0.34 nM, whereas fully methylated target sequences were bound no more tightly than non-specific DNA ( KdS ≈12.5 nM). As a consequence, M.Hae III can bind at various sites to DNA fragments with different affinities. These low dissociation constants suggest the possible co-existence of multiple copies of the M.Hae III protein bound both covalently and non-covalently to the same cognate modified DNA molecule in emulsion.
In order to analyze the influence of non-covalent M.Hae III-DNA complexes on the outcome of affinity selection experiments, three different DNA templates were prepared: (i) DNA comprising a cross-linking site and coding for M.Hae III-CaM-6xHis tag [CaM = calmodulin from X. laevis ( Neri et al. , 1995 )], (ii) DNA as in (i) but without cross-linking site and (iii) DNA coding for M.Hae III-EDB-6xHis tag [EDB = Extra domain B of fibronectin ( Zardi et al. , 1987 )] including a cross-linking site. For expression of the M.Hae III fusion proteins in emulsion and after extraction of the water phases, DNA-M, 100 ng of each template was used. Hae III-CaM-6xHis tag fusions were captured on streptavidin coated magnetic beads that had been previously coated with a calmodulin-binding peptide ( Montigiani et al. , 1996 ) and blocked with biotinylated short double stranded DNA molecules. This blocking protocol was more effective in reducing background recovery of DNA templates than biotin or DNA alone ( data not shown ). After washing, DNA templates still bound to the beads were amplified by PCR (amplicon length≈1700 bp) without prior elution from the magnetic beads. The outcome of the selections was analyzed on an agarose gel (Fig. 3 ). Only DNA molecules containing the coding sequence for M.Hae III-CaM-6xHis tag fusion protein and the covalent cross-linking site were recovered from the affinity selection experiment, whereas DNA molecules coding for the same M.Hae III fusion protein but lacking the modified sequence 5′-GGFC-3′ and DNA templates encoding an irrelevant M.Hae III fusion protein were not selected. Therefore, we concluded that a covalent linkage of DNA and protein is needed for an efficient recovery of DNA molecules via the binding specificity of the encoded M.Hae III fusion protein.
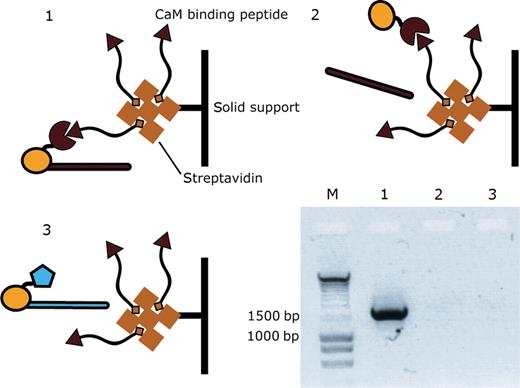
Covalent linkage of the DNA template coding for M.Hae III-CaM-6xHis tag protein is necessary for its efficient recovery in affinity selection experiments. Three different DNA templates were prepared: (1) DNA encoding M.Hae III-CaM (red) with 5′-GGFC-3′ cross-linking site; (2) DNA encoding M.Hae III-CaM without 5′-GGFC-3′ cross-linking site and (3) DNA encoding M.Hae III-EDB (blue) with 5′-GGFC-3′ cross-linking site. After expression of each template in three separate emulsions and extraction of the water phase, DNA-M.Hae III-CaM fusions were captured on magnetic streptavidin beads coated with CaM-binding peptide ( Montigiani et al. , 1996 ). The beads were washed, suspended in water and DNA molecules still bound to the beads were amplified by PCR without prior elution from the beads. The PCR products (amplicon length≈1700 bp) were loaded on an agarose gel that was stained with ethidium bromide to visualize the DNA. DNA molecules containing the covalent attachment site were only recovered if the DNA encoded a M.Hae III fusion protein with the appropriate binding specificity.
