




Abstract
In Alzheimer disease (AD), amyloid-β (Aβ) oligomer is suggested to play a critical role in imitating neurodegeneration, although its pathogenic mechanism remains to be determined. Recently, the cellular prion protein (PrPC) has been reported to be an essential co-factor in mediating the neurotoxic effect of Aβ oligomer. However, these previous studies focused on the synaptic plasticity in either the presence or the absence of PrPC and no study to date has reported whether PrPC is required for the neuronal cell death, the most critical element of neurodegeneration in AD. Here, we show that Prnp−/− mice are resistant to the neurotoxic effect of Aβ oligomer in vivo and in vitro. Furthermore, application of an anti-PrPC antibody or PrPC peptide prevents Aβ oligomer-induced neurotoxicity. These findings are the first to demonstrate that PrPC is required for Aβ oligomer-induced neuronal cell death, the pathology essential to cognitive loss.
INTRODUCTION
Recently, it has been demonstrated that the specific binding of amyloid-β (Aβ) oligomer to cellular prion protein (PrPC) is essential for synaptic toxicity reflected in loss of long-term potentiation (LTP) (1). Moreover, ablation of PrPC enhances cognitive function in transgenic mice overexpressing mutant amyloid precursor protein (APP) genes (APPswe and PS1ΔE9) preventing premature death and memory impairment (2). However, other reports questioned these findings by noting that lack of PrPC did not prevent Aβ oligomer-mediated synaptic toxicity or cognitive impairment (3–5). Thus, while our recent study (6), along with others, confirms the physical interaction between Aβ and PrPC, it remains unclear whether PrPC is essential to neurotoxicity of Aβ oligomer.
The apparent conflict may be due to the assays used, for previous reports all focused on synaptic plasticity (1,3,5,7,8), rather than neuronal cell death, the final pathway defining Alzheimer disease (AD). Here, we report that neuronal cell death induced by synthetic Aβ oligomer was prevented by reducing or eliminating PrPC, or blocking the binding between PrPC and Aβ oligomer using either a PrPC antibody or a decoy PrPC peptide. These findings strongly suggest that PrPC is required for Aβ-induced neuronal cell death. This is the first demonstration that the PrPC/Aβ interaction is necessary for neuronal cell loss, the pathology underlying cognitive decline in AD.
RESULTS
PrPC is required for Aβ oligomer-induced neurotoxicity in slice culture
We first examined the effect of Aβ oligomer on neuronal cell death in hippocampal slice cultures prepared from wild-type (WT) or Prnp−/− mice (Fig. 1A and B) to test whether PrPC is necessary for Aβ oligomer-mediated neuronal cell death. Consistent with previous studies (9), Aβ oligomer (500 nm) induced significant neuronal cell death in WT samples, as measured by propidium iodide (PI) uptake (Fig. 1C). However, the neuronal cell death induced by Aβ oligomer was dramatically decreased in slice cultures prepared from Prnp−/− mice (Fig. 1C). This result was confirmed by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay which demonstrated the increase in cell death by Aβ oligomer in WT mice but not in Prnp−/− mice (Fig. 1D). In addition, activation of caspase-3 after Aβ oligomer treatment was attenuated in slices from Prnp−/− mice (Fig. 1E). Thus, PrPC is required for Aβ oligomer-induced neurotoxicity.
