




Abstract
Autosomal dominant cancer predisposition genes for common cancers such as breast cancer and colorectal cancer have been well recognized for over a decade. Monoallelic mutations in these genes are associated with high risks of adult-onset cancer. In recent years, it has become apparent that biallelic mutations in some of these genes, such as BRCA2 , MSH2 and MLH1 , result in distinctive phenotypes, including childhood cancer predisposition. Conversely, it has also become evident that some genes which cause autosomal recessive cancer predisposition syndromes such as Fanconi anaemia and ataxia-telangiectasia are associated with modestly increased risks of adult cancers in monoallelic mutation carriers. These observations raise interesting implications with respect to the identification and phenotypic characterization of cancer predisposition genes.
INTRODUCTION
The identification of constitutionally mutated cancer predisposition genes has been a cornerstone of cancer genetics leading to fundamental clinical and biological insights ( 1 ). These genes were primarily identified through positional cloning or candidate gene analyses in familial cancer syndromes and developmental disorders associated with cancer. Each newly identified gene was associated with a distinctive autosomal dominant or recessive cancer syndrome ( 2 , 3 ). Perhaps surprisingly, it was not until relatively recently that it was appreciated that clinical phenotypes can occur in biallelic (i.e. homozygous or compound heterozygous) mutation carriers of autosomal dominant cancer syndromes and in monoallelic (i.e. heterozygous) mutation carriers of autosomal recessive cancer syndromes (Table 1 ). In this review, we discuss the DNA repair genes, BRCA2 , MSH2 , MLH1 , MSH6 , PMS2 , ATM , BRIP1 and PALB2 , which exemplify this phenomenon. It is likely that further genes associated with different phenotypes in monoallelic and biallelic mutation carriers will be recognized both for cancer syndromes and more widely in human disease. However, the identification and phenotypic characterization of such genes may require large-scale, meticulous clinical and molecular investigations.
Cancer genes associated with distinct phenotypes in monoallelic and biallelic mutation carriers
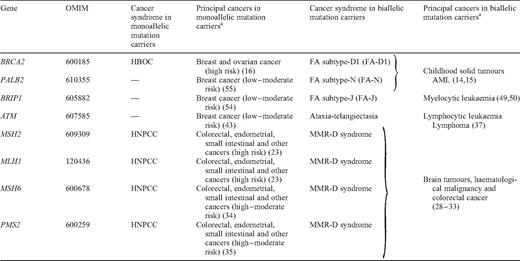
Cancer genes associated with distinct phenotypes in monoallelic and biallelic mutation carriers
GENES WITH DISTINCT HIGH-RISK CANCER PHENOTYPES IN MONOALLELIC AND BIALLELIC MUTATION CARRIERS
BRCA2
BRCA2 exemplifies this class of gene, which is associated with high cancer risks and distinctive phenotypes in both monoallelic and biallelic mutation carriers. BRCA2 was identified by linkage analysis and positional cloning in 1995 using familial breast cancer pedigrees with multiple cases of breast cancer in successive generations ( 4 , 5 ). Monoallelic truncating mutations confer high risks of breast and ovarian cancers and more modest risks of some other cancers ( 6 ). Such mutations are generally rare, being present in approximately one in 800 of the UK population, although certain founder mutations occur at appreciable prevalence in some populations, for example, 6174delT in the Ashkenazim and 999del5 in Iceland ( 7–9 ).
It was not until 2002 that biallelic BRCA2 mutation carriers were identified by candidate gene analyses in Fanconi anaemia (FA) patients ( 10 ). FA is a recessive condition associated with progressive bone marrow aplasia, multiple congenital abnormalities and predisposition to malignancies, particularly leukaemia and solid tumours of the head and neck, oesophagus and vulva ( 11–13 ). At the cellular level, FA is a chromosomal fragility syndrome and cells from FA patients are hypersensitive to DNA intrastrand cross-linking agents. Howlett et al. ( 10 ) considered BRCA2 a candidate FA gene because BRCA2 null cells exhibit sensitivity to cross-linking agents and mice with abrogated BRCA2 function have some features of FA. They identified biallelic BRCA2 mutations in patients with FA subtype D1 (FA-D1) and over 20 such individuals have now been reported in the literature. The phenotype of biallelic BRCA2 mutations is distinctive and results in a severe form of FA with high risks of childhood cancer, particularly Wilms tumour, brain tumours and acute myelogenous leukaemia ( 10 , 14 , 15 ). Breast and ovarian cancers have not been reported in biallelic mutation carriers, but this may primarily be because FA-D1 is associated with very high mortality and almost all known individuals with the condition have died in childhood.
The mutation spectrum in monoallelic and biallelic BRCA2 mutation carriers appears to differ, although the extent and significance of these differences are unclear ( 14 , 15 ). In the initial report, it was suggested that individuals with biallelic mutations would only be viable if they carried at least one ‘hypomorphic’ allele, such as a missense mutation or a truncating mutation close to the 5′ terminus ( 10 ). Subsequently, this was disproved and several FA-D1 patients with biallelic early truncating mutations have been reported ( 14 , 15 ). Currently, the available data indicate that mutations that cause breast cancer in monoallelic mutation carriers also cause FA-D1 in biallelic mutation carriers and there is no compelling evidence that variants that do not predispose to breast cancer can cause FA-D1. However, there are provocative data suggesting some limitations in the combinations of BRCA2 mutations that are viable. For example, the spectrum of mutations in FA-D1 patients is not consistent with the spectrum predicted from the prevalence of mutations in breast cancer families; in particular, there is an over-representation of mutations in exons 7 and 8 in FA-D1 patients (Breast Cancer Information Core database, www.nhgri.nih.gov/intramura_research/lab_transfer/bic ). Furthermore, there is currently no known FA-D1 patient with biallelic truncating mutations in exon 11, despite it accounting for nearly 50% of the coding sequence and the high frequency of truncating exon 11 mutations in familial breast cancer pedigrees. Finally, despite the appreciable population prevalence of 6174delT (which is in exon 11) in the Ashkenazim, no homozygous carrier of this mutation has been reported. Interestingly, genotype–phenotype analyses of BRCA2 breast cancer pedigrees mirror the observations in biallelic BRCA2 mutation cases, with different cancer risks associated with monoallelic truncating mutations in exon 11 when compared with mutations 3′ or 5′ of this exon ( 16 , 17 ). Over the ensuing years, it should become possible to confirm whether these associations are robust. If confirmed, it will challenge current concepts regarding BRCA2 structure–function relationships and will require innovative research to unravel the molecular explanations for these unusual genotype–phenotype associations.
Mismatch repair genes
The mismatch repair (MMR) genes MLH1 , MSH2 , MSH6 and PMS2 similarly result in distinct clinical conditions in monoallelic and biallelic mutation carriers. These genes were first implicated in cancer predisposition through investigations of families with hereditary non-polyposis colorectal cancer (HNPCC). In 1993, a hypermutation phenotype similar to that observed in DNA MMR deficient bacteria and yeasts was identified in tumours occurring in individuals from HNPCC families, suggesting that the causative genes were involved in MMR ( 18 ). Over the next 2 years, searches for human orthologues of the bacterial and yeast MMR genes combined with linkage analyses and positional cloning in HNPCC families led to the identification of MSH2 and MLH1 ( 19–22 ). Monoallelic mutations in these genes account for 60–80% of classical HNPCC pedigrees ( 23 ). They confer an increased risk of a variety of tumours in adulthood, principally colorectal carcinoma. A number of other human MMR genes have since been identified. Monoallelic mutations in two of these, PMS2 and MSH6 , have been identified in families with HNPCC ( 24 , 25 ). In 1995, monoallelic mutations in PMS2 and MLH1 were reported in individuals who developed brain tumours and colorectal carcinoma at a young age ( 26 ). These individuals also had constitutional microsatellite instability, suggesting that a covert second mutation might be present ( 27 ), but it was not until 2004 that biallelic mutations in some of these individuals were finally uncovered ( 28–32 ).
Over the last few years, over 40 individuals with biallelic mutations in the MMR genes MLH1 , MSH2 , MSH6 and PMS2 have been reported ( 28–33 ). The phenotypes associated with defects in these genes overlap considerably and we have used the term ‘mismatch repair-deficiency (MMR-D) syndrome’ to describe the condition, which is characterized by a greatly increased risk of malignancy from early childhood together with hyperpigmented and in some cases hypopigmented skin lesions. Unlike other DNA repair syndromes, growth parameters are usually within the normal range and, prior to the onset of malignancy, affected children have typically not required medical attention. Haematological and primary brain malignancies, which occur rarely in HNPCC patients, are the most frequently reported cancers in MMR-D syndrome, but colorectal carcinoma is also common and occurs at much younger ages than in monoallelic mutation carriers. Despite the clinical overlap of biallelic and monoallelic mutation carriers, the diagnosis of MMR-D is often missed, delayed or mistaken for other conditions, particularly neurofibromatosis type-1. As is the case for FA-D1, biallelic carriers of MMR gene mutations will often present with cancer before their parents and other carrier relatives and thus a history of HNPCC may not be apparent to aid diagnosis.
The cancer risks associated with monoallelic mutations in these genes differ, with higher risks of colorectal cancer associated with mutations in MSH2 and MLH1 than with MSH6 or PMS2 mutations ( 34 , 35 ). In contrast, the phenotypes associated with biallelic mutations in these four genes are similar in severity and spectrum, although it is notable that all four supratentorial primitive neuroectodermal tumours occurred in children with biallelic PMS2 mutations ( 36 ). There is also no evidence that the mutation spectrum differs in monoallelic and biallelic mutation carriers, although the small number of MMR-D patients currently limits formal genotype–phenotype or gene–phenotype analyses.
GENES CAUSING CHILDHOOD CANCER SYNDROMES IN BIALLELIC MUTATION CARRIERS AND MODEST INCREASED RISK OF CANCER IN MONOALLELIC MUTATION CARRIERS
ATM is the paradigm for this class of gene, but two additional examples, BRIP1 and PALB2 , were identified last year, each underlying distinct subtypes of FA.
ATM
Biallelic ATM inactivating mutations result in the childhood disorder ataxia-telangiectasia. This is an autosomal recessive condition characterized by progressive cerebellar ataxia, conjunctival telangiectasia, sensitivity to ionizing radiation and increased risk of malignancy, particularly lymphoid cancers in childhood ( 37 ). Early mortality is common in ataxia-telangiectasia and, therefore, the phenotypic characteristics of adults are not readily apparent. The gene was identified by classic linkage mapping and positional cloning ( 38 , 39 ). Certain mutations, resulting in a milder phenotype and compatible with longer survival, have been documented. The most notable of these is T7271G, which was identified in an unusual Orkney family with two affected individuals, homozygous for T7271G, both of whom had mild ataxia-telangiectasia and breast cancer. Their mother, an obligate heterozygous carrier of the mutation, also developed breast cancer, suggesting that monoallelic ATM mutation carriers might be at increased risk of the disease ( 40 ). Extensive, systematic, epidemiological analyses of relatives of ataxia-telangiectasia cases over the past two decades have consistently supported this hypothesis, demonstrating that female relatives are at 2–4-fold increased risk of breast cancer ( 41 , 42 ). Molecular confirmation of these epidemiological observations was published last year and demonstrated that the mutations that cause ataxia-telangeictasia in biallelic mutation carriers overall confer an ∼2-fold increased risk of breast cancer in monoallelic mutation carriers ( 43 ). There is currently no compelling evidence that mutations that do not cause ataxia-telangiectasia can confer susceptibility to cancer in heterozygotes, despite many studies to evaluate such variants ( 44 ). Importantly, the modest increase in breast cancer risk associated with monoallelic ATM mutations does not result in families with multiply affected individuals with breast cancer that segregates with a mutation and therefore could not have been identified by linkage analysis of breast cancer families. The phenotype of biallelic ATM mutations and analyses of ataxia-telangiectasia families was, therefore, crucial in identifying this breast cancer susceptibility gene.
FA genes, BRIP1 and PALB2
Unlike ataxia-telangiectasia, FA is a highly heterogeneous condition, with 13 subtypes currently recognized, 12 of which have been attributed to known genes ( 45 , 46 ). Breast cancer has not been demonstrated to occur at increased frequency in FA patients overall and epidemiological surveys of FA families have not revealed an increased cancer risk in relatives of FA patients ( 47 ). Such analyses primarily report on the commoner subtypes, FA-A, FA-C and FA-G, which together account for > 85% of the cases ( 11 ).
As described earlier, in 2002, biallelic BRCA2 mutations were shown to underlie FA-D1 ( 10 ). This led to renewed interest in the potential association of monoallelic mutations in FA genes with cancer susceptibility. Mutational screening of the genes underlying FA-A, FA-C, FA-D2, FA-E, FA-F and FA-G in familial breast cancer cases not due to BRCA1 or BRCA2 did not reveal any mutations ( 48 ), consistent with epidemiological surveys of all subtypes together. Thus, the association with breast cancer appears to be limited to certain FA genes. Eight of the known FA proteins form a nuclear core complex that mediates monoubiquitination and activation of FANCD2. Activated FANCD2 is translocated to DNA repair foci where it cooperates with other components to facilitate DNA repair (Fig. 1 ) ( 45 ). Until 2005, BRCA2 was the only known FA gene that encodes a protein that acts downstream of D2. Recently, two new FA genes, BRIP1 and PALB2 , were identified ( 46 , 49–51 ). BRIP1 (for BRCA-interacting protein 1) encodes a DEAH helicase that interacts with the BRCT domain of BRCA1 and affects BRCA1-dependent DNA repair and checkpoint functions ( 52 ). Linkage and complementation analyses together with positional cloning identified biallelic BRIP1 mutations as the cause of FA subtype J (FA-J) ( 49 , 50 ). PALB2 (for ‘partner and localizer of BRCA2’) encodes a protein that interacts with and facilitates the nuclear localization and stability of BRCA2 and is required for some of its functions in homologous recombination and double-strand break repair ( 53 ). Recently, biallelic PALB2 mutations were shown to cause a new sybtype of FA, FA-N ( 46 , 51 ).
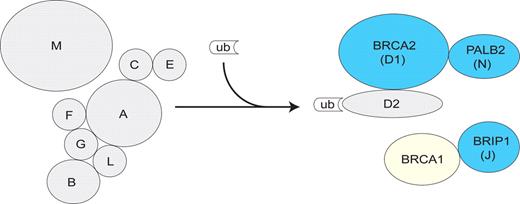
Schematic representation of the FA/BRCA pathway showing the components for which monoallelic and/or biallelic mutations are known to result in a cancer predisposition phenotype. Both monoallelic and biallelic mutations in BRCA2, BRIP1 and PALB2 (indicated with blue symbols) result in clinical phenotypes. Only biallelic/recessive mutations in the other FA genes (grey) and only monoallelic mutations in BRCA1 (yellow) are known to cause clinical phenotypes. The FA core complex comprises eight components (FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL and FANCM) and is essential for the ubiquitination and activation of FANCD2 following DNA damage. Activated FANCD2 translocates to DNA repair foci, colocalizing with DNA damage response proteins such as BRCA2 to participate in homology-directed repair. PALB2, BRIP1 and BRCA1 also function downstream of FANCD2.
We mutationally screened BRIP1 and PALB2 in familial breast cancer cases not due to BRCA1 or BRCA2 and demonstrated that the genes confer modest increased risks of breast cancer (2–3-fold) similar to CHEK2 and ATM ( 43 , 54–56 ). Thus, all three of the known FA genes that act downstream of FANCD2 also confer susceptibility to breast cancer (Fig. 1 ). However, despite this functional similarity, there are phenotypic differences between these genes that indicate further unexplained complexities in the association of FA genes with breast cancer susceptibility (Fig. 2 ). The clinical and cellular phenotypes of biallelic mutations in BRCA2 and PALB2 are nearly identical, causing severe chromosomal instability and high risks of childhood solid tumours, whereas FA-J is associated with classical FA and none of the reported FA-J cases developed childhood solid tumours ( 14 , 46 , 49 , 50 ). Conversely, the heterozygote phenotypes are most similar for BRIP1 and PALB2 and only BRCA2 is associated with high cancer risks in monoallelic mutation carriers ( 16 , 54 , 55 ). The molecular explanations for these similarities and differences are not known.
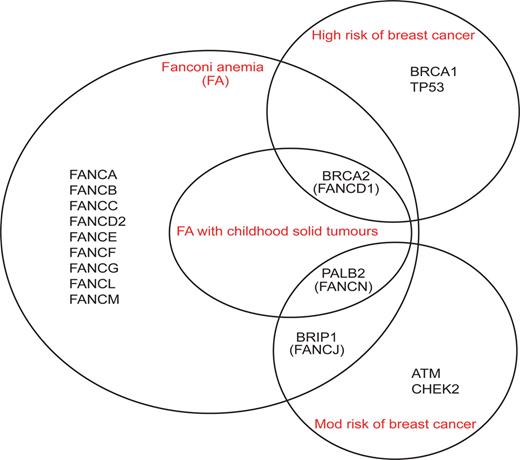
Phenotypes associated with DNA repair genes that are known to cause FA and/or breast cancer predisposition. BRCA2 , PALB2 and BRIP1 can cause both phenotypes: monoallelic mutations confer increased risks of breast cancer and biallelic mutations cause FA. Biallelic mutations in BRCA2 and PALB2 cause a severe form of FA associated with greatly increased risks of childhood solid tumours and severe chromosomal instability, whereas mutations in BRIP1 cause classical FA. Monoallelic mutations in BRCA2 are associated with greatly increased risks of breast cancer, whereas mutations in PALB2 and BRIP1 confer modestly increased risks. FA, Fanconi anemia; Mod, moderate.
As for ATM , linkage analysis in familial breast cancer could not identify BRIP1 or PALB2 as breast cancer susceptibility genes. For these genes, epidemiological analyses of FA and/or investigation of FA-J or FA-N families would also be unlikely to provide strong evidence of a link with breast cancer because of the extensive genetic heterogeneity of FA and the rarity of these subtypes. Hence, the identification of BRIP1 and PALB2 as breast cancer predisposition genes was dependent on correctly judging them to be candidates, together with large-scale mutational screening of breast cancer cases enriched for unknown susceptibility alleles.
IDENTIFICATION OF PHENOTYPES ASSOCIATED WITH BIALLELIC AND MONOALLELIC MUTATIONS
Identification of the clinical phenotypes associated with biallelic and monoallelic mutations in the same gene can pose significant challenges. At the present time, it is very likely that there is an under-appreciation of the phenotypic spectrum associated with many genes. However, it is also clear that there has been injudicious extrapolation of phenotypic associations for some genes, particularly in relation to conjectures on the clinical consequences of common missense and non-coding gene variants.
Dominant cancer predisposition genes: finding the biallelic phenotypes
There are many autosomal dominant cancer syndromes for which no individuals with biallelic mutations have been reported ( 2 ). There are broadly three possible explanations for this: (i) biallelic mutations are not associated with additional phenotypic consequences over and above that associated with monoallelic mutations; (ii) biallelic mutations are associated with unrecognized phenotypes and (iii) biallelic mutations result in embryonic lethality.
Intuitively, one might anticipate that biallelic mutations of cancer predisposition genes, most of which are key players in basic biological processes, would have important clinical consequences and, therefore, the first of these explanations is unlikely. In contrast, there is considerable redundancy in many of these pathways, which might mitigate the effects of such mutations. It is also possible that, in some circumstances, haploinsufficiency and/or dominant negative effects (for tumour suppressor genes) or dominant effects (for proto-oncogenes) result in monoallelic mutations being as severe as biallelic mutations. However, it is probable that biallelic mutations in most dominant cancer predisposition genes result in unrecognized phenotypes or embryonic lethality, although it is difficult to predict which for any given gene. Mutation prevalence will also be a critical factor in our ability to identify the clinical syndromes caused by biallelic mutations, which will occur very rarely for most of the genes. A high index of clinical suspicion, together with extensive mutation screening in a broad selection of samples, particularly from individuals with clinical features seen in known recessive cancer syndromes (such as childhood cancer, small stature, skeletal and pigmentary abnormalities) will likely be required to identify biallelic phenotypes of dominant cancer predisposition genes.
Recessive cancer predisposition genes: finding the monoallelic phenotypes
There are several autosomal recessive cancer genes for which a cancer risk in monoallelic carriers has not been identified ( 2 ). Again, there are three broad explanations for this: (i) monoallelic mutations are not associated with an increased risk of cancer; (ii) monoallelic mutations are associated with unrecognized risks of the cancers seen in biallelic mutation carriers, although perhaps at lower penetrance and (iii) monoallelic mutations are associated with unrecognized risks of different cancers to those seen in biallelic mutation carriers. Genes fulfilling each of these categories are already known, as described earlier. It is quite possible that many recessive cancer genes are not associated with risks of cancer or other diseases in heterozygotes. Although it should be possible to demonstrate that mutations are not associated with high or moderate risks, it is difficult to unequivocally show that a gene is associated with no risk. Proving that a gene is associated with small increases in risk can also be challenging. Extensive, systematic extension of families affected with the recessive cancer syndrome may be helpful, as for ataxia-telangiectasia. However, if a condition is very rare, and/or very heterogeneous, this approach may not have power to formally demonstrate increases in risk, particularly for common cancers. It is very likely that very large analyses of cancer patients for mutations in the gene will be required. Using samples enriched for genetic susceptibility factors, for example, from individuals with a family history or multifocal disease, or using isolated populations in which founder mutations exist at appreciable population prevalence may help in identifying or clarifying the role of monoallelic mutations in cancer susceptibility. However, it is very likely that large-scale mutational surveys will be required for the majority of genes.
CONCLUSIONS
The last few years have revealed new insights into the clinical phenotypes associated with a number of well-known and newly recognized cancer predisposition genes. The future likely promises further discoveries related to cancer genes and more broadly in human disease. These insights provide novel information and avenues for research regarding the complexity of biological functions that are clearly present for many disease susceptibility genes. They also hint at unexpected intricacies in the effects of differentially positioned truncating mutations in genes. Currently, it appears that the mutations that are pathogenic in biallelic mutation carriers are the same as those that are pathogenic in monoallelic mutation carriers. It is certainly possible that different classes of mutation may be associated with different/additional phenotypic effects. However, caution must be exercised in attributing pathogenicity to variants not known to cause the primary condition associated with a given gene, particularly rare missense variants, which will inevitably be discovered in mutational surveys.
Extension of the phenotypic spectrum of a given gene may also have important clinical sequelae for affected families. For example, diagnosis of biallelic BRCA2 mutations or MMR-D syndrome directly affects management of the patient and extended family. In particular, it has important implications for the treatment of childhood cancers that occur with high frequency in individuals with these conditions. The potential for both increased understanding of the biological basis of cancer predisposition and improved management of affected individuals suggests that the challenging work that will be required to identify the full range of phenotypes associated with cancer predisposition genes will be worthwhile.
ACKNOWLEDGEMENTS
This work was supported by the Institute of Cancer Research (UK). R.H.S. is supported by a grant which forms part of the Michael and Betty Kadoorie Cancer Genetics Research Programme.
Conflict of Interest statement . The authors declare no conflicts of interests.

{kind=link}
{kind=link}