Elution of DNA-protein fusions at basic pH
Selection experiments with protein libraries generated using covalent DNA display technology are commonly performed using biotinylated antigen captured on streptavidin coated magnetic beads. However, when PCR amplification of selected DNA molecules was carried out directly on the magnetic beads without prior elution, we always observed unspecific PCR products ( data not shown ). Additionally, high concentration of magnetic beads reduced the performance of the PCR. For these reasons, we chose to insert an elution step in the protocol, using a buffer compatible with subsequent PCR amplification of the eluted DNA templates. In phage display, elution from antigen immobilized on solid supports is often performed at very basic pH (e.g. 100 mM triethylamine; Silacci et al. , 2005 ). However, both triethylamine and NaOH interfere with PCR ( data not shown ). By contrast, DNA amplification by PCR worked reliably in presence of KOH. For this reason, we investigated the feasibility of a quantitative elution of DNA-protein fusions from antigen immobilized on magnetic beads, using KOH solutions. Two DNA templates encoding M.Hae III-Flag tag and M.Hae III-CaM-6xHis tag (negative control), respectively, were expressed in two separate solutions. Subsequently, DNA-protein fusions were captured on magnetic streptavidin beads coated with biotinylated α-Flag tag antibody. After the last washing step, 1 µl of beads was taken to measure the number of DNA molecules still bound to the beads by real-time PCR. Then, the samples were split into four and the beads were resuspended in either 1, 3, 6 or 10 mM KOH. After 2 min, the eluates were removed from the beads and the KOH was neutralized in PCR buffer. The number of DNA molecules present in each sample was determined by real-time PCR (Table I ). M.Hae III-Flag tag-DNA fusions could be eluted quantitatively with yields ranging from 65% to 79% (with DNA molecules on beads = 100%). Some unspecifically bound M.Hae III-CaM-6xHis tag-DNA adducts were eluted at KOH concentrations higher than 3 mM. A 6 mM KOH solution has a pH value of 11.7. Such a basic pH should ensure that even high-affinity interactions between protein variants and immobilized antigen can be broken and, therefore, the use of 6 mM KOH could provide a general route to elute DNA-protein fusions bound to a solid support via the binding specificity of the protein.
Elution of M.Hae III-DNA fusions using different concentrations of KOH
| DNA template | Sample | Numbers of DNA molecules a | Standard deviation a | Flag/CaM b |
|---|---|---|---|---|
| M.Hae III-Flag | Beads c | 3.4 × 10 5 | 8.6 × 10 3 | — |
| M.Hae III-CaM | Beads c | 7.2 × 10 1 | 1.8 × 10 1 | 4 722 |
| M.Hae III-Flag | 1 mM KOH d | 2.8 × 10 5 | 4.3 × 10 4 | — |
| M.Hae III-CaM | 1 mM KOH | n.d. | n.d. | ≥280 000 |
| M.Hae III-Flag | 3 mM KOH | 2.5 × 10 5 | 3.3 × 10 4 | — |
| M.Hae III-CaM | 3 mM KOH | n.d. | n.d. | ≥250 000 |
| M.Hae III-Flag | 6 mM KOH | 2.7 × 10 5 | 5.8 × 10 3 | — |
| M.Hae III-CaM | 6 mM KOH | 2.9 × 10 1 | 5.0 × 10 0 | 9 310 |
| M.Hae III-Flag | 10 mM KOH | 2.2 × 10 5 | 2.2 × 10 4 | — |
| M.Hae III-CaM | 10 mM KOH | 7.0 × 10 1 | 2.0 × 10 1 | 3 143 |
| DNA template | Sample | Numbers of DNA molecules a | Standard deviation a | Flag/CaM b |
|---|---|---|---|---|
| M.Hae III-Flag | Beads c | 3.4 × 10 5 | 8.6 × 10 3 | — |
| M.Hae III-CaM | Beads c | 7.2 × 10 1 | 1.8 × 10 1 | 4 722 |
| M.Hae III-Flag | 1 mM KOH d | 2.8 × 10 5 | 4.3 × 10 4 | — |
| M.Hae III-CaM | 1 mM KOH | n.d. | n.d. | ≥280 000 |
| M.Hae III-Flag | 3 mM KOH | 2.5 × 10 5 | 3.3 × 10 4 | — |
| M.Hae III-CaM | 3 mM KOH | n.d. | n.d. | ≥250 000 |
| M.Hae III-Flag | 6 mM KOH | 2.7 × 10 5 | 5.8 × 10 3 | — |
| M.Hae III-CaM | 6 mM KOH | 2.9 × 10 1 | 5.0 × 10 0 | 9 310 |
| M.Hae III-Flag | 10 mM KOH | 2.2 × 10 5 | 2.2 × 10 4 | — |
| M.Hae III-CaM | 10 mM KOH | 7.0 × 10 1 | 2.0 × 10 1 | 3 143 |
a DNA molecules detected by real-time PCR in 1 µl of sample with standard deviation (3 measurements); b ratio of M.Hae III-Flag tag/M.Hae III-CaM-6xHis tag DNA molecules in corresponding samples; c Magnetic beads after last washing step; and d fractions eluted with KOH at indicated concentrations.
