




Abstract
Electrocorticographic (ECoG) activity was recorded for up to 129 h from 12 acutely brain-injured human patients using six platinum electrodes placed near foci of damaged cortical tissue. The method probes ECoG activity in the immediate vicinity of the injured cortex and in adjacent supposedly healthy tissue. Six out of twelve patients displayed a total of 73 spontaneous episodes of spreading depression of the ECoG. Of the remaining 6 patients 1 displayed an episode of synchronous depression of ECoG during surgery. Using the same electrodes we also measured the slow potential changes (SPC) (0.005–0.05 Hz) to test the hypothesis that the ECoG depressions were identical to Leao's cortical spreading depression (CSD), and to be able to record peri-infarct depolarisations (PIDs) in electrically ‘silent’ cortical tissue. Changes in the SPC indicate depolarization of brain tissue. For the analysis, the SPCs were enhanced by calculating the time integral of the ECoG signal. Spreading ECoG depressions were accompanied at every single recording site by stereotyped SPCs, which spread across the cortical mantle at 3.3 (0.41–10) mm/min (median, range), i.e. at the same speed of spread as the depression of the ECoG activity. The amplitude of the SPCs was 0.06–3 mV. In 4 out of 6 patients the ECoG recovered spontaneously. In 2 patients we subsequently recorded recurrent SPCs, but without recovery of the initial ECoG background activity until 2–5 h later. This represents the first direct recording of PIDs in acutely injured human brain. Evidence from this and our previous study of 14 brain-injured patients suggests that CSDs in acute brain disorders occur at higher incidence in patients <30 years (83%) than above (33%). CSD was recorded in 4 out of 5 traumatic brain injury patients, and in 2 out of 7 patients with spontaneous haemorrhages. We conclude that the spreading ECoG depressions recorded in patients are identical to CSDs recorded in animal experiments. We furthermore provide direct electrophysiological evidence for the existence of PIDs and hence a penumbra in the human brain. We hypothesize that the depolarization events might contribute to tissue damage in acute disorders in the human brain.
Introduction
We investigated the occurrence in patients with two types of severe, but transient depolarization of the cerebral cortex. These phenomena are well characterized in animal experiments, but their occurrence in the human brain is disputed. First, cortical spreading depression (CSD) is a wave of pervasive failure of brain ion homeostasis that transiently interrupts cortical function in the intact brain (Leao, 1944b; Martins-Ferreira et al., 2000). It has been considered until recently an experimental phenomenon that requires to be specifically ‘induced’. Secondly, peri-infarct depolarizations (PIDs) are ‘spontaneous’ waves that propagate through the penumbra region of cortical infarcts or traumatized cortex into normally perfused tissue where they take on the characteristic features of CSD (Nedergaard and Hansen 1993; Nilsson et al., 1993; Strong et al., 1996). CSD in the intact, normally perfused brain does not lead to cell death (Nedergaard and Hansen 1988), while recurrent PIDs in the injured brain are associated with increases in final infarct volume (Gill et al., 1992; Mies et al., 1993; Back et al., 1996; Busch et al., 1996; Takano et al., 1996). Moreover, the infarct increases stepwise for each PID (Busch et al., 1996). The incidence of CSDs and PIDs in experimental animals can be strongly attenuated by appropriate blockers of glutamate receptors (Marrannes et al., 1988; Park et al., 1988; Lauritzen and Hansen, 1992; Hossmann, 1994; Ohta et al., 2001). Accordingly, treatment with N-methyl-d-aspartate (NMDA)-receptor antagonists significantly reduced the final infarct volume in experimental focal ischaemia (Mies et al., 1993). This implied a possible new treatment to limit brain injury, but clinical studies using these neuroprotective agents have so far been disappointing (Heiss et al., 1999; Lees et al., 2000; Sacco et al., 2001). One explanation for this failure may be that only a subset of patients with acute brain disorders develops CSDs or PIDs, and that only this group will benefit from treatment such as NMDA-receptor antagonists. If so, it is important to document to what extent these phenomena occur in acute brain injury, and to identify patient groups that are prone to develop CSDs or PIDs.