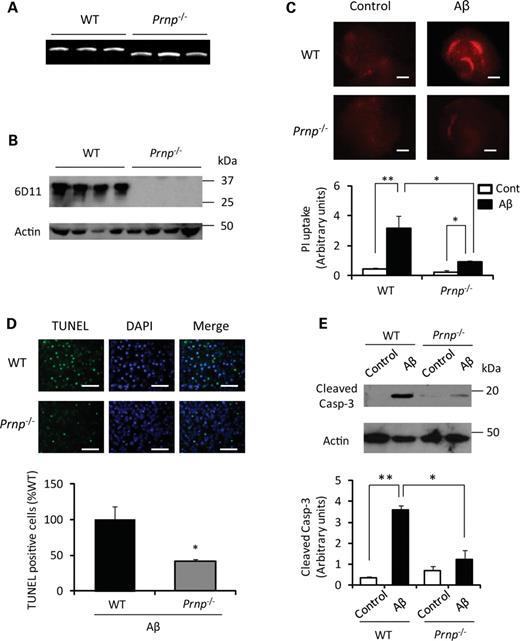
PrPC is essential for Aβ oligomer-induced neurotoxicity. (A) DNA was extracted from WT and Prnp−/− mice and each genotype was identified by PCR with the primer sets specifically detecting each genotype as described in the previous study (20). WT yields a 1100 bp PCR product and Prnp−/− yields a 850 bp PCR product. (B) Total protein (20 μg) from whole brain of the indicated genotype was analyzed by immunoblot with anti-PrP antibody (6D11). Immunoblot analysis shows the expected glycoforms of PrPC in WT samples migrating between 25–37 kDa, and no band in Prnp−/− samples. (C) The slices were treated with Aβ1–42 oligomer or the control reverse Aβ42–1 peptide (500 nm) in the presence of PI (red) for 48 h. Representative pictures showed that Aβ oligomer-induced PI uptake was significantly reduced in the slice cultures from Prnp−/− mice. The PI uptake was quantitatively analyzed (n= 5). Slices from WT or Prnp−/− mice with the control Aβ42–1 peptide show no difference. Scale bar, 500 μm (**P< 0.01 or *P< 0.05). (D) The number of TUNEL-positive cells (green) was also significantly reduced in Prnp−/− hippocampal slice culture (n= 5). Scale bar, 100 μm (*P< 0.05). (E) Representative western blot data show that the expression of active caspase-3 is significantly reduced in Prnp−/−slices after a 24 h treatment with Aβ oligomer (n= 4) (**P< 0.01 or *P< 0.05).
Blocking the PrPC-Aβ oligomer interaction inhibits neurotoxicity
To block the PrPC-Aβ oligomer interaction, we treated the slice cultures with the anti-PrPC93–109 antibody 6D11 (10 µg/ml), which binds to the critical region of PrPC/Aβ binding (amino acids 93–109), and examined whether the antibody could prevent neuronal cell death induced by Aβ oligomer. A previous report showed that the 6D11 antibody blocked the binding of Aβ oligomer to PrPC (1), while another anti-PrPC antibody, 6H4, which recognizes PrPC144–152, failed to block Aβ oligomer/PrPC binding. In the hippocampal slice cultures prepared from WT mice, pretreatment with 6D11 significantly reduced PI uptake and caspase-3 activation following treatment with Aβ oligomer, while pretreatment with normal mouse IgG or 6H4 antibody failed to reduce either marker of cell death (Fig. 2A and B). We extended the competitive inhibition approach by using specific PrPC peptides either containing the Aβ-binding region or not (6). Consistent with the antibody blocking experiment, addition of the peptide corresponding to PrPC98–107 (Pep29, 500 nm) dramatically reduced the neurotoxicity of Aβ oligomer in the hippocampal slice cultures (Fig. 2C and D). In contrast, the peptide corresponding to PrPC213–230 (Pep56) had no effect on Aβ oligomer-induced neurotoxicity (Fig. 2C and D). Further, addition of either peptide without Aβ oligomer treatment had no effect on cell death (Fig. 2C and D). Nissl staining confirmed that co-application of the peptide corresponding to PrPC98–107 with Aβ oligomer prevented neuronal loss (data not shown). We also tested whether a caspase-inhibitor (Z-VAD-FMK) can prevent the cell death and found that the caspase inhibitor significantly prevented neuronal cell death (Fig. 3A) corresponding to the level of caspase-3 activation (Fig. 3B). These results confirmed that PrPC/Aβ binding is necessary for Aβ oligomer-mediated neurotoxicity.
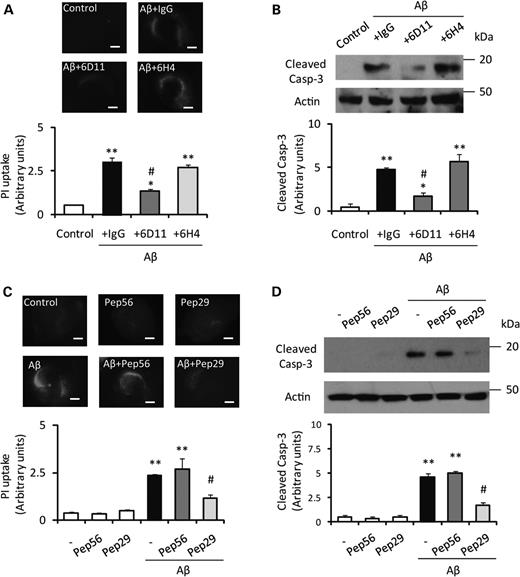
Aβ oligomer-induced neuronal cell loss is prevented by blocking the PrPC-Aβ interaction with PrPC-specific antibodies or peptides. (A) The intensity of PI in slices treated with Aβ oligomer after the addition of 6D11 antibody, control immunoglobulin G (IgG) or 6H4 1 h before Aβ oligomer treatment (n= 5). The PI updake is significantly reduced by 6D11 antibody but not by either IgG or 6H4 antibody. Scale bar, 500 μm (**P< 0.01 versus control #P< 0.05 versus +IgG). (B) Pretreatment with 6D11 suppressed the activation of caspase-3 induced by Aβ oligomer (n= 4) (**P< 0.01 versus control #P< 0.05 versus +IgG). (C) Co-treatment of synthesized peptide-29, corresponding to PrPC98–107, significantly prevents Aβ oligomer-induced PI uptake (n= 5). Scale bar, 500 μm (**P< 0.01 versus control #P< 0.05 versus non-peptide). (D) Co-treatment with peptide-29 suppressed the activation of caspase-3 induced by Aβ oligomer (n= 4) (**P< 0.01 versus control #P< 0.05 versus non-peptide).
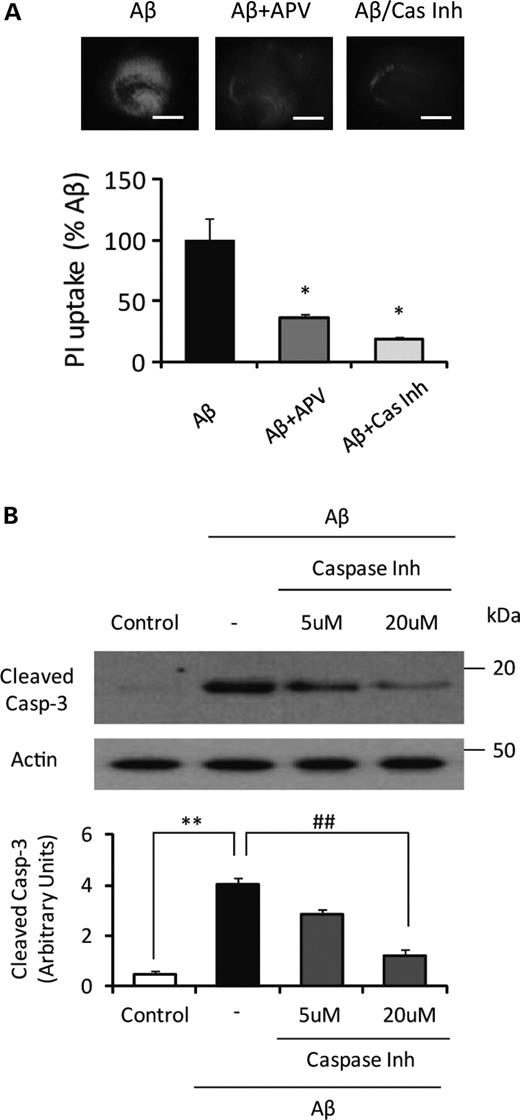
NMDA antagonist (APV) and caspsase inhibitor (Z-VAD-FMK) treatment significantly attenuate Aβ oligomer-induced PI uptake. (A) The slices were treated with Aβ1–42 oligomer (500 nm). APV (20 μm) or Z-VAD-FMK (Cas Inh) (20 μm) was co-applied with Aβ oligomer (n= 5). Both APV and Z-VAD-FMK dramatically reduced the level of PI uptake induced by Aβ1–42 oligomer. Scale bar, 500 μm (*P< 0.05 versus Aβ). (B) Pretreatment of caspase inhibitor, Z-VAD-FMK (20 μm), significantly reduced Aβ oligomer-induced caspase-3 activation (n= 3) (**P< 0.01 versus control, ##P< 0.01 versus non- inhibitor).
In the previous studies, the link between PrPC and N-methyl-D-aspartic acid (NMDA) receptor has been demonstrated (10,11), suggesting the potential role of PrPC in NMDA receptor-mediated excitotoxicity. Therefore, we tested whether the inhibition of NMDA receptor can prevent neuronal cell death in our experimental model. Indeed, (2R)-amino-5-phosphonovaleric acid (APV), a NMDA antagonist, significantly reduced the level of neuronal cell death induced by Aβ oligomer (Fig. 3A).