Elution of M.Hae III-DNA fusions using different concentrations of KOH
| DNA template | Sample | Numbers of DNA molecules a | Standard deviation a | Flag/CaM b |
|---|---|---|---|---|
| M.Hae III-Flag | Beads c | 3.4 × 10 5 | 8.6 × 10 3 | — |
| M.Hae III-CaM | Beads c | 7.2 × 10 1 | 1.8 × 10 1 | 4 722 |
| M.Hae III-Flag | 1 mM KOH d | 2.8 × 10 5 | 4.3 × 10 4 | — |
| M.Hae III-CaM | 1 mM KOH | n.d. | n.d. | ≥280 000 |
| M.Hae III-Flag | 3 mM KOH | 2.5 × 10 5 | 3.3 × 10 4 | — |
| M.Hae III-CaM | 3 mM KOH | n.d. | n.d. | ≥250 000 |
| M.Hae III-Flag | 6 mM KOH | 2.7 × 10 5 | 5.8 × 10 3 | — |
| M.Hae III-CaM | 6 mM KOH | 2.9 × 10 1 | 5.0 × 10 0 | 9 310 |
| M.Hae III-Flag | 10 mM KOH | 2.2 × 10 5 | 2.2 × 10 4 | — |
| M.Hae III-CaM | 10 mM KOH | 7.0 × 10 1 | 2.0 × 10 1 | 3 143 |
| DNA template | Sample | Numbers of DNA molecules a | Standard deviation a | Flag/CaM b |
|---|---|---|---|---|
| M.Hae III-Flag | Beads c | 3.4 × 10 5 | 8.6 × 10 3 | — |
| M.Hae III-CaM | Beads c | 7.2 × 10 1 | 1.8 × 10 1 | 4 722 |
| M.Hae III-Flag | 1 mM KOH d | 2.8 × 10 5 | 4.3 × 10 4 | — |
| M.Hae III-CaM | 1 mM KOH | n.d. | n.d. | ≥280 000 |
| M.Hae III-Flag | 3 mM KOH | 2.5 × 10 5 | 3.3 × 10 4 | — |
| M.Hae III-CaM | 3 mM KOH | n.d. | n.d. | ≥250 000 |
| M.Hae III-Flag | 6 mM KOH | 2.7 × 10 5 | 5.8 × 10 3 | — |
| M.Hae III-CaM | 6 mM KOH | 2.9 × 10 1 | 5.0 × 10 0 | 9 310 |
| M.Hae III-Flag | 10 mM KOH | 2.2 × 10 5 | 2.2 × 10 4 | — |
| M.Hae III-CaM | 10 mM KOH | 7.0 × 10 1 | 2.0 × 10 1 | 3 143 |
a DNA molecules detected by real-time PCR in 1 µl of sample with standard deviation (3 measurements); b ratio of M.Hae III-Flag tag/M.Hae III-CaM-6xHis tag DNA molecules in corresponding samples; c Magnetic beads after last washing step; and d fractions eluted with KOH at indicated concentrations.
Selection of proteins binding to MSA from a library of FynSH3 variants
We created a phagemid library with six randomized amino acids in the RT-Src-loop of FynSH3 domain (Fig. 4 ). This library was used in selections using phage display with MSA as a target protein. From this selection, a clone named G4 was isolated that bound to MSA with very low affinity. It was possible to detect binding in a phage ELISA, but ELISA experiments performed with the soluble domain failed to yield weak signals even at concentrations > 100 µM (discussed later). For this reason, we aimed at affinity maturing the clone G4 using covalent DNA display technology. Starting from G4, a library of FynSH3 mutants was created, in which the n-Src-loop was extended to six randomized amino acid residues. The library DNA coding for the FynSH3 mutants was assembled by PCR with DNA fragments coding for M.Hae III and the regulatory sequences necessary for efficient expression in vitro . Using this library, which contained 6 × 10 7 different FynSH3 variants, several rounds of selections were performed using covalent DNA display.
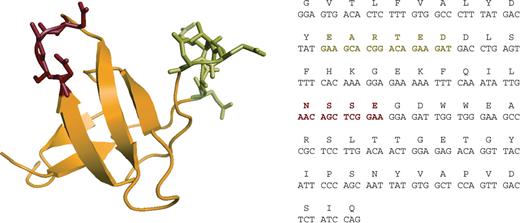
Sequence and structure of the FynSH3 domain. (left) Structure of the FynSH3 domain (pdb file 1M27 prepared with the software ‘Pymol’ (www.pymol.org). The RT-Src-loop is colored in green and the n-Src-loop in red. (right) DNA and amino acid sequence of the FynSH3 domain. The amino acid sequence is conserved in man, mouse, rat and monkey (gibbon).
A key step in the selection protocol is the amplification of the isolated DNA molecules by PCR with both high efficiency and fidelity. In order to enhance the efficiency of the PCR reaction and to avoid the introduction of undesired mutations in the constant parts of the DNA template (M.Hae III gene and regulatory DNA sequences), only the relatively short DNA fragments coding for the FynSH3 mutants were amplified by PCR. Full-length DNA templates amenable to the following round of selection were then produced by two PCR assemblies (Fig. 5 ). This DNA amplification strategy allowed the robust recovery of selected DNA sequences for at least five rounds of selection ( data not shown ).
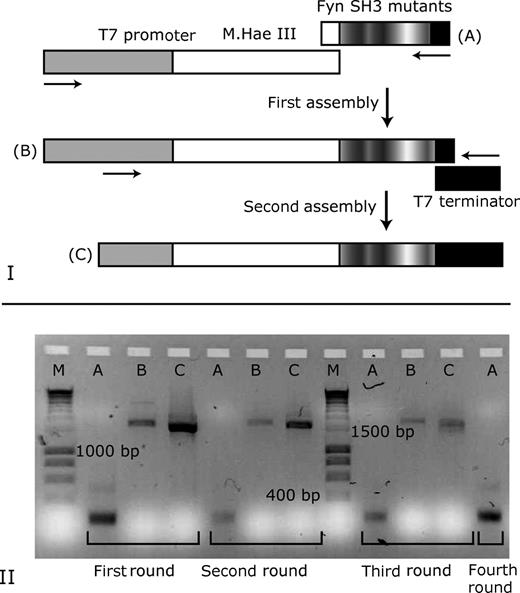
Strategy for the amplification of DNA molecules selected by covalent DNA display. (I) Selected DNA fragments encoding the FynSH3 variants (A) were amplified by PCR. Subsequently, full-length DNA templates were prepared by a two-step procedure: first, the FynSH3 DNA fragments were assembled with a DNA fragment comprising the gene for M.Hae III and the T7 promotor (B). Subsequently, a second PCR assembly of the DNA fragments (B) with the T7 terminator was performed to obtain DNA templates (C) that can be used for the next round of selection. (II) Agarose gel showing ethidium bromide-stained DNA fragments recovered after selection rounds one to four. A: DNA fragments encoding FynSH3 variants. B: DNA fragments after first assembly. C: full-length DNA fragments were obtained after second PCR assembly and were used for the subsequent round of selection. M: DNA marker.
After two or four rounds of selection, the recovered FynSH3 protein variants were cloned to allow for cytoplasmic expression of the soluble domains with a C-terminal 6xHis tag in E. coli . In addition, 92 clones were picked randomly from the original affinity maturation library to examine the abundance of binding clones in the library before selection. Individual bacterial colonies were grown in a 96-well plate, and after induction of protein expression, the cells were lysed. The lysates were then tested by ELISA for the presence of a MSA binding FynSH3 mutant. For this purpose, a 96-well plate was coated with MSA and, after addition of the lysates to the individual wells, binding proteins were detected with an α-6xHis tag-horseradish peroxidase (HRP) immunoconjugate. None of the FynSH3 variants randomly picked from the affinity maturation library was found to bind MSA. In contrast, about 30% of the clones isolated by covalent DNA display produced a strong ELISA signal, and thus some of them were analyzed by sequencing (Table II ). The third (His) and the fifth residue (Leu) of the n-Src-loop were completely conserved in all analyzed sequences. The clones isolated after two rounds of selection had similar amino acid residues in the n-Src-loop, which were encoded by different codons on the DNA level. In contrast, one amino acid sequence was predominantly found in the n-Src-loop of the clones isolated after four rounds of selection, and these amino acids were encoded by the same set of codons. A further difference between the clones isolated from two and four rounds of selection, respectively, was the occurrence of framework mutations, which were exclusively found in clones B8, C7 and D12 that were isolated after four rounds of selection (Table II ).
Sequences of selected FynSH3 mutants binding to MSA
| Clone | sequence RT-loop | sequence src-loop |
|---|---|---|
| Fyn SH3 wt | D Y E A R T E D D L | I L N S S E – – G D |
| G4 parental | D Y Y S A G F G D L | I L N S S E – – G D |
| Affinity maturation (two rounds of selection) | ||
| B2 | D Y Y S A G F G D L | I L R P H K L T G D |
| F8 | D Y Y S A G F G D L | I L R P H P L A G D |
| H3 | D Y Y S A G F G D L | I L R P H H L A G D |
| Affinity maturation (four rounds of selection) | ||
| A2, E9 (+7 others) | D Y Y S A G F G D L | I L G R H Y L A G D |
| B8 a , C7 a , E2 a | D Y Y S A G F G D L | I L G R H Y L A G D |
| C10 | D YH S A G F G D L | I L G R H Y L A G D |
| D12 | D Y Y S A G F G D L | I L R R H P L A G D |
| Clone | sequence RT-loop | sequence src-loop |
|---|---|---|
| Fyn SH3 wt | D Y E A R T E D D L | I L N S S E – – G D |
| G4 parental | D Y Y S A G F G D L | I L N S S E – – G D |
| Affinity maturation (two rounds of selection) | ||
| B2 | D Y Y S A G F G D L | I L R P H K L T G D |
| F8 | D Y Y S A G F G D L | I L R P H P L A G D |
| H3 | D Y Y S A G F G D L | I L R P H H L A G D |
| Affinity maturation (four rounds of selection) | ||
| A2, E9 (+7 others) | D Y Y S A G F G D L | I L G R H Y L A G D |
| B8 a , C7 a , E2 a | D Y Y S A G F G D L | I L G R H Y L A G D |
| C10 | D YH S A G F G D L | I L G R H Y L A G D |
| D12 | D Y Y S A G F G D L | I L R R H P L A G D |
Bold letters indicate framework residues outside the randomized loops. a Clones with framework point mutations: B8: Ile29Ala; C7: Glu49Gly, Ala59Val; D12: Asp38Gly; E2: Thr50Ala.
Sequences of selected FynSH3 mutants binding to MSA
| Clone | sequence RT-loop | sequence src-loop |
|---|---|---|
| Fyn SH3 wt | D Y E A R T E D D L | I L N S S E – – G D |
| G4 parental | D Y Y S A G F G D L | I L N S S E – – G D |
| Affinity maturation (two rounds of selection) | ||
| B2 | D Y Y S A G F G D L | I L R P H K L T G D |
| F8 | D Y Y S A G F G D L | I L R P H P L A G D |
| H3 | D Y Y S A G F G D L | I L R P H H L A G D |
| Affinity maturation (four rounds of selection) | ||
| A2, E9 (+7 others) | D Y Y S A G F G D L | I L G R H Y L A G D |
| B8 a , C7 a , E2 a | D Y Y S A G F G D L | I L G R H Y L A G D |
| C10 | D YH S A G F G D L | I L G R H Y L A G D |
| D12 | D Y Y S A G F G D L | I L R R H P L A G D |
| Clone | sequence RT-loop | sequence src-loop |
|---|---|---|
| Fyn SH3 wt | D Y E A R T E D D L | I L N S S E – – G D |
| G4 parental | D Y Y S A G F G D L | I L N S S E – – G D |
| Affinity maturation (two rounds of selection) | ||
| B2 | D Y Y S A G F G D L | I L R P H K L T G D |
| F8 | D Y Y S A G F G D L | I L R P H P L A G D |
| H3 | D Y Y S A G F G D L | I L R P H H L A G D |
| Affinity maturation (four rounds of selection) | ||
| A2, E9 (+7 others) | D Y Y S A G F G D L | I L G R H Y L A G D |
| B8 a , C7 a , E2 a | D Y Y S A G F G D L | I L G R H Y L A G D |
| C10 | D YH S A G F G D L | I L G R H Y L A G D |
| D12 | D Y Y S A G F G D L | I L R R H P L A G D |
Bold letters indicate framework residues outside the randomized loops. a Clones with framework point mutations: B8: Ile29Ala; C7: Glu49Gly, Ala59Val; D12: Asp38Gly; E2: Thr50Ala.