We recently described repeated waves of transient depression of electrocorticographic (ECoG) activity in brain-injured patients. The waves spread across the cortical mantle with the same speed as CSD in animal experiments (Strong et al., 2002). The data suggested that CSD or CSD-like events occurred in a large proportion of patients with acute, focal brain damage. The recordings supposedly represented events of severe cortical depolarization, but the frequency range that was used did not allow detection of slow waves of tissue depolarization. In animals, an ∼20 mV negative direct current slow potential change (SPC) lasting ∼1 min is the hallmark of CSD and PID (Leao, 1951; Koroleva and Bures, 1996). This phenomenon invariably accompanies the ECoG suppression and reflects the dramatic changes of the intra- and extracellular ion concentrations which are an integral part of the massive depolarization of neurons and glia characteristic of CSD (Kraig and Nicholson, 1978; Hansen and Zeuthen, 1981; Somjen, 2001). The demonstration of SPCs in the human brain would furnish proof that the depressions of cortical activity we have observed are indeed CSD. Furthermore, recording of SPC in electrically silent tissue would enable us to demonstrate the occurrence of PIDs.
In animals, an intracortical glass microelectrode connected to a direct current (DC) amplifier against a remote reference is a straightforward way to record the SPC. However, long-term intracranial DC recordings easily become unstable in humans, unless measures are taken against this. We here describe a methodology that allowed us to record SPCs from human cerebral cortex for up to 129 h. Our data provide direct and definite evidence for the existence of PIDs in the acutely injured human brain and demonstrate that the ECoG depressions observed previously are identical to Leao's CSD (Leao, 1944b).
Methods
Patient recruitment and clinical care
The research monitoring protocol was approved by the local research ethics committee (London). After a clinical decision had been made that surgery was required, clinical and research consents were obtained. In 12 consecutive patients with traumatic (n = 5) or spontaneous (n = 7) intracranial haematomas and/or subarachnoid haemorrhage requiring craniotomy, we placed an ECoG recording strip on the cortex accessible from the craniotomy. The lead cable from the strip was tunnelled under the skin and exteriorized through a stab wound to allow removal of the strip at end of monitoring without re-opening the wound. In three of the patients, a microdialysis probe was inserted next to the ECoG strip. The results of microdialysis in these and in nine other patients have been published separately (Parkin et al., 2005). After surgery, patients were transferred to the intensive care unit (ICU), where core variables were monitored continuously (arterial and intracranial pressures and arterial oxygen saturation (SaO2, pulse oximetry). The intracranial pressure transducer (Codman) was located on the same side as the craniotomy and usually in the cortical parenchyma near the ECoG strips (haematoma cases). These variables were logged continuously into the same data set as the ECoG data.
All patients were ventilated, at least initially, but were paralysed only exceptionally, and sedation was maintained with propofol (n = 10), or with fentanyl and midazolam (n = 5). In eight of these patients sedation was withdrawn during monitoring. In three of the most recent patients, insulin therapy was used by the intensive care team, aiming to keep plasma glucose within a target range of 3.5–5.5 mM.
Location of cortical recordings
If possible, the electrode strip was placed radiating away from the lesioned area so that the electrodes would be in contact with presumably viable cortex (Fig. 1). In some trauma patients, the lesions were widespread, and supposedly viable tissue was chosen for strip placement between injured areas. In most cases, part of the electrode strip was positioned outside the area of cortex exposed by craniotomy so that placement of the entire strip on a single gyrus, although usually attempted, could not be verified.