PrPC dependence of Aβ oligomer–mediated neurotoxicity in vivo
We also established similar findings in an animal model. Intra-hippocampal injection of Aβ oligomer has been shown to result in profound memory impairment and neuronal apoptosis in vivo (12). We injected Aβ oligomer into the hippocampus of WT or Prnp−/− mice and analyzed neuronal cell death. Neuronal cell loss was evident in WT mice injected with Aβ oligomer 20 days after injection. Importantly, consistent with the previous results in the hippocampal slice culture model, neuronal cell death was almost completely eliminated in Prnp−/− mice when compared with WT mice (Fig. 4A and B), thus confirming the role of PrPC in Aβ oligomer-induced neuronal cell death in vivo.
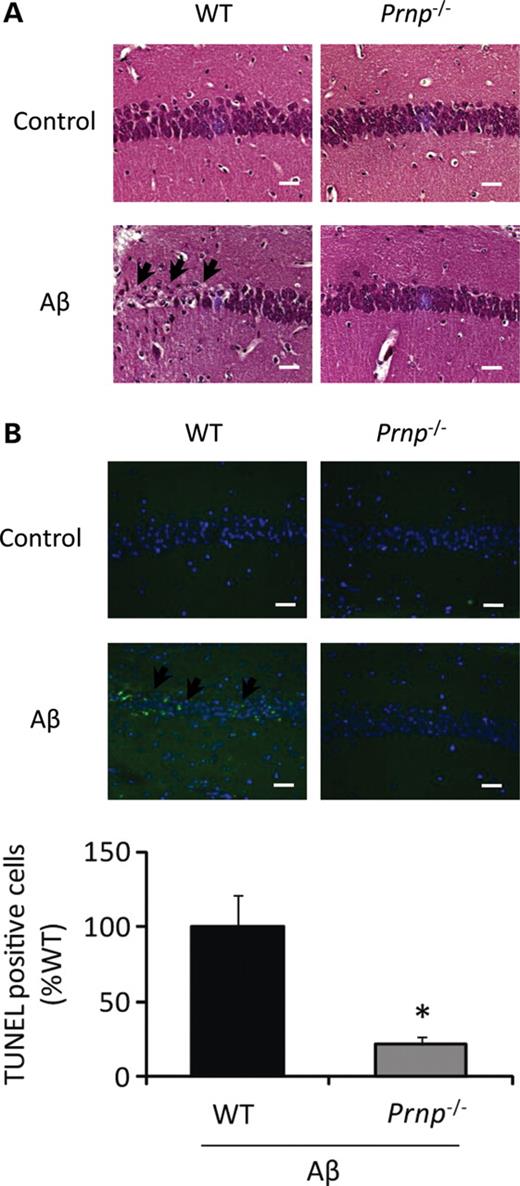
PrPC is essential for Aβ oligomer-induced neurotoxicity in vivo. (A) Prnp−/− and WT mice were sacrificed and brain tissues stained with H&E at 20 days after Aβ oligomer injection. Neuronal cell loss in hippocampus was evident in WT mice injected with Aβ oligomer (arrows) but not in Prnp−/− mice. Scale bar, 100 μm. (B) The number of TUNEL-positive cells (arrows) in hippocampus was dramatically reduced in Prnp−/− mice after Aβ oligomer injection compared with WT mice (n= 5). Scale bar, 100 μm (*P< 0.05).
DISCUSSION
The present study strongly supports a critical role for PrPC in mediating the neurotoxic effect of Aβ oligomer. While previous research focused on the effect of Aβ oligomer in synaptic impairment in vitro, we provide additional convincing in vitro and in vivo evidences that the PrPC/Aβ interaction is necessary for triggering neuronal cell loss. In a recent study supporting this view, Resenberger et al. (11) demonstrated that overexpression of PrPC in neuronal cell cultures increased vulnerability to the neurotoxic effects of various β-sheet-rich conformers, including Aβ. Our results provide further support for this conclusion based on the results obtained using a knockout of PrPC, or blocking the PrPC-Aβ interaction by the use of PrPC-specific antibodies or peptide under more physiological conditions (summarized in Fig. 5).
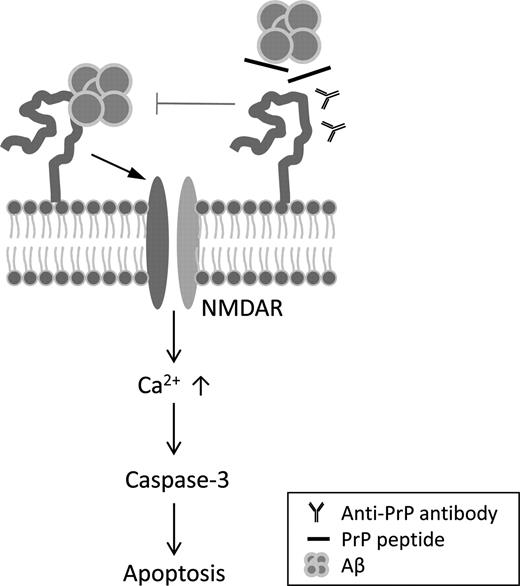
Hypothetical model for Aβ oligomer-induced neurotoxic signaling through PrPC. Aβ oligomer binding to PrPC at plasma membrane activates NMDA receptors and subsequent caspase-3-dependent neuronal cell death. Treatment with anti-PrPC antibody or competitive PrPC peptides prevents the activation of NMDA receptor, suggesting that PrPC/Aβ oligomer interaction is a key mechanism of Aβ oligomer-induced neurotoxicity.
The pretreatment with the antibody 6D11, which binds PrPC93–109, prevents neuronal cell death by Aβ oligomer, while another anti-PrPC antibody, 6H4, which recognizes PrPC144–152, failed to block Aβ oligomer-induced neuronal toxicity. Furthermore, consistent with the antibody experiments, addition of the peptide corresponding to residues PrPC98–107 reduced the neurotoxicity of Aβ oligomer in the hippocampal slice cultures, whereas the peptide corresponding to residues PrPC213–230 had no effect on the Aβ oligomer-induced neurotoxicity. These data indicate that it is the essential region PrPC98–107 in PrPC that mediates Aβ oligomer-PrPC interaction. This binding site is similar to the sequence (amino acids 95–105) identified in a previous study (1) which showed that the treatment with antibody binding this region prevented the interaction and Aβ oligomer-induced LTP (1) and improved cognitive deficits in aged AD transgenic mice (13). More recent studies also confirmed that an anti-PrP antibody targeted to PrPC93–102 blocks LTP induced by Aβ-containing AD brain extract (7,8). Collectively, our results strongly suggest that PrPC98–107 contains the critical amino acid sequence for Aβ oligomer-induced synaptic impairment and neuronal cell death.
While the current data support the pathological role of PrPC/Aβ interaction, three independent studies failed to confirm a critical role of PrPCin vivo and in vitro (3–5). The exact reason for this discrepancy needs to be examined, but differences in the experimental conditions, such as the specific transgenic animal model or in the preparation of Aβ oligomer, might be possible reasons. Additionally, PrPC is apparently not the only cellular surface protein that interacts with Aβ oligomer, since elimination of PrPC only reduces Aβ oligomer binding by 50% to cultured hippocampal neurons (1). Several putative receptor sites have been proposed to mediate neutrotoxic signaling of Aβ oligomer, such as the receptor of advanced glycation end product (14), NMDA (11), insulin (15) and p75 neutrophin receptor (16). Consistent with this result, our data showed that blocking of PrPC/Aβ interaction, either by application of an anti-PrP antibody or competitive peptides, inhibits ∼60% of Aβ oligomer-induced neuronal cell loss. These results further support the idea that other neurotoxic signaling pathways, which are independent of PrPC, may contribute to neurotoxicity.
A previous report suggested that NMDA receptor-mediated excitotoxicity might be the downstream mechanism of Aβ neurotoxicity (11), which was also confirmed in our study. Although further studies will be required to elucidate the pathological mechanism(s) in detail, a mechanistic link between Aβ-PrPC and the NMDA receptor for neurotoxicity is further supported by the previous finding that an NMDA antagonist prevents Aβ-induced neuronal loss and disruption of synaptic plasticity (17). In addition, Aβ oligomer was found to directly or indirectly bind NMDA receptor (18) and PrPC is also reported to interact with the NR2D subunit, which is a key regulatory subunit of the NMDA receptor (10). Collectively, these data suggest that regulation of NMDA receptor function may contribute to the neuroprotective effect of blocking the binding of Aβ oligomer to PrPC. Furthermore, there is indirect evidence that PrPC binding by Aβ oligomer colocalizes with both mGlu5 (glutamate metabotropic subtype 5) and NMDA receptors (18). Thus, the binding of PrPC/Aβ oligomer may promote cross-linking of glutamate receptors. Interestingly, a recent study found that Aβ oligomer increases the localization of PrPC to the cell surface by increasing its trafficking (19). Thus, Aβ oligomer may induce the formation of ectopic signaling platforms by recruiting PrPC at the plasma membrane (18). Future studies are needed to clarify the detailed mechanisms by which PrPC mediates Aβ-induced neurodegeneration. In addition, the effect of familial mutations in PrPC and overexpression of PrPC on Aβ-induced neurodegeneration remains to be determined.
In conclusion, we found that Prnp−/− mice are more resistant to the neurotoxic effect of Aβ oligomer than WT mice in both in vivo and in vitro models. Furthermore, the application of a specific anti-PrPC antibody or competitive PrPC peptide, which block Aβ/PrPC binding, rescues Aβ oligomer-induced neuronal cell death, demonstrating the requirement for PrPC in Aβ oligomer-induced neurotoxicity. Our results strongly support the concept that PrPC contributes to neurotoxic signaling induced by Aβ oligomer, and mediates neuronal cell death.
MATERIALS AND METHODS
Mouse strains
Prnp−/− mice (Zürich І) (20) backcrossed onto the FVB/N background were obtained from George Carlson, McLaughlin Research Institute, Great Falls, Montana.
Preparation of Aβ oligomer
Soluble Aβ oligomer was prepared from synthetic peptide as described previously (21).
Preparation of hippocampal slice cultures
Organotypic hippocampal slice cultures were prepared as described (22). Briefly, hippocampal slice cultures were prepared from 7–10-day-old mouse pups. Four hundred micrometer slices were cut using a McIlwain tissue chopper and transferred to Millicell (Millipore Corp.) membrane inserts (0.4 μm).
Assessment of neuronal cell death
To determine neuronal cell death in the hippocampal slices, PI was added to the slice culture medium. Images were acquired through an AxioCam camera on an Axiovert 200M microscope (Zeiss). Nissl staining was also performed for routine histochemical and morphological analyses. TUNEL staining followed a previous protocol (23). Western blots for detection of caspase-3 were performed as previously described (24).
Intrahippocampal injection of Aβ oligomer
FVB mice (2–3-month old; Jackson Laboratories) were anaesthetized with pentobarbital and placed in a stereotaxic frame. Injection was made using a 10 μl microsyringe (Hamilton). One microliter of Aβ oligomer, 50 μm in phosphate buffered saline (PBS), was injected into the left hippocampus. Control animals were prepared identically and injected with the same concentration of Aβ42–1 in PBS (reversed sequence of Aβ1–42). Injections were made at stereotaxic coordinates of Bregma; antereoposterior (AP) = 2.3 mm, mediolateral (ML) = 2.5 mm, doroventral (DV) = −2.5 mm. This corresponds to a site in the dorsal hippocampus in the apical dendritic zones of the CA1 region near the hippocampal fissure. Mice were sacrificed 20 days after injection, brains dissected out, fixed in 10% buffered formalin and paraffin embedded.
Statistical analysis
Data were expressed as the means ± SE; the number of independent experiments is indicated in the corresponding figure legend. Differences between groups were examined for statistical significance using one-way analysis of variance with an unpaired Student's t-test. A P-value <0.05 indicated a statistically significant difference.
FUNDING
This work was supported by the National Institutes of Health (AG028679 to H.G.L. and NS062787 to W.Q.Z.); the Alzheimer's Association (NIRG-09-132727 to H.G.L.); and the Dr Robert M. Kohrman Memorial Fund (to X.W.Z.).
ACKNOWLEDGEMENTS
The authors are grateful to Dr Jan Langeveld for providing PrP peptides.
Conflict of Interest statement: G.P. is a consultant for Takeda Pharmaceuticals and Neurotez and owns stock options in Neurotez and Voyager. X.Z. was a consultant for and received grant support from Medivation. M.A.S. was a consultant for Anavex Life Sciences Corporation, Eisai, Medivation, Neurotez, and Takeda Pharmaceuticals; owned stock options in Aria Neurosciences, Neurotez, Panancea and Voyager, and received lecture fees from GSK, Medivation and Pfizer.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}