Clones B2, F8, H3, E9 (as A2), C7 and C10 were expressed in shake flasks and purified by affinity chromatography. Expression yields varied significantly between 1 and 29 mg/l bacterial culture (Table III ). Using the purified proteins, the binding specificity of the clones was examined by ELISA. MSA, HSA, RSA, BSA, OVA and MPBS (2%) were coated on plastic, and after addition of the purified FynSH3 mutants, bound proteins were detected with α-6xHis tag-HRP immunoconjugate (Fig. 6 ). All clones clearly bound to MSA. C7, E9, B2 and H3 cross-reacted with BSA and also weakly bound to RSA. Binding of the parental clone used for affinity maturation (G4) or of FynSH3 wild-type protein to any of the tested antigens was not detected.
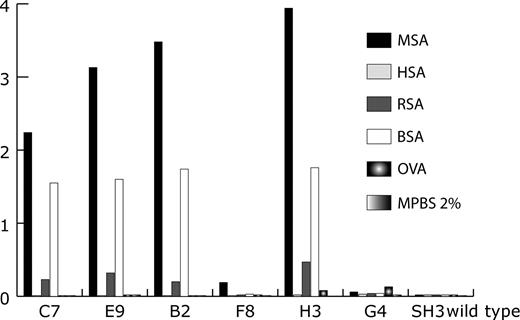
Binding specificity of some clones isolated by covalent DNA display. Several antigens were coated on plastic followed by the addition of purified FynSH3 mutants. Bound FynSH3 variants were detected with α-6xHis tag-HRP immunoconjugate. Clones B2, F8 and H3 were isolated after two rounds of selection. Clones C7 and E9 were selected after four rounds. G4 is the starting clone for the construction of the affinity maturation library. MPBS (2%): 2% milk (w/v) in PBS.
Expression yield of some clones selected by covalent DNA display
| Clone | Expression yield (mg/l) a |
|---|---|
| Fyn SH3 wt | 60 |
| G4 parental | 58 |
| Affinity maturation (two rounds of selection) | |
| B2 | 29 |
| F8 | 8 |
| H3 | 23 |
| Affinity maturation (four rounds of selection) | |
| E9 | 11 |
| C7 | 4 |
| C10 | 1 |
| Clone | Expression yield (mg/l) a |
|---|---|
| Fyn SH3 wt | 60 |
| G4 parental | 58 |
| Affinity maturation (two rounds of selection) | |
| B2 | 29 |
| F8 | 8 |
| H3 | 23 |
| Affinity maturation (four rounds of selection) | |
| E9 | 11 |
| C7 | 4 |
| C10 | 1 |
a Expression under non-optimized conditions in shake flasks.
Expression yield of some clones selected by covalent DNA display
| Clone | Expression yield (mg/l) a |
|---|---|
| Fyn SH3 wt | 60 |
| G4 parental | 58 |
| Affinity maturation (two rounds of selection) | |
| B2 | 29 |
| F8 | 8 |
| H3 | 23 |
| Affinity maturation (four rounds of selection) | |
| E9 | 11 |
| C7 | 4 |
| C10 | 1 |
| Clone | Expression yield (mg/l) a |
|---|---|
| Fyn SH3 wt | 60 |
| G4 parental | 58 |
| Affinity maturation (two rounds of selection) | |
| B2 | 29 |
| F8 | 8 |
| H3 | 23 |
| Affinity maturation (four rounds of selection) | |
| E9 | 11 |
| C7 | 4 |
| C10 | 1 |
a Expression under non-optimized conditions in shake flasks.
Determination of apparent dissociation constants by ELISA and surface plasmon resonance (BIAcore)
For the determination of apparent dissociation constants of MSA binding FynSH3 mutants, the clones G4 (parental clone), C7, and E9 were produced by cytoplasmic expression in E. coli and then purified by affinity chromatography on Ni 2+ -NTA resin. For the ELISA, FynSH3 variants were added at different concentrations to the wells of a MSA coated 96-well plate and bound proteins were detected using an α-6xHis tag-HRP immunoconjugate (Fig. 7 A). Binding of the parental clone G4 to MSA was not detectable even at a protein concentration as high as 180 µM. In the case of clones E9 and C7, however, binding was apparent at protein concentrations below 100 nM (Fig. 7 A) and 50% of maximal signal intensity was reached at 140 nM (E9) and 240 nM (C7), respectively.
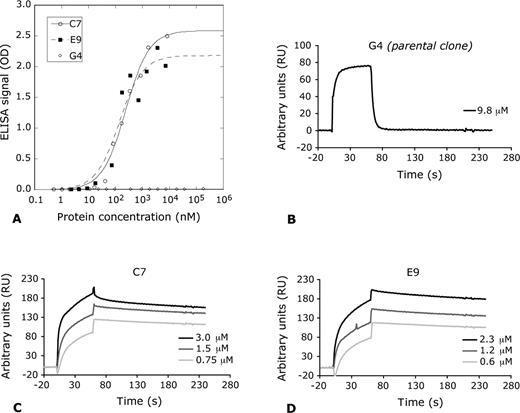
Determination of apparent dissociation constants of isolated MSA binding FynSH3 variants. (A) Clones C7, E9 and the parental clone of the affinity maturation G4 were added to MSA coated wells of a 96-well plate at different concentrations. Bound proteins were detected using an α-6xHis tag-HRP immunoconjugate. Binding of G4 (open diamond) to MSA was not detectable, whereas clones C7 (open circle), E9 (closed diamond) and reach 50% of the maximal signal intensity at concentrations of 240 and 140 nM, respectively. (B–D) BIAcore sensograms of the MSA binding FynSH3 derivatives G4 (parental clone), C7 and E9. C7 and E9 were injected at three different concentrations.
Apparent dissociation constants were also determined by surface plasmon resonance. Purified FynSH3 derivatives G4, C7 and E9 were used for real-time interaction analysis on a BIAcore chip coated with MSA (Fig. 7 C and D), revealing apparent dissociation constants ( KD ) of 35 nM (C7) and 26 nM (E9), at the antigen density used. Binding of the parental clone G4 to the MSA coated chip could not be detected even at concentrations of G4 of 9.8 µM (Fig. 7 B).
Discussion
DNA templates can be used for the expression of M.Hae III fusion proteins in the compartments of a water-in-oil emulsion. The resulting proteins can be covalently linked in situ to their encoding DNA template, yielding an irreversible and covalent linkage between the genotype (DNA) and the corresponding phenotype (protein). This allows the selection of DNA molecules by virtue of the binding specificity of their encoded protein.
We have applied an optimized protocol for covalent DNA display to the isolation of variants of the FynSH3 with high binding affinity to MSA. After four rounds of selection, about 40% of the tested clones were positive in ELISA screening ( data not shown ). Among these clones, one DNA sequence was predominant (11 clones out of 12 sequenced), but more diverse sequences of binding clones could be detected at earlier rounds of panning, in full analogy to what is often observed with phage display libraries.
In our previous work ( Bertschinger and Neri, 2004 ), we had speculated that several M.Hae III fusion proteins could in principle be non-covalently bound to the DNA molecule in addition to the covalently linked protein, thus leading to background recovery of DNA molecules in the absence of the covalent cross-linking site. In this article, using covalent DNA display of calmodulin, we could clearly demonstrate that selection for capture with a calmodulin-binding peptide is only possible in the presence of a direct covalent bond between DNA and M.Hae III-calmodulin (Fig. 3 ). However, we have observed low background levels of non-covalent DNA-protein complexes, whenever certain M.Hae III-peptide fusions, characterized by high expression levels, were used in selection experiments ( data not shown ). In any case, the non-covalent association of M.Hae III to DNA does not appear to interfere with the isolation of well-behaved binding proteins from a library (Fig. 6 ).
The faithful recovery of DNA information corresponding to selected binding proteins is a challenge common to most in vitro selection/amplification protein engineering methodologies. Early attempts to amplify full-length DNA segments (spanning the entire expression cassette) had failed to provide reliable results after more than two rounds of panning ( data not shown ). This technical limitation could be solved by PCR amplification of the DNA segment corresponding to the binding protein, followed by a PCR-assembly-based reconstitution of the expression cassette for the next round of panning (Fig. 5 ). This selective PCR amplification strategy limits the probability of inserting undesired mutations in the methylase gene and in the accessory regulatory sequences. However, insertions and deletions might occur during PCR-assembly reactions, especially at the junctions of the DNA fragments to be assembled.
The stable and covalent linkage between DNA and its encoded protein in covalent DNA display might be especially advantageous for affinity maturation and optimization of biophysical properties of therapeutic proteins. Because the methodology takes place fully in vitro , transformation of living cells is not necessary during library construction and, therefore, this technology might allow the rapid isolation of binding proteins. Library preparation can be done within two days using standard PCR protocols and, in analogy to phage display, one round of selection can be performed in 2 days (day 1: library expression and affinity purification, day 2: amplification of the isolated DNA molecules). Due to the need for compartmentalization, the size of the library to be processed in 1 ml of emulsion is limited to about 10 10 , which is in the same range as large phage display libraries ( Silacci et al. , 2005 ), but significantly smaller than the libraries that can be processed with ribosome display ( Mattheakis et al. , 1994 ), mRNA display ( Roberts and Szostak, 1997 ) or CIS display ( Odegrip et al. , 2004 ) (up to 10 14 ). However, rather than working with very large libraries, we favor an approach where the directed evolution of proteins is achieved by repeating several times the two steps of (i) creating genetic diversity and (ii) isolating protein variants with the desired biologic activity. In such a procedure, the libraries are generated starting from the best protein candidate selected in the preceding evolutionary cycle.
To our knowledge, two alternative fully in vitro selection technologies have been described in the literature that allow a covalent linkage between protein and the encoding genotype. The first of these technologies is mRNA display, which was later further developed into cDNA display ( Weng et al. , 2002 ). Even though it has been possible to isolate binding peptides and proteins using mRNA display, the methodology is relatively time consuming. First, the mRNA templates must be ligated to a puromycin containing oligonucleotide, which is followed by the purification of the ligation products and translation in vitro . Unfortunately, by the end of a standard translation reaction, only a small amount of the in vitro synthesized protein is converted to its mRNA-protein fusion ( Liu et al. , 2000 ).
The yield of mRNA-peptide fusions can be increased by post-translational addition of Mg 2+ and K + , use of a flexible linker of the correct length (puromycin oligonucleotides) and long incubations (12–48 h) at room temperature (with metal ions) or low temperatures (−20°C) (without metal ions) ( Liu et al. , 2000 ). Once synthesized, mRNA-protein fusions should be purified in order to generate a cDNA/mRNA hybrid fusion product using reverse transcriptase before the selective step ( Liu et al. , 2000 ). If the affinity selection step is directly performed after translation using the mRNA-protein fusion/translation lysate mixture, RNA aptamers binding the selection target could rather be selected than peptides.
In a recent publication ( Reiersen et al. , 2005 ), a further technology has been published, where a direct and covalent linkage of template DNA to its encoded protein has been proposed exploiting the cis -nicking activity of the endonuclease P2A from the bacteriophage P2. However, no data visualizing the formation of covalent protein-DNA fusions have been published, and expression levels of P2A-scFv in vitro were low. It was estimated, that only ∼3% of the input DNA molecules were able to produce full-length P2A-scFv protein ( Reiersen et al. , 2005 ).
For this reason, we believe that covalent DNA display may represent a useful alternative to existing selection technologies since it allows the direct covalent cross-linking of DNA with its encoded protein within short time (2–3 h) with a yield of ∼50%. Additionally, the valence of display might be increased by inclusion of more than one cross-linking site (5′-GGFC-3′) in the DNA library templates by PCR.
It will be interesting to investigate the influence of different experimental conditions during selection (e.g. stringency of washing, temperature) and DNA amplification (e.g. introduction of random mutations by error-prone PCR) on the outcome of selections with covalent DNA display. Furthermore, it will be important to see how covalent DNA display will perform in selections starting from naïve protein libraries.
Mutants of the FynSH3 domain, capable of specific binding to protein targets of pharmaceutical interest, may be particularly suitable for biomedical applications, in light of the protein identity among different species. Indeed, we have observed that even FynSH3 mutants with 12 mutations compared to the parental wild-type protein are not immunogenic in mice, even after repeated administrations (Grabulovski et al ., 2006).
We believe that covalent DNA display represents a useful addition to the protein engineer's toolbox, with the potential to deliver good-quality binding proteins for biomedical applications.
References
Edited by Philipp Holliger
Author notes
These authors equally contributed to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}