





Abstract
Background. Otsuka Long-Evans Tokushima Fatty (OLETF) rats genetically develop diabetes which is associated with hypertension. In preliminary studies, urinary excretions of L-PGDS (lipocaline-type prostaglandin D synthase) increase before diabetic nephropathy obviously develops, and this may predict progression of renal injury following diabetes. In the present study, we attempted to define whether urinary excretions of L-PGDS behave as the predictor of development of diabetic nephropathy in OLETF rats.
Methods. We investigated alterations of urinary L-PGDS excretions during the establishment of diabetes and assessed the relationship between the L-PGDS excretions and renal function in OLETF rats. Furthermore, we treated OLETF rats with troglitazone and analysed the effects on L-PGDS metabolisms. Urinary L-PGDS was measured by immunoenzyme assay and the occurrence of L-PGDS and its mRNA in the kidney was assessed by immunohistochemistry and a PCR method.
Results. Urinary excretions of L-PGDS were significantly higher in OLETF rats than non-diabetic Long-Evans Tokushima Otsuka (LETO) rats. The excretions age-dependently increased in OLETF and this increase appeared to be due to increased glomerular permeability to L-PGDS. Messenger RNA and antigenicity of L-PGDS were demonstrated in renal tissue; however, the de novo synthesis of L-PGDS mRNA seemingly contributed to urinary L-PGDS excretions much less than glomerular filtration. Multiple regression analysis revealed that urinary L-PGDS was determined by urinary protein excretions, and not by high blood pressure per se. Conversely, urinary proteinuria in the established diabetic nephropathy was predicted by urinary L-PGDS excretions in the early stage of diabetes.
Conclusions. Urinary excretions of L-PGDS are likely to reflect the underlying increase in glomerular permeability. This property may be useful to predict forthcoming glomerular damage following diabetes in OLETF rats.
Introduction
Otsuka Long-Evans Tokushima Fatty (OLETF) rats, which were isolated by Nakayama et al. from an outbred colony of Long-Evans rat strain, are a genetic model of spontaneous non-insulin-dependent diabetes mellitus [1–3]. OLETF rats exhibit a mild degree of insulin resistance from about 24 weeks after birth. OLETF rats older than 30 weeks show significant proteinuria compared with non-diabetic, control Long-Evans Tokushima Otsuka (LETO) rats. The proteinuria in OLETF progressively increases with ageing. Pathological changes of the kidney in OLETF rats older than 40 weeks are characterized by extra-cellular matrix accumulation in the mesangium, thickening of the glomerular basement membranes and glomerulosclerosis, all of which correspond to diabetic glomerulopathy in humans [1–3]. Thus, this strain of rats has been acknowledged as a useful model for investigating the pathogenesis of organ injury following diabetes.
We have recently reported that urinary excretions of lipocaline-type prostaglandin D2 synthase (L-PGDS) increase in the early stage of diabetes in humans, and the excretions decline with the control of blood glucose levels by hospitalization [4,5]. Moreover, the cross-sectional studies have revealed that in some diabetics, urinary L-PGDS excretions are greater than non-diabetic subjects although urinary albumin excretions are within the normal range [4,5]. These data strongly suggest that L-PGDS in urine might be useful to predict alterations of glomerular permeability in the early stage of diabetes.
Serum albumin molecules are negatively charged in blood. L-PGDS molecules are expected to take a negative charge in blood as well considering the isoelectrical point. The anion charges of L-PGDS interfere in part with passing through the negative-charged basement membranes of glomeruli. On the other hand, L-PGDS, an acidic monomeric protein, has relatively smaller molecular weight (MW) of 20 000–31 000 Da than serum albumin (MW 80 000 Da). Considering such properties, it seems probable that derangement of anion-charge barrier in damaged glomeruli more likely increases permeability to L-PGDS molecules than serum albumin. Moreover, it is reported that decreased filtration of L-PGDS due to impaired glomerular filtration rate due to chronic renal diseases is associated with a rise in serum L-PGDS concentrations [6,7].
These studies strongly suggest that L-PGDS in urine reflects glomerular permeability in the early stage of diabetes and hypertension, and this would be useful to predict forthcoming proteinuria following diabetes or hypertension [4,5,8]. Thus, in the present study, to test the hypothesis that urinary L-PGDS excretions in the early diabetes predict the progression of diabetic nephropathy, we investigated alterations of urinary L-PGDS excretions for about 12 months during establishment of diabetes and diabetic nephropathy, and assessed the relationship between urinary L-PGDS excretions and forthcoming decline of renal function using diabetic OLETF and the non-diabetic, control LETO rats.
Materials and methods
Protocol of the experiment
Forty-one OLETF rats were used that had been bred at Tokushima Research Institute, Otsuka Pharmaceutical Co., Ltd, Tokushima, Japan [1–3]. These inbred rats were isolated from an outbred colony of Long-Evans rats that had been purchased in 1982 from Charles River Canada (St Constant, Quebec, Canada). For comparison, 16 LETO rats were also offered from the same institute. All rats were fed a regular chow (Oriental Koubo Co., Ltd, Tokyo, Japan) containing 7.6 g of water, 24.6 g of coarse protein, 5.6 g of lipids, 52.8 g of carbohydrates and 360 kcal per 100 g of chow.
The OLETF rats aged 6 weeks were assigned to three groups; (1) 13 untreated rats; (2) 14 rats treated with low-dose troglitazone (0.5 mg/ml of water) and (3) 14 rats treated with high-dose troglitazone (1 mg/ml of water). The untreated OLETF and 16 LETO rats were given water alone. Water and regular chow were provided ad libitum during the study. The rats were maintained until when they were 48 weeks old.
Systolic blood pressures (BPs) were taken using the standard tail-cuff method every 4 weeks throughout the experiment [9,10]. 24 h urine was collected every 4 weeks during the experiment. The rats were placed in metabolic cages for three consecutive days, and urine obtained on the last day was utilized to determine urinary variables. On the last day of the experiment, the rats were fasted for 18 h, and then, blood specimens and the organs of interest were obtained under Brevital anesthesia (50 mg/kg body weight). The right kidney was utilized for morphological investigation. The left kidney was quickly plunged into liquid nitrogen for measuring L-PGDS mRNA levels. Plasma, 24 h urine and the left kidney were stored at −80°C until assayed. The procedure of the animal study was in accordance with the institutional guidelines for experimental animals of University of Tokyo.
Determination of L-PGDS
Plasma and urinary L-PGDS were measured by an enzyme-linked immunosorbent assay method (ELISA) using a polyclonal antibody, as described previously [4,5]. In brief, the samples were incubated at 30°C for 90 min in Maxi Sorp F96 microtiter plates (Nalge Nunc, Roskilde, Denmark) precoated with the anti-L-PGDS polyclonal antibody (3 µg/ml) at 4°C overnight. After a wash, the plates were incubated at 30°C with a biotinylated polyclonal antibody (1.4 µg/ml) for 90 min. The microtiter plates were washed three times. Thereafter, we added, to each well of the plates, streptavidin-HRP (horse-radish peroxidase) conjugates and 2,2′azino-bis (3-ethylbenzothiazoline)-6-sulfonic acid solution (Roche Diagnostics, Mannheim, Germany). The reaction was stopped by adding 1.5% oxalic acid. The plates were read on a Spectra-Max 250 microplate reader (Molecular Devices, Sunny Vale, CA, USA) at 405–492 nm. The intra- and inter-assay coefficients of variations were 3.2 and 5.7%, respectively.
Determination of various parameters
Creatinine concentration was measured by a creatinine autoanalyzer (Beckman Instrument Japan, Tokyo, Japan). Urinary protein concentration was determined by a Bio-Rad Protein Assay kit (Nihon Bio-Rad Laboratories, Tokyo, Japan). Microalbumin in urine was measured by the standard competitive ELISA method (Albuwell M, Exocell Inc., Philadelphia, PA) [11]. N-acetyl-β-d-glucosaminidase activity (NAG) was determined using sodio-meta-cresolsulfonphthaleinyl N-acetyl-β-d-glucosaminide as a substrate (NAG assay kit, Shionogi Pharmaceutical Co., Osaka, Japan).