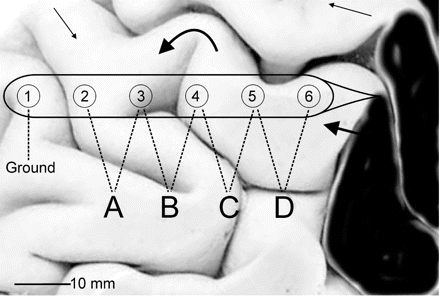
Schematic diagram of electrode strip placement on the cortex with respect to injury and gyri. Electrode 6 is positioned over viable cortex next to contused tissue (black area). The visible part of the strip is placed along a gyrus and slipped under the edge of cut dura and bone (not shown). Thus, Electrodes 1–4 are usually not visible. Signals from Electrodes 2–6 are amplified in a differential montage to derive data channels A to D. Thick arrows indicate possible course of a CSD along a gyrus, curving around sulci and reaching the electrode strip at different angles. In this example, the time required for the accompanying SPC to get from Electrodes 4 to 3 will be long, resulting in a low apparent velocity compared with Electrodes 5–4. Thin arrows indicate how a CSD spreading from another part of the lesion may eventually reach the electrode strip from the other end.
Electrocorticography
Four active data channels were acquired from a 6-electrode (linear array) subdural strip (Wyler; platinum, 5-mm diameter, 10-mm interval between electrode centres; Ad-Tech Medical, Racine, WI) that was connected to two 2-channel pre-amplifiers (Bioamps, ADInstruments, New South Wales) in a sequential bipolar fashion, with Electrode 1 (most remote from lesion) used as ground (Fig. 1). Thus, the recordings were from a strip of cortex of ∼4 cm in length and yielded four ECoG channels, named A, B, C and D. The ECoG was recorded continuously for periods of up to 129 h. The lower frequency limit of the alternating current (AC) amplifiers was 0.02 Hz (29% attenuation), but frequencies as low as 0.002 Hz were detectable (90% attenuation). Accordingly, a monophasic change of the DC potential lasting for 1–2 min could be recorded albeit with a distorted form, since the AC amplifier returned the derivative of the DC signal.
Data collection
The ECoG data were digitized (100–200 Hz per channel) with a Powerlab 16SP analogue/digital converter (ADInstruments) and recorded with Chart software (ADInstruments) and a laptop computer (Powerbook, Apple). Arterial blood pressure and oxygen saturation, intracranial blood pressure and optional microdialysis data were digitized simultaneously in the same system and files. Analysis was performed on a PC using Chart software.
Data analysis and integration
Each transient period of ECoG amplitude reduction or loss was first examined according to the criteria reported previously (Strong et al., 2002). Recovery of the locally generated ECoG activity was gradual, and isolated bursts occurred in the depressed part of the curve, before continuous activity resumed. The time for recovery of local activity was arbitrarily defined to the time point when bursts occurred continuously at intervals shorter than 20 s.
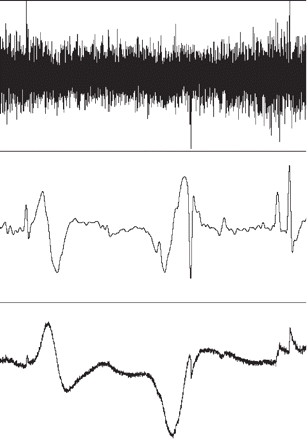
We noticed that the baseline level of the unfiltered ECoG signal slowly deviated predominantly in a negative direction for 1–3 min just at the onset of the ECoG depressions. To amplify this slow baseline deviation, and to reverse the derivative transformation caused by the AC-filtration, we calculated the time integral of the ECoG signal followed by a subtraction of the general slope of the integral (300 s time constant decay) (Figs 2 and 3). This summarized and amplified even small deviations of the baseline if lasting for ∼>10 s, while the AC changes in the conventional EEG range (0.5–30 Hz) summated to ∼0 because of phase cancellation. The procedure resulted in visualization of an SPC, which developed over 1–3 min synchronous with the spreading ECoG depressions. The SPCs had opposite phases in adjacent channels which made it possible to assign each component of the SPC to a single electrode (Fig. 2). Here it is important to note that a bipolar montage as used in the present study is made up of sequential linkages of channels of equispaced electrodes along the electrode strip. Each channel is connected to one pair of active electrodes and displays the potential difference between them. If a localized change of the SPC occurs at or near the electrode common to both channels, the signal will deflect in opposite direction (phase reversal). This demonstrates the active locus of the SPC. The two components of the SPC, one from each of the electrodes contributing to a channel, may be more or less clearly separated depending of the recovery time of the SPC (Figs 2 and 4). The signal component derived from one single electrode has a steep mono- or biphasic component preceded and followed by slight deviations from the baseline. In two patients, we observed recurrent stereotyped SPCs spreading between channels during a prolonged period of depressed ECoG (Fig. 5). These episodes were classified as PIDs (Table 1).
![Recording of CSD in the injured human cortex over a period of 40 min (Patient no. 4). Upper four traces show the ECoG recorded in a differential chain from five electrodes (full scale 1 mV). A 50–80% reduction of the ECoG amplitude spreads from Channel A to C, while a 25% reduction of amplitude is seen in Channel D. Middle four traces show the same curves upon integration of the signal (full scale 4 mVs) to enhance the slow potential changes. Each depression in the upper traces is accompanied by a stereotyped baseline change reflecting an SPC. Electrode numbers are indicated to mark the onset of depolarization at that particular electrode as evidenced by phase reversal. Thus, the SPC occurring exclusively in Channel A compares to a depolarizing event at Electrode 2 alone, while a phase reversal between Channels A and B corresponds to an event at Electrode 3, etc. Lower four traces show the power of the 0.5–70 Hz band of the ECoG signal [full scale 0.03 mV2 (Channels A and D) or 0.12 mV2 (Channels B and C)]. The traces are amplified a little out of range to visualize loss of amplitude. The reduced activity of Channel D is better appreciated here than in the raw ECoG signal of the upper traces.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/brain/129/3/10.1093/brain/awh716/2/m_awh716f2.gif?Expires=1716462130&Signature=VbN~Ecan72H8UKHuuOZR2qRnyT1Zy7KzCORwce3aN2di1dMV1nsVBB~mltfe4V3no3CRqNvGsq1bewigKM~5XecITVTFCVhIVjCyTTa59g4FRa4gaTb4jLfJefItEl~XE~wnsoxS70pGKNBuIRUnOmW-UxT350w2ghp8XznutVXdiudasVQz8MHXUKRkwQFmuhJLmkpDWHWF1KZVp6PTjjYMzAc9rMpEKH96TxObtK9Dm0x77XydgvmfIhamU~5Gv9ASmDp0P3HgN89bXBhCpJ1i31rmIB63mkHZ9xWkWoaTAb0SnNh-4fNsibnbqIu554qJIVApzkFkwK7MWtV5VQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Recording of CSD in the injured human cortex over a period of 40 min (Patient no. 4). Upper four traces show the ECoG recorded in a differential chain from five electrodes (full scale 1 mV). A 50–80% reduction of the ECoG amplitude spreads from Channel A to C, while a 25% reduction of amplitude is seen in Channel D. Middle four traces show the same curves upon integration of the signal (full scale 4 mVs) to enhance the slow potential changes. Each depression in the upper traces is accompanied by a stereotyped baseline change reflecting an SPC. Electrode numbers are indicated to mark the onset of depolarization at that particular electrode as evidenced by phase reversal. Thus, the SPC occurring exclusively in Channel A compares to a depolarizing event at Electrode 2 alone, while a phase reversal between Channels A and B corresponds to an event at Electrode 3, etc. Lower four traces show the power of the 0.5–70 Hz band of the ECoG signal [full scale 0.03 mV2 (Channels A and D) or 0.12 mV2 (Channels B and C)]. The traces are amplified a little out of range to visualize loss of amplitude. The reduced activity of Channel D is better appreciated here than in the raw ECoG signal of the upper traces.
Low-pass filtering versus integral: 20-min recording from Patient no. 4. (Upper and lower traces are details from Fig. 2, only Channel D is shown here). Upper trace, baseline changes of the raw ECoG signal (full