Histological examination of the kidney
Half of each kidney removed was immediately fixed in 4% paraformaldehyde and 4% sucrose in 100 mM sodium phosphate (pH 7.4) on ice for 1 h and then with Bouin's solutions (pH 3.8) for 3 days at 4°C. Sagittal slices were cut and embedded in paraffin. Sections 3 µm in thickness were cut and stained with Azan and periodic acid-Schiff (PAS) stains. At least 50 glomeruli were examined in each specimen. Glomerular sclerotic lesions were intensely stained by PAS. The severity of these lesions was graded according to the percentage of the glomeruli involved: 0, no lesions (Group 0); 1+, 1–25% (Group 1); 2+, 26–50% (Group 2); 3+, 51–75% (Group 3) and 4+, 76–100% (Group 4). An overall glomerulosclerosis score was calculated by multiplying the severity score (0 to 4+) by the percentage of glomeruli affected and totalling the five values [12].
To localize immunohistochemically L-PGDS molecules in the kidney, we used the polyclonal antibody against mouse L-PGDS [13]. In brief, deparaffinized sections were pretreated with 0.3% H2O2 in methanol for 15 min at room temperature to eliminate endogenous peroxidase activity, then digested with 0.3% (w/v) pepsin (Sigma-Aldrich Japan, Tokyo, Japan) in 0.01 mol/l HCl for 5 min at room temperature in order to unmask the antigens. Thereafter, non-specific binding sites were blocked for 1 h with 10% normal goat serum in sodium phosphate buffered saline (PBS), pH 7.4, containing 0.1% (v/v) Triton X-100 and 0.1% sodium azide. After incubation overnight at 4°C with rabbit polyclonal anti-mouse L-PGDS antibody (10 000-fold dilution) in PBS containing 1.0% (v/v) normal goat serum and 0.1% (v/v) Triton X-100, the sections were serially incubated with biotinylated anti-rabbit IgG antibody for 1 h at room temperature. A Vectastain Elite avidin-biotin-peroxidase kit (Vector Laboratories) with a H2O2-supplemented 3,3′-diaminobenzidine substrate was used according to the manufacturer's protocol. Tissues were counterstained with haematoxylin. For the control experiments, pre-immune rabbit IgG was used as the primary antibody. The absorbed antibody was prepared by incubation of the polyclonal antibody with excess amounts of the recombinant rat L-PGDS (1 mg/ml) at 4°C overnight and used as another control. To identify the cell types in the renal tissue, we also immunostained tissue sections with monoclonal antibodies against CD34 (Nichirei), von Willebrand factor (DAKO Japan, Co., Ltd, Kyoto, Japan), CD68 (DAKO), and smooth muscle α-actin (DAKO) to identify myofibroblasts, endothelial cells, macrophages and smooth muscle cells, respectively.
L-PGDS mRNA in the kidney
Total RNA was extracted from the rat kidney by the guanidinium thiocyanate–phenol–chloroform method using ISOGEN (Nippon Gene, Tokyo, Japan). First strand cDNA was transcribed from 1 µg total RNA of rat kidney with random primers and avian myeloblastosis virus reverse transcriptase (TAKARA BIO INC., Shiga, Japan). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was used as an internal control. The sequence-specific primers used were as follows: rat L-PGDS forward primer, 5′-CAGGAAAAACCAGTGTGAGACC-3′; rat L-PGDS reverse primer, 5′-AGAGGGTGGCCATGCGGAAG-3′; rat GAPDH forward primer, 5′-TGAACGGGAAGCTCACTGG-3′; and rat GAPDH reverse primer, 5′-TCCACCACCCTGTTGCTGTA-3′.
For semiquantitative PCR, we amplified the cDNA by using a Light Cycler and LightCycler™ FastStart DNA Master SYBR Green I kit (Roche Diagnostics, Mannheim, Germany) [14]. The reactions were cycled 40 times with denaturation at 95°C for 15 s, annealing for 5 s at 56°C for L-PGDS or at 60°C for GAPDH, and elongation at 72°C for 10 s. Fluorescence for L-PGDS and GAPDH were detected after elongation at a temperature of 87°C. Quantification data were analysed with LightCycler analysis software. All PCR products were visualized with UV light after electrophoresis in 1% agarose gel containing ethidium bromide to verify that only specific amplificaion had occurred and subsequently sequenced to confirm their origin from the intended mRNAs.
Statistical analysis
The values were expressed as mean±SE. All statistical analyses were performed using STATISTICA software version 6.0 (StatSoft, Tulsa, OK) running on a Windows XP computer system. The differences were assessed by using ANOVA following post hoc analysis (Scheffe test), Student's t-test or Wilcoxon's test, multivariate analysis or analysis of Kaplan–Meier product-limit method. Probability values of <0.05 were defined as significant.
Results
Basal characteristics
One OLETF rat assigned to the untreated group and one of the high-dose treatment group died during the experiment. Hence, these two rats were excluded from the analyses.
Water intake was measured in the last week of the therapeutic period. The doses of troglitazone, which were estimated using amounts of water intake and the respective drug concentrations, were 15.6±1.4 mg/day/kg body weight for the low-dose troglitazone treatment and 23.7±1.8 mg/day/kg body weight for the high-dose troglitazone treatment. The doses accorded with its therapeutic range reported previously.
Changes of body weight were shown in Figure 1. The OLETF rats gained body weight in an age-dependent manner throughout the study. The untreated and treated OLETF rats exhibited higher body weight than the control LETO. However, there were no differences among the treated and untreated OLETF rats. Systolic BPs were demonstrated in Figure 2. In the untreated OLETF rats systolic BP significantly increased along with ageing up to 169±5 mmHg at the age of 48 weeks. The troglitazone treatments significantly attenuated the BP rising throughout the experiment. The BPs in the non-diabetic LETO rats slightly increased along with ageing; however, the BP levels remained within a normal range almost throughout the experiment.
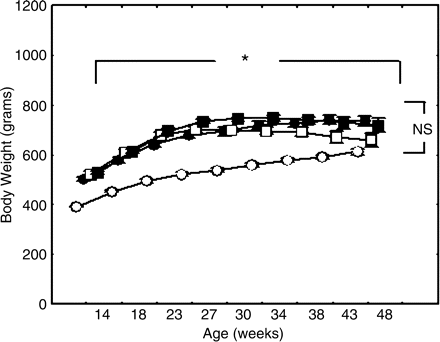
Alterations in body weight. Time-dependent alterations in body weight in the respective experimental groups are shown. The body weights increased along with the ageing in the OLETF rat groups; however, there were no differences in body weight among the three OLETF groups. In contrast, the body weight in the non-diabetic LETO rats was less than the untreated OLETF rats throughout the study. The differences were analysed using two-way ANOVA. *The body weight significantly increased along with the ageing among the four experimental groups (P<0.02). NS, no differences in body weight among the three OLETF rat groups. Open circle, non-diabetic rata (LETO); solid circle, untreated OLETF rats; open square, 0.05% troglitazone-treated OLETF rats, and solid square, 0.1% troglitazone-treated OLETF rats.
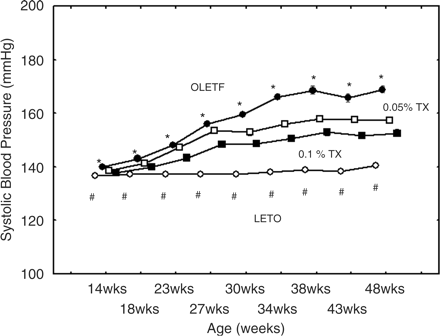
Systolic blood pressures increased along with the ageing of the untreated OLETF rats (P<0.0001)(solid circles), and this was significantly attenuated with troglitazon trreatments (low dose indicated with open squares amd the high dose with solid squares. *The differences among the OLETF rat groups were analyzed at the respective ages by two-way ANOVA (P<0.001). Rising of systolic blood pressures of the LETO group was very slight. #P<0.0025 vs the untreated OLETF rats. OLETF, untreated OLETF rats; 0.05%TX, 0.05% troglitazone-treated OLETF; 0.1%TX, 0.1% troglitazone treated OLETF; and LETO, control LETO rats.
Glucose metabolism
The time points when glucosuria (≥5 mg/dl) occurred were shown using Kaplan–Meier analysis in Figure 3. The onset of glucosuria was presented as a completed (uncensored) time point marked by circles or squares. The censored time points were presented as plus letters at the age of 48 weeks. The urinary excretions of glucose were far below its detection limit in the untreated OLETF aged 14 or 18 weeks. Thereafter, the number of rats showing glucosuria increased, and the troglitazone treatments delayed the onset of glucosuria. In the LETO rats, only one exhibited more than 5 mg/dl glucosuria throughout the experiment.
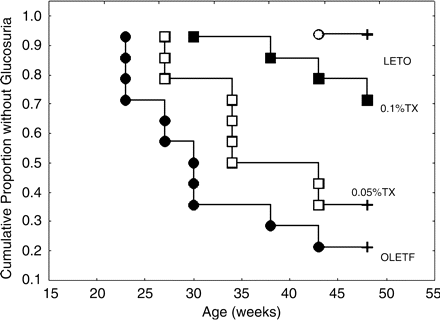
Kaplan–Meier analysis of onset of glucosuria. The onset of glucosuria (5 mg/dl) was analysed using Kaplan–Meier analysis. Only one rat in the LETO group developed glucosuria during the experiment. In the untreated OLETF rats, the number of rats developing the glucosuria stepwisely increased from 23 weeks after birth. Total number of valid observations: 57, uncensored as indicated by circles and squares: 25 (43.86%) and censored as indicated by plus: 32 (56.14%), chi-square = 22.975, df = 3, P<0.00004. Censored mark in 0.1%TX group overlapped the square at the age of 48 weeks. LETO, non-diabetic, control rats; OLETF, untreated OLETF rats; 0.05%TX, 0.05% troglitazone-treated OLETF rats; 0.1%TX, 0.1% troglitazone-treated OLETF.
In the untreated OLETF rats aged 48 weeks, the plasma glucose concentrations were higher than the non-diabetic LETO rats, and conversely, the insulin levels were lower in the untreated OLETF than the non-diabetic LETO rats (P = 0.0001, P = 0.0082, respectively) (Figure 4). The troglitazone treatments did not alter the plasma glucose concentrations; however, the insulin levels tended to decrease (untreated vs 0.05%TX, P = 0.0125).
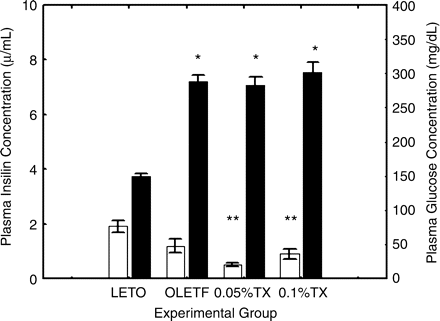
Glucose and insulin levels. Plasma glucose concentrations (right columns) were significantly higher in the untreated OLETF rats than non-diabetic LETO aged 48 weeks. The troglitazone treatments did not affect the glucose levels; however, plasma insulin levels (left columns) tended to decrease with the treatments. The differences were analysed using one-way ANOVA followed by post hoc analysis of Scheffe. The group differences were significant for glucose (P<0.0001) and insulin (P<0.0001). *P<0.0001, **P<0.001 vs LETO (Scheffe test). LETO, non-diabetic, control rats; OLETF, untreated OLETF rats; 0.05%TX, 0.05% troglitazone-treated OLETF rats; 0.1%TX, 0.1% troglitazone-treated OLET. The open columns represent plasma insulin levels on the left scale and the solid columns plasma glucose levels on the right scale.
Plasma L-PGDS levels
The plasma concentrations of L-PGDS tended to increase in the untreated OLETF rats, as compared with those of the non-diabetic LETO (50.14±4.22 vs 43.26±1.19 ng/ml, P = 0.054), and this increase was reversed toward the control levels with the high-dose troglitazone treatment [47.02±2.55 ng/ml for low dose and 40.40±1.52 ng/ml for high dose, P = 0.010 (vs untreated OLETF)].
Urinary L-PGDS excretions and variables indicating renal injuries
In the non-diabetic LETO rats aged 14 weeks, the urinary L-PGDS excretions were as great as 5.92±1.72 µg/day. In the untreated OLETF rats at the same age, which were presumed to be before the onset of type 2 diabetes, exhibited 61% higher urinary excretions of L-PGDS than the non-diabetic LETO (9.52±1.86 µg/day, P<0.0001) (upper graph of Figure 5). The excretions of L-PGDS in the untreated OLETF became greater along with their ageing. Ultimately, the difference between the untreated diabetic OLETF and non-diabetic LETO increased by 96% at the age of 48 weeks (10.39±0.27 vs 20.37±1.19 µg/day, P<0.0001). The high-dose troglitazone treatments significantly decreased urinary excretions of L-PGDS in OLETF rats aged 43 and 48 weeks.
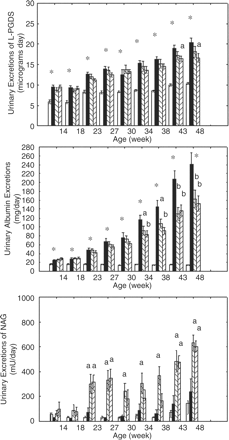
Urinary excretions of L-PGDS, albumin and NAG. The upper graph shows the time-dependent urinary excretions of L-PGDS, the middle the urinary excretions of albumin, and the lower the urinary excretions of NAG. The differences were analysed by one- and two-way ANOVA followed by post hoc analysis of Scheffe. Group effects were statistically significant (P<0.0001) for urinary L-PGDS, urinary albumin and urinary NAG excretions. *P<0.0001 vs non-diabetic LETO. aP<0.05 and bP<0.001 vs the untreated OLETF rats. Open column, non-diabetic LETO; solid column, untreated OLETF; slashed column (left high), 0.05% troglitazone-treated OLETF; and slashed column (right high), 0.1% troglitazone-treated OLETF rats.
Creatinine clearance was smaller in OLETF rats compared to LETO rats (3612±323 vs 4525±137 ml/day, P<0.05). The troglitazone treatments significantly improved creatinine clearance rate, as compared with the untreated OLETF (4433±213 for low-dose and 4230±211 ml/day for high dose, P<0.05 and <0.05 vs the untreated OLETF, respectively).
As a marker of glomerular permeability in diabetes, we examined the alterations of urinary excretions of albumin (middle graph of Figure 5). The excretions of albumin were significantly higher in the untreated OLETF rats aged 18 weeks, which would be in the pre-diabetic state, than the non-diabetic LETO. In the older rats, the excretions strikingly increased, and the difference between the untreated OLETF and the non-diabetic LETO became much greater. The troglitazone treatments attenuated the increases of albuminuria in OLETF rats.
As a marker of tubular injury, we investigated urinary excretions of NAG (lower graph of Figure 5). There were no differences in the urinary excretions of NAG between the untreated OLETF and non-diabetic LETO throughout the study, thereby suggesting that the tubular injury might be minimal in the renal injury associated with diabetes in OLETF rats. Surprisingly, however, the troglitazone treatments significantly increased the urinary excretions of NAG in rats aged 26 weeks and older, as compared with the untreated OLETF rats.
To determine glomerular filtration rate of L-PGDS, clearance rate of L-PGDS from glomeruli (Cpgds) was estimated and expressed as the ratio (%) to creatinine clearance rate (Ccr). While the ratio of albumin clearance (Calb/Ccr) was almost below its detection limit in the non-diabetic LETO, the Cpgds/Ccr was significant in the non-diabetic LETO. The order of Cpgds was approximately 50 times as great as Calb (Figure 6). Moreover, when diabetes advanced, the Cpgds/Ccr was significantly increased by 147% in the untreated OLETF rats, as compared with the non-diabetic LETO aged 48 weeks (13.38 vs 5.43%, P<0.001). The troglitazone treatments tended to decrease both Cpgds/Ccr and Calb/Ccr. The clearance of NAG was increased in the untreated OLETF, as compared with the non-diabetic LETO, and interestingly, the troglitazone tended to augment this ratio.
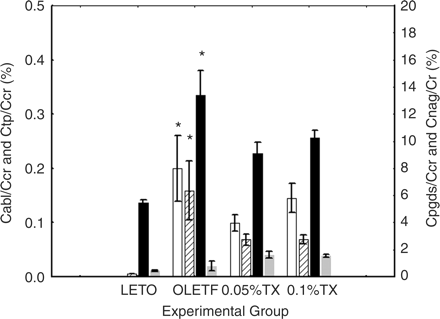
Glomerular filtration rate L-PGDS. Glomerular clearance rates of albumin (Calb), total protein (Ctp), L-PGDS (Cpgds), and NAG (Cnag) were demonstrated in the ratio to creatinine clearance rate (Ccr). The differences were analysed using one-way ANOVA followed by post hoc analysis of Scheffe. Group differences were <0.001 for Calb, <0.01 for Ctp, <0.0001 for Cpgds and <0.01 for Cnag. *P<0.01 vs non-diabetic LETO. LETO, non-diabetic, control rats; OLETF, untreated OLETF rats; 0.05%TX, 0.05% troglitazone-treated OLETF rats; and 0.1%TX, 0.1% troglitazone-treated OLETF. Open column, Calb/Ccr on the left scale; slashed column, Ctp/Ccr on the left scale; solid column, Cpgds/Ccr on the right scale; and dark gray column, Cnag/Ccr on the right scale.
Histological examination
We investigated the histological alterations in the kidney. The control LETO rats exhibited glomeruli with normal appearance (Figure 7a). In contrast, the glomeruli of OLETF exhibited mesangial sclerosis and PAS-positive nodular lesions with various degress of severity, which correspond to the lesions characteristic of diabetic nephropathy (Figure 7b and c). The number of glomeruli with severe sclerosis was significantly greater in the untreated OLETF rats than the non-diabetic LETO. The troglitazone treatments attenuated such sclerotic lesions (Figure 7c and d).
![Representative glomerular lesions. Glomerular lesions of the rats aged 48 weeks were stained by PAS. The micrograph (a) represents a normal glomerulus in the non-diabetic LETO. A representative glomerulus of the untreated, diabetic OLETF exhibited segmental or global sclerosis [micrograph (b)] and PAS-positive nodular lesions as indicated by an arrow in micrograph (b). Basically, the troglitazone-treated OLETF rats represented glomerular lesions similar to the untreated OLETF as shown in micrograph (c) for the low-dose and in micrograph (d) for the high-dose, but the severity differed between the untreated and troglitazone-treated OLETF.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/ndt/21/4/10.1093_ndt_gfk009/1/m_gfk009f7.gif?Expires=1716894448&Signature=T1s7GeldvzhZkm049rDCJgWVLH63ljF5DWfXC0bl4WKn~hSunb0MNKuUeGgTvX5iZ35yub5SBFYRgYB3f0seGz0-KteuNaUx8he-6sxr6D9m561EVTw1rkNWwIWP~Ie6Ol5zU60RntLoKx8Ar774yddnaKGQI-LMe4iDP2AuPwjrRAUy4IzkqSKWSIAOjxtNQqhcR-RJR5xLRsdSVLuqw3leP8vkTbaxSXEbKijt9MLPaiJ3fU8bLgndBU-NURiHHZ4ZObMtXRxSDhUr-aFRm0DvaP-ZXH~Z3bk0AUm0bPxB8Ua3NJhcVFVR7NEGIWk-fW1KxncmZMOGm92yxp2hnQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Representative glomerular lesions. Glomerular lesions of the rats aged 48 weeks were stained by PAS. The micrograph (a) represents a normal glomerulus in the non-diabetic LETO. A representative glomerulus of the untreated, diabetic OLETF exhibited segmental or global sclerosis [micrograph (b)] and PAS-positive nodular lesions as indicated by an arrow in micrograph (b). Basically, the troglitazone-treated OLETF rats represented glomerular lesions similar to the untreated OLETF as shown in micrograph (c) for the low-dose and in micrograph (d) for the high-dose, but the severity differed between the untreated and troglitazone-treated OLETF.
The lesions in glomeruli were semiquantitatively assessed according to the method reported previously [9,10]. The score ranges from 0 (absolutely normal glomeruli) to 400 (all glomeruli exhibit obsolescence). The untreated OLETF rats exhibited higher glomerular sclerosis score than the non-diabetic LETO (226±10 vs 80±10, P = 0.001), and this increase was attenuated with the troglitazone treatments [189±13 for low-dose OLETF, and 192±10 for high-dose OLETF, P = 0.019 and P = 0.034 (vs untreated OLETF), respectively].
L-PGDS mRNA in the kidney
To disclose the role of de novo synthesis in L-PGDS in urine, we determined L-PGDS mRNA in the kidney, using semiquantitative PCR. In the renal medullae, there was no difference in the mRNA levels between the non-diabetic LETO and untreated OLETF aged 48 weeks (Figure 8).
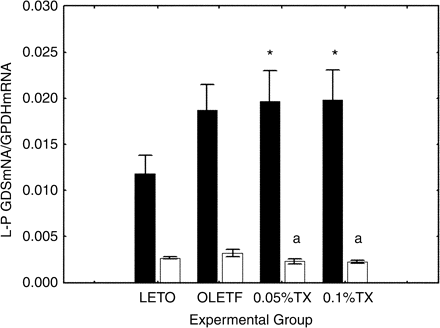
L-PGDS mRNA in the kidney. L-PGDS mRNA was seminquantitatively assessed in the renal cortex (solid columns) and the medulla (open columns). The group differences were assessed using one-way ANOVA followed by post hoc analysis of Scheffe. The group differences were statistically significant for the renal cortex (P<0.05), but not for the renal medulla. *P<0.05 vs the control LETO, aP<0.05 vs the untreated OLETF. LETO, non-diabetic control rats; OLETF, untreated OLETF rats; 0.05%TX, 0.05% troglitazone-treated OLETF rats; and 0.1%TX, 0.1% troglitazone-treated OLETF.
In contrast, in the cortex, the mRNA levels tended to increase in the untreated OLETF aged 48 weeks, as compared with the non-diabetic LETO (P = 0.074). However, the increase was not changed with the troglitazone treatments.
Immunohistochemistry of L-PGDS in the kidney
We attempted to localize L-PGDS antigenicity in the kidney. In the 48-week-old OLETF rats, L-PGDS antigens were detected in the proximal tubules in the deep cortex and outer medulla, but not in the inner medullary and papillary regions. The signals were not detected in the glomeruli (Figure 9). The signals disappeared when the antigenicity was blocked using L-PGDS antigens before the staining.
![Localization of L-PGDS antigenicity in the kidney. We localized the L-PGDS antigenicity in the kidney of OLETF rats aged 48 weeks using immunoenzyme staining as described in the text. The signals were detected in the proximal tubules in the deep cortex and the outer medulla, as shown in micrograph (a). In contrast, the signals were not found in the glomeruli per se. It was noted that the signals were not found in the thin-limb of Henle's loop in the outer medulla, as shown in micrograph (b). The signals in the tubules were blocked by pretreatment of amounts of L-PGDS prior to the staining, suggesting that the expression of L-PGDS antigenicity was a specific finding [micrograph (c)]. The signals were also detected in urine casts, suggesting the presence of the antigenicity in urine per se [micrograph (d)].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/ndt/21/4/10.1093_ndt_gfk009/1/m_gfk009f9.gif?Expires=1716894448&Signature=XpSzHF7BAMJruPLzAg-8OJU7Lg7lMd5QnQw~7xkNj8fJeJjEPg4yq6WLJWoinpUH5nt6xXIZSXO3r3RZHKLX9jhO2NXwkA-g1TJwJL76fW4kUtLa2GbzuEP2uP9GDZ7cQvj-kQViJDNCl7bYEPmq9FLxbK-zpTLRtpPWEy6O9SER5Rv8VpRNbNS7TK8Y6Z-OjPoRcPK5siA2ozrpxp0ysVbKFvMmhhQ3Ws-b4O6vKgm0pZb1Di6lEYimuvaDyFJ~taleXL-54Zr5pHogcRkSDvc0d9yDTBewTKoAEkpXErUiaU098VdhsJIvNBfXKhLD32SywB4tYnh1oUzis9tbaQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Localization of L-PGDS antigenicity in the kidney. We localized the L-PGDS antigenicity in the kidney of OLETF rats aged 48 weeks using immunoenzyme staining as described in the text. The signals were detected in the proximal tubules in the deep cortex and the outer medulla, as shown in micrograph (a). In contrast, the signals were not found in the glomeruli per se. It was noted that the signals were not found in the thin-limb of Henle's loop in the outer medulla, as shown in micrograph (b). The signals in the tubules were blocked by pretreatment of amounts of L-PGDS prior to the staining, suggesting that the expression of L-PGDS antigenicity was a specific finding [micrograph (c)]. The signals were also detected in urine casts, suggesting the presence of the antigenicity in urine per se [micrograph (d)].
Multivariate analysis of urinary L-PGDS
To investigate the correlations between the urinary excretions of L-PGDS in the early diabetes and alterations of renal function following established diabetes at 48 weeks after birth, we analysed the data using the Pearson's bivariate correlation method. The urinary L-PGDS excretions in the rats aged 14 weeks before the onset of diabetes significantly correlated with the variables including albuminuria, systolic BP, urinary excretions of total protein (UTP), urinary excretions of NAG (UNAG) and glomerular sclerosing (GS) score in the rats aged 48 weeks (Table 1). The higher the urinary L-PGDS excretions at the age of 14 weeks, the higher the variables indicating renal function at the age of 48 weeks.
Pearson's correlation analysis
| PGDS-14 weeks | PGDS-18 weeks | PGDS-43 weeks | |
|---|---|---|---|
| PGDS-14 weeks | 1.00 | 0.70* | 0.60* |
| PGDS-18 weeks | 0.70* | 1.00 | 0.61* |
| PGDS-23 weeks | 0.67* | 0.76* | 0.74* |
| PGDS-27 weeks | 0.65* | 0.74* | 0.75* |
| PGDS-30 weeks | 0.55* | 0.54* | 0.64* |
| PGDS-34 weeks | 0.55* | 0.53* | 0.84* |
| PGDS-38 weeks | 0.55* | 0.58* | 0.90* |
| PGDS-43 weeks | 0.60* | 0.61* | 1.00 |
| PGDS-48 weeks | 0.60* | 0.57* | 0.81* |
| Alb-14 weeks | 0.59* | 0.41* | 0.47* |
| Alb-18 weeks | 0.50* | 0.55* | 0.63* |
| Alb-23 weeks | 0.52* | 0.56* | 0.73* |
| Alb-27 weeks | 0.47* | 0.37* | 0.71* |
| Alb-30 weeks | 0.46* | 0.42* | 0.65* |
| Alb-34 weeks | 0.48* | 0.47* | 0.68* |
| Alb-38 weeks | 0.49* | 0.47* | 0.77* |
| Alb-43 weeks | 0.47* | 0.55* | 0.70* |
| Alb-48 weeks | 0.54* | 0.56* | 0.69* |
| Systolic BP | 0.43* | 0.51* | 0.67* |
| UTP | 0.53* | 0.47* | 0.72* |
| SCr | −0.15 | −0.23 | −0.08 |
| cor mRNA | 0.15 | 0.32 | 0.33* |
| med mRNA | −0.09 | 0.01 | −0.09 |
| pNAG | 0.05 | 0.11 | −0.06 |
| Ccr | −0.18 | −0.11 | −0.31 |
| UNAG | 0.30* | 0.26 | 0.39* |
| GS | 0.51* | 0.49* | 0.73* |
| PGDS-14 weeks | PGDS-18 weeks | PGDS-43 weeks | |
|---|---|---|---|
| PGDS-14 weeks | 1.00 | 0.70* | 0.60* |
| PGDS-18 weeks | 0.70* | 1.00 | 0.61* |
| PGDS-23 weeks | 0.67* | 0.76* | 0.74* |
| PGDS-27 weeks | 0.65* | 0.74* | 0.75* |
| PGDS-30 weeks | 0.55* | 0.54* | 0.64* |
| PGDS-34 weeks | 0.55* | 0.53* | 0.84* |
| PGDS-38 weeks | 0.55* | 0.58* | 0.90* |
| PGDS-43 weeks | 0.60* | 0.61* | 1.00 |
| PGDS-48 weeks | 0.60* | 0.57* | 0.81* |
| Alb-14 weeks | 0.59* | 0.41* | 0.47* |
| Alb-18 weeks | 0.50* | 0.55* | 0.63* |
| Alb-23 weeks | 0.52* | 0.56* | 0.73* |
| Alb-27 weeks | 0.47* | 0.37* | 0.71* |
| Alb-30 weeks | 0.46* | 0.42* | 0.65* |
| Alb-34 weeks | 0.48* | 0.47* | 0.68* |
| Alb-38 weeks | 0.49* | 0.47* | 0.77* |
| Alb-43 weeks | 0.47* | 0.55* | 0.70* |
| Alb-48 weeks | 0.54* | 0.56* | 0.69* |
| Systolic BP | 0.43* | 0.51* | 0.67* |
| UTP | 0.53* | 0.47* | 0.72* |
| SCr | −0.15 | −0.23 | −0.08 |
| cor mRNA | 0.15 | 0.32 | 0.33* |
| med mRNA | −0.09 | 0.01 | −0.09 |
| pNAG | 0.05 | 0.11 | −0.06 |
| Ccr | −0.18 | −0.11 | −0.31 |
| UNAG | 0.30* | 0.26 | 0.39* |
| GS | 0.51* | 0.49* | 0.73* |
PGDS, urinary excretions of L-PGDS at respective ages; Alb, urinary excretions of albumin; systolic BP, systolic blood presure at the age of 48 weeks; UTP, urinary excretions of total protein at the age of 48 weeks; cor mRNA, L-PGDS mRNA in the renal cortex; med mRNA, L-PGDS mRNA in the medullar; pNAG, plasma levels of NAG; Ccr, creatinine clearance; UNAG, urinary excretions of NAG at the age of 48 weeks; and GS, glomerulo sclerosing scale as defined in the text. Marked correlations are significant at P<0.05.
Pearson's correlation analysis
| PGDS-14 weeks | PGDS-18 weeks | PGDS-43 weeks | |
|---|---|---|---|
| PGDS-14 weeks | 1.00 | 0.70* | 0.60* |
| PGDS-18 weeks | 0.70* | 1.00 | 0.61* |
| PGDS-23 weeks | 0.67* | 0.76* | 0.74* |
| PGDS-27 weeks | 0.65* | 0.74* | 0.75* |
| PGDS-30 weeks | 0.55* | 0.54* | 0.64* |
| PGDS-34 weeks | 0.55* | 0.53* | 0.84* |
| PGDS-38 weeks | 0.55* | 0.58* | 0.90* |
| PGDS-43 weeks | 0.60* | 0.61* | 1.00 |
| PGDS-48 weeks | 0.60* | 0.57* | 0.81* |
| Alb-14 weeks | 0.59* | 0.41* | 0.47* |
| Alb-18 weeks | 0.50* | 0.55* | 0.63* |
| Alb-23 weeks | 0.52* | 0.56* | 0.73* |
| Alb-27 weeks | 0.47* | 0.37* | 0.71* |
| Alb-30 weeks | 0.46* | 0.42* | 0.65* |
| Alb-34 weeks | 0.48* | 0.47* | 0.68* |
| Alb-38 weeks | 0.49* | 0.47* | 0.77* |
| Alb-43 weeks | 0.47* | 0.55* | 0.70* |
| Alb-48 weeks | 0.54* | 0.56* | 0.69* |
| Systolic BP | 0.43* | 0.51* | 0.67* |
| UTP | 0.53* | 0.47* | 0.72* |
| SCr | −0.15 | −0.23 | −0.08 |
| cor mRNA | 0.15 | 0.32 | 0.33* |
| med mRNA | −0.09 | 0.01 | −0.09 |
| pNAG | 0.05 | 0.11 | −0.06 |
| Ccr | −0.18 | −0.11 | −0.31 |
| UNAG | 0.30* | 0.26 | 0.39* |
| GS | 0.51* | 0.49* | 0.73* |
| PGDS-14 weeks | PGDS-18 weeks | PGDS-43 weeks | |
|---|---|---|---|
| PGDS-14 weeks | 1.00 | 0.70* | 0.60* |
| PGDS-18 weeks | 0.70* | 1.00 | 0.61* |
| PGDS-23 weeks | 0.67* | 0.76* | 0.74* |
| PGDS-27 weeks | 0.65* | 0.74* | 0.75* |
| PGDS-30 weeks | 0.55* | 0.54* | 0.64* |
| PGDS-34 weeks | 0.55* | 0.53* | 0.84* |
| PGDS-38 weeks | 0.55* | 0.58* | 0.90* |
| PGDS-43 weeks | 0.60* | 0.61* | 1.00 |
| PGDS-48 weeks | 0.60* | 0.57* | 0.81* |
| Alb-14 weeks | 0.59* | 0.41* | 0.47* |
| Alb-18 weeks | 0.50* | 0.55* | 0.63* |
| Alb-23 weeks | 0.52* | 0.56* | 0.73* |
| Alb-27 weeks | 0.47* | 0.37* | 0.71* |
| Alb-30 weeks | 0.46* | 0.42* | 0.65* |
| Alb-34 weeks | 0.48* | 0.47* | 0.68* |
| Alb-38 weeks | 0.49* | 0.47* | 0.77* |
| Alb-43 weeks | 0.47* | 0.55* | 0.70* |
| Alb-48 weeks | 0.54* | 0.56* | 0.69* |
| Systolic BP | 0.43* | 0.51* | 0.67* |
| UTP | 0.53* | 0.47* | 0.72* |
| SCr | −0.15 | −0.23 | −0.08 |
| cor mRNA | 0.15 | 0.32 | 0.33* |
| med mRNA | −0.09 | 0.01 | −0.09 |
| pNAG | 0.05 | 0.11 | −0.06 |
| Ccr | −0.18 | −0.11 | −0.31 |
| UNAG | 0.30* | 0.26 | 0.39* |
| GS | 0.51* | 0.49* | 0.73* |
PGDS, urinary excretions of L-PGDS at respective ages; Alb, urinary excretions of albumin; systolic BP, systolic blood presure at the age of 48 weeks; UTP, urinary excretions of total protein at the age of 48 weeks; cor mRNA, L-PGDS mRNA in the renal cortex; med mRNA, L-PGDS mRNA in the medullar; pNAG, plasma levels of NAG; Ccr, creatinine clearance; UNAG, urinary excretions of NAG at the age of 48 weeks; and GS, glomerulo sclerosing scale as defined in the text. Marked correlations are significant at P<0.05.
To assess the cause–consequence relationship, firstly, we extracted independent predictors of urinary L-PGDS excretions from various parameters indicating renal damage using multiple regression analyses. The urinary L-PGDS excretions at the age of 48 weeks were independently determined by UTPs (B = 0.0387, P = 0.0012) and creatinine clearance rate (B = 0.0024, P = 0.0315). Systolic BP was not an independent predictor of urinary L-PGDS excretions (B = 0.0256, P = 0.7615).
Secondly, we attempted to reveal predictors of variables indicating renal damage, i.e. urinary excretions of total protein or GS score in the OLETF rats aged 48 weeks (Tables 2 and 3). The urinary excretions of L-PGDS in the rats aged 14 weeks were a strong predictor of the UTP in the rats aged 48 weeks. However, we could not find the same relationship between the urinary L-PGDS excretions and GS score aged 48 weeks.
Multiple regression summary for urinary excretion of total protein
| β | SE | β | SE | t(51) | P-level | |
|---|---|---|---|---|---|---|
| Intercept | −43.6584 | 49.63708 | −0.879553 | 0.383227 | ||
| PGDS-14 weeks | 0.358558 | 0.173488 | 16.3164 | 7.89470 | 2.066756 | 0.043852 |
| PGDS-18 weeks | 0.008775 | 0.170965 | 0.4221 | 8.22421 | 0.051327 | 0.959266 |
| Alb-14 weeks | −0.119507 | 0.210524 | −2.4626 | 4.33821 | −0.567663 | 0.572755 |
| Alb-18 weeks | 0.472996 | 0.209720 | 4.9445 | 2.19234 | 2.255370 | 0.028430 |
| Nag-14 weeks | 0.030396 | 0.108818 | 0.0280 | 0.10006 | 0.279331 | 0.781121 |
| Nag-18 weeks | 0.047144 | 0.108357 | 0.0444 | 0.10211 | 0.435077 | 0.665342 |
| β | SE | β | SE | t(51) | P-level | |
|---|---|---|---|---|---|---|
| Intercept | −43.6584 | 49.63708 | −0.879553 | 0.383227 | ||
| PGDS-14 weeks | 0.358558 | 0.173488 | 16.3164 | 7.89470 | 2.066756 | 0.043852 |
| PGDS-18 weeks | 0.008775 | 0.170965 | 0.4221 | 8.22421 | 0.051327 | 0.959266 |
| Alb-14 weeks | −0.119507 | 0.210524 | −2.4626 | 4.33821 | −0.567663 | 0.572755 |
| Alb-18 weeks | 0.472996 | 0.209720 | 4.9445 | 2.19234 | 2.255370 | 0.028430 |
| Nag-14 weeks | 0.030396 | 0.108818 | 0.0280 | 0.10006 | 0.279331 | 0.781121 |
| Nag-18 weeks | 0.047144 | 0.108357 | 0.0444 | 0.10211 | 0.435077 | 0.665342 |
Dependent variable is urinary excretion of total protein at the age of 48 weeks. Independent variables are urinary excretions of L-PGDS at the age of 14 weeks (PGDS-14 weeks) and of 18 weeks (PGDS-18 weeks), urinary excretions of albumin at the age of 14 weeks (Alb-14 weeks) and 18 weeks (Alb-18 weeks), and urinary excretions of NAG at the age of 14 weeks (NAG-14 weeks) and 18 weeks (NAG-18 weeks).
Multiple regression summary for urinary excretion of total protein
| β | SE | β | SE | t(51) | P-level | |
|---|---|---|---|---|---|---|
| Intercept | −43.6584 | 49.63708 | −0.879553 | 0.383227 | ||
| PGDS-14 weeks | 0.358558 | 0.173488 | 16.3164 | 7.89470 | 2.066756 | 0.043852 |
| PGDS-18 weeks | 0.008775 | 0.170965 | 0.4221 | 8.22421 | 0.051327 | 0.959266 |
| Alb-14 weeks | −0.119507 | 0.210524 | −2.4626 | 4.33821 | −0.567663 | 0.572755 |
| Alb-18 weeks | 0.472996 | 0.209720 | 4.9445 | 2.19234 | 2.255370 | 0.028430 |
| Nag-14 weeks | 0.030396 | 0.108818 | 0.0280 | 0.10006 | 0.279331 | 0.781121 |
| Nag-18 weeks | 0.047144 | 0.108357 | 0.0444 | 0.10211 | 0.435077 | 0.665342 |
| β | SE | β | SE | t(51) | P-level | |
|---|---|---|---|---|---|---|
| Intercept | −43.6584 | 49.63708 | −0.879553 | 0.383227 | ||
| PGDS-14 weeks | 0.358558 | 0.173488 | 16.3164 | 7.89470 | 2.066756 | 0.043852 |
| PGDS-18 weeks | 0.008775 | 0.170965 | 0.4221 | 8.22421 | 0.051327 | 0.959266 |
| Alb-14 weeks | −0.119507 | 0.210524 | −2.4626 | 4.33821 | −0.567663 | 0.572755 |
| Alb-18 weeks | 0.472996 | 0.209720 | 4.9445 | 2.19234 | 2.255370 | 0.028430 |
| Nag-14 weeks | 0.030396 | 0.108818 | 0.0280 | 0.10006 | 0.279331 | 0.781121 |
| Nag-18 weeks | 0.047144 | 0.108357 | 0.0444 | 0.10211 | 0.435077 | 0.665342 |
Dependent variable is urinary excretion of total protein at the age of 48 weeks. Independent variables are urinary excretions of L-PGDS at the age of 14 weeks (PGDS-14 weeks) and of 18 weeks (PGDS-18 weeks), urinary excretions of albumin at the age of 14 weeks (Alb-14 weeks) and 18 weeks (Alb-18 weeks), and urinary excretions of NAG at the age of 14 weeks (NAG-14 weeks) and 18 weeks (NAG-18 weeks).
Multiple regression summary for GS scale
| β | SE | β | SE | t(50) | P-level | |
|---|---|---|---|---|---|---|
| Intercept | 59.34988 | 32.17712 | 1.844475 | 0.071042 | ||
| PGDS-14 weeks | 0.189104 | 0.177556 | 5.57367 | 5.23329 | 1.065042 | 0.291973 |
| PGDS-18 weeks | 0.115079 | 0.171068 | 3.58300 | 5.32622 | 0.672709 | 0.504230 |
| Alb-14 weeks | 0.098419 | 0.217304 | 1.35556 | 2.99301 | 0.452909 | 0.652573 |
| Alb-18 weeks | 0.355637 | 0.211535 | 2.41920 | 1.43896 | 1.681220 | 0.098957 |
| Nag-14 weeks | −0.016056 | 0.108548 | −0.00957 | 0.06471 | −0.147912 | 0.883007 |
| Nag-18 weeks | 0.100080 | 0.107474 | 0.06107 | 0.06558 | 0.931200 | 0.356225 |
| β | SE | β | SE | t(50) | P-level | |
|---|---|---|---|---|---|---|
| Intercept | 59.34988 | 32.17712 | 1.844475 | 0.071042 | ||
| PGDS-14 weeks | 0.189104 | 0.177556 | 5.57367 | 5.23329 | 1.065042 | 0.291973 |
| PGDS-18 weeks | 0.115079 | 0.171068 | 3.58300 | 5.32622 | 0.672709 | 0.504230 |
| Alb-14 weeks | 0.098419 | 0.217304 | 1.35556 | 2.99301 | 0.452909 | 0.652573 |
| Alb-18 weeks | 0.355637 | 0.211535 | 2.41920 | 1.43896 | 1.681220 | 0.098957 |
| Nag-14 weeks | −0.016056 | 0.108548 | −0.00957 | 0.06471 | −0.147912 | 0.883007 |
| Nag-18 weeks | 0.100080 | 0.107474 | 0.06107 | 0.06558 | 0.931200 | 0.356225 |
Dependent variable is GS scale at the age of 48 weeks. Independent variables are urinary excretions of L-PGDS at the age of 14 weeks (PGDS-14 weeks) and of 18 weeks (PGDS-18 weeks), urinary excretions of albumin at the age of 14 weeks (Alb-14 weeks) and 18 weeks (Alb-18 weeks), and urinary excretions of NAG at the age of 14 weeks (NAG-14 weeks) and 18 weeks (NAG-18 weeks).
Multiple regression summary for GS scale
| β | SE | β | SE | t(50) | P-level | |
|---|---|---|---|---|---|---|
| Intercept | 59.34988 | 32.17712 | 1.844475 | 0.071042 | ||
| PGDS-14 weeks | 0.189104 | 0.177556 | 5.57367 | 5.23329 | 1.065042 | 0.291973 |
| PGDS-18 weeks | 0.115079 | 0.171068 | 3.58300 | 5.32622 | 0.672709 | 0.504230 |
| Alb-14 weeks | 0.098419 | 0.217304 | 1.35556 | 2.99301 | 0.452909 | 0.652573 |
| Alb-18 weeks | 0.355637 | 0.211535 | 2.41920 | 1.43896 | 1.681220 | 0.098957 |
| Nag-14 weeks | −0.016056 | 0.108548 | −0.00957 | 0.06471 | −0.147912 | 0.883007 |
| Nag-18 weeks | 0.100080 | 0.107474 | 0.06107 | 0.06558 | 0.931200 | 0.356225 |
| β | SE | β | SE | t(50) | P-level | |
|---|---|---|---|---|---|---|
| Intercept | 59.34988 | 32.17712 | 1.844475 | 0.071042 | ||
| PGDS-14 weeks | 0.189104 | 0.177556 | 5.57367 | 5.23329 | 1.065042 | 0.291973 |
| PGDS-18 weeks | 0.115079 | 0.171068 | 3.58300 | 5.32622 | 0.672709 | 0.504230 |
| Alb-14 weeks | 0.098419 | 0.217304 | 1.35556 | 2.99301 | 0.452909 | 0.652573 |
| Alb-18 weeks | 0.355637 | 0.211535 | 2.41920 | 1.43896 | 1.681220 | 0.098957 |
| Nag-14 weeks | −0.016056 | 0.108548 | −0.00957 | 0.06471 | −0.147912 | 0.883007 |
| Nag-18 weeks | 0.100080 | 0.107474 | 0.06107 | 0.06558 | 0.931200 | 0.356225 |
Dependent variable is GS scale at the age of 48 weeks. Independent variables are urinary excretions of L-PGDS at the age of 14 weeks (PGDS-14 weeks) and of 18 weeks (PGDS-18 weeks), urinary excretions of albumin at the age of 14 weeks (Alb-14 weeks) and 18 weeks (Alb-18 weeks), and urinary excretions of NAG at the age of 14 weeks (NAG-14 weeks) and 18 weeks (NAG-18 weeks).
Discussion
OLETF rats were originally introduced as a rat model of genetic spontaneous diabetes by Kawano et al. [15]. The responsible genetic regions have been mapped to chromosome X and 14. Its profile of glucose metabolism has been well characterized thus far by many investigators. Through these efforts, it is recognized that this strain exhibits increased albuminuria from 18 weeks of age and increased haemoglobin A1c from 22–30 weeks of age. Glomerular lesions start as an increase of glomerular area and mesangial expansion at 22 weeks, and then the mesangial lesions progress to mesangial sclerosis, and exudative lesions are found at 36 weeks. The diabetes enters insulin-dependent phase at 54 weeks of age and thereafter [16,17]. It is also revealed that slightly higher excretion of albumin at 6–18 weeks of age is due to a decrease of anion-charge in the basement membranes and massive amount of albumin excretion is associated with loss of charge selective property [17]. Glomerular lesions at the stage of insulin-dependent diabetes are characterized by loss of charge-selective property and glomerular thickening, mesangial expansion and sclerosis with exudative lesions and nodular lesions, which are acknowledged as glomerular damage quite similar to diabetic nephropathy [18,19].
To accomplish the long-term study, we very carefully handled the animals without any stress, and avoided sampling of blood specimen. For this experimental design we utilized urinary glucose excretion for assessing diabetes status. Such an age-dependent progression of diabetes is useful to investigate the pathophysiological implications of urinary L-PGDS in the establishment of nephropathy following diabetes. In fact, the glucosuria in the untreated OLETF rats was detected at the age of 18–23 weeks, and then the number of rats with glucosuria increased as shown in Figure 3. The troglitazone treatments significantly shifted this trend towards the right on the Kaplan–Meier glucosuria analysis, thereby suggesting that the onset of diabetes was delayed. These data were apparently in accord with previous reports [9–11]. Moreover, we demonstrated glomerular lesions of segmental or global sclerosis with PAS-positive deposits, which are characteristic of lesions seen in diabetic nephropathy. These data suggested that the untreated OLETF rats exhibited diabetes and had indeed diabetic nephropathy in 48 weeks after birth.
This rat strain exhibits rising of BP around 18 weeks of age and this particular time corresponds to the time when insulin resistance develops. In fact, the association of BP rising and diabetes has been reported in other models of diabetes including fructose feeding [20] and Goto Kanzaki (GK) genetic rat model for type 2 diabetes [21]. The literature suggests that diabetes and hypertension are linked in pathophysiological mechanisms underlying and it is difficult to discuss separately the effects of hyperglycaemia and BP rising on kidney damage in diabetic state. In fact, it has been reported that hypertension superimposed on type 2 diabetes in GK strain induces progressive nephropathy. In this sense, we would not necessarily neglect the influence of BP rising on kidney damage, thereby increasing urinary L-PGDS excretions in OLETF. In fact, we found a bivariate correlation between urinary L-PGDS excretions and systolic BP aged 48 weeks; however, multiple regression analyses revealed that urinary L-PGDS excretions were determined by renal damage presented by urinary protein excretions, but not by the higher BPs. This might suggest that urinary L-PGDS excretions were more closely related to the damaged glomeruli per se rather than the causes of the damage, i.e. high BP or diabetic state.
The most important finding in the present study was that in the untreated OLETF rats, the urinary excretions of L-PGDS markedly increased, as compared with the urinary albumin excretions in the very early diabetes. This would be due to increased permeability of the basement membranes. To make this point clear, we estimated the glomerular clearance rate. The control, non-diabetic LETO rats were expected to have normal permeability of glomeruli. However, the glomerular clearance rate of L-PGDS was more than 50 times as great as the clearance rate of albumin. Considering these findings, L-PGDS easily passed through the basement membranes, and this might contribute to the predictability of underlying alterations of the glomerular permeability in diabetes.
More interestingly, we demonstrated that the urinary L-PGDS excretions in the early diabetes well correlated to the subsequent alterations of variables indicating renal injury following diabetes. The higher the excretions of L-PGDS in 14-week-old rats the greater the proteinuria or the glomerular lesions in 48-week-old rats. This predictability was confirmed by multiple regression analysis. The increase in urinary excretions of L-PGDS was probably due to an increase in the glomerular permeability to large molecules, which might be an integral component of diabetic nephropathy in the aged OLETF rats.
In the present study, we did not have data on the plasma glucose levels in the untreated OLETF rats aged 14 weeks. According to the literature from other laboratories [15–19], OLETF rats exhibit increased plasma glucose concentrations around 20 weeks after birth, the OLETF rats aged 14 weeks were presumed to have normal or near normal levels of LETO rats. If this is true, the increased permeability to L-PGDS might be prior to the abnormality of glucose metabolism. This strongly suggests that in the OLETF rats, the permeability of the basement membranes is intrinsically greater than the control LETO rats. The exact mechanisms remain to be elucidated; however, this seems quite interesting to understand the susceptibility of diabetics to kidney damage. We determined L-PGDS mRNA levels to examine whether L-PGDS was biosynthesized in the kidney and if so, to assess how much the de novo synthesis contributed to the urinary excretions of L-PGDS. There was no difference in the mRNA levels in the medulla between the LETO and the untreated OLETF rats. In contrast, in the cortex, the L-PGDS mRNA levels tended to be greater in the untreated OLETF than the LETO rats. This indicated that the urinary excretions of L-PGDS in the OLETF rats might be attributed in part to the de novo synthesis. However, since multiple regression analysis revealed that the L-PGDS mRNA was not an independent determinant of the urinary excretions of L-PGDS in 48-week-old OLETF rats, the contribution of the de novo synthesis to the urinary excretions appeared to be small in the present study design.
The pathophysiological aspect of urinary L-PGDS in the progression of renal injury in diabetes is beyond the scope of the present study. However, recent studies highlighted the role of L-PGDS in the cardiovascular system. Eguchi et al. have demonstrated that L-PGDS is expressed in several cells in the cardiovascular system, and the signals are marked particularly in the atherosclerotic lesions [22]. L-PGDS is rich in mast cells infiltrating into the atherosclerotic lesions. L-PGDS enhances PGD2, and this eicosanoid affects haemodynamics in the kidney. In Dahl salt-sensitive rats susceptible to hypertensive injury in the kidney, PGD2 decreases in the renal tissues [23]. Moreover, PGD2 and its metabolites reduce inducible nitric oxide synthase (iNOS) mRNA stimulated by interleukin-1β [24]. NO following expressions of iNOS play a pathophysiological role as reported in diabetic nephropathy [25]. In fact, Shirahase et al. [26] have demonstrated in vivo that the pretreatment with PGD2 attenuates organ damage induced by lipopolysaccharide endotoxin shock. The inhibition of cytokine-mediated NO synthesis by enhanced PGD2/L-PGDS in the kidney possibly attenuates progression of the kidney damage. Accordingly, it seemed probable that the increase in L-PGDS in the kidney is a sort of adaptation mechanism against the kidney injury in diabetes.
Finally, the urinary excretions of L-PGDS markedly increased, as compared with the urinary albumin excretions in the very early diabetes in OLETF rats. Urinary excretions of L-PGDS are likely to reflect the underlying increase in glomerular permeability. This property may be useful to predict forthcoming glomerular injury following diabetes in OLETF rats. Some studies using the transgenic and knockout mice are now under way, and the data from these experiments would address the pathophysiological implications of urinary L-PGDS more directly.
The authors acknowledge Noriko Ohshima and Kozo Sasaki for technical assistance. They also thank Otsuka Pharmaceutical Co., Ltd, Tokushima, Japan for the donation of OLETF and LETO rats and also thank Sankyo Pharmaceutical Co., Ltd, Osaka, Japan for the donation of troglitazone.
Conflict of interest statement. None declared.
References
Nakayama N, Sato T, Asahi Y, Toide K, Yabuucthi Y. The insulin resistance in Otsuka Long-Evans Tokushima Fatty (OLETF), a new spontaneous NIDDM model rat strain. In: Huh KB, Shinn SH, Kaneko T, eds.
Kawano K, Hirashima T, Mori S, Natori T. OLETF (Otsuka Long-Evans Tokushima Fatty) rat; a new NIDDM rat strain.
Hirashima T, Kawano K, Mori S, Matsumoto K, Natori T. A diabetogenic gene (ODB-1) assigned to the X-chromosome in OLETF rats.
Hirawa N, Uehara Y, Ikeda T et al. Urinary prostaglandin D synthase (β-trace) excretion increases in the early-stage of diabetes mellitus.
Hamano K, Totsuka Y, Ajima M et al. Blood sugar control reverses the increase in urinary excretion of prostaglandin D synthase in diabetic patients.
Hoffmann A, Nimtz M, Conradt S. Molecular characterization of beta-trace protein in human serum and urine: a potential diagnostic marker for renal disease.
Melegos DN, Grass L, Pierratos A, Diamandis EP. Highly elevated levels of prostaglandin D synthase in the serum of patients with renal failure.
Hirawa N, Uehara Y, Yamakado M et al. Lipocalin-type prostaglandin D synthase in essential hypertension.
Uehara Y, Hirawa N, Numabe A et al. Angiotensin converting enzyme inhibition delays onset of glucosuria with regression of renal injuries in genetic rat model of non-insulin dependent diabetes mellitus.
Uehara Y, Hirawa N, Kawabata Y et al. Angiotensin II subtype-1 receptor antagonists improve hemodynamic and renal changes without affecting glucose metabolism in genetic rat model of non-insulin dependent diabetes mellitus.
Uusitupa M, Sitonen O, Penttila K. Proteinuria in newly diagnosed type 2 diabetic patients.
Hirawa N, Uehara Y, Kawabata Y et al. Long-term inhibition of renin-angiotensin system sustains memory function in aged Dahl rats.
Gerena RL, Eguchi N, Urade Y, Killian GJ. Stage and region-specific localization of lipocalin-type prostaglandin D synthase in the adult murine testis and epididymis.
Kawano K, Hirashima T, Mori S, Saitoh Y, Kurosumi M, Natori T. Spontaneous long-term hyperglycemic rat with diabetic complications, Otsuka Long-Evans Tokushima Fatty (OLETF) strain.
Yagi K, Kim S, Wanibuchi H, Yamashita T, Yamamura Y, Iwao H. Characteristics of diabetes, blood pressure, and cardiac and renal complications in Otsuka Long-Evans Tokushima Fatty rats.
Fukazawa Y, Watanabe Y, Inaguma D, Hotta N. Evaluation of glomerular lesion and abnormal urinary findings in OLETF rats resulting from a long-term diabetic state.
Toyota E, Ogasawara Y, Fujimoto K et al. Global heterogeneity of glomerular volume distribution in early diabetic nephropathy.
Nagai Y, Yao L, Kobori H et al. Temporary angiotensin II blockade at the prediabetic stage attenuates the development of renal injury in type 2 diabetic rats.
Kotchen TA, Reddy S, Zhang HY. Increasing insulin sensitivity lowers blood pressure in the fructose-fed rat.
Cheng ZJ, Vakonen T, Tikkanen I et al. Endothelial dysfunction and salt-sensitive hypertension in spontaneously diabetic Goto-Kakizaki rats.
Eguchi Y, Eguchi N, Oda H et al. Expression of lipocalin-type prostaglandin D synthase (β-trace) in human heart and its accumulation in the coronary circulation of angina patients.
Tobian L, Uehara Y, Iwai J. Prostaglandin alterations in barely hypertensive Dahl S rats.
Nagoshi H, Uehara Y, Kanai F et al. Prostaglandin D2 inhibits inducible nitric oxide synthase expression in vascular smooth muscle cells.
Craven PS, DeRubertis FR, Melhem M. Nitric oxide in diabetic nephropathy.
Author notes
1Department of Medicine #2, Yokohama City University, Yokohama, 2Biochemistry Research Laboratory, Central Research Institute, Maruha Corporation, Tsukuba, 3Core Research for Evolution Science and Technology, Japan Science and Technology Corporation and Department of Molecular Behavioral Biology, Osaka Bioscience Institute, Osaka, 4The Department of Clinical Laboratory Medicine and Institute of Medical Science, Dokkyo University School of Medicine, Mibu and 5Health Service Center, University of Tokyo, Tokyo, Japan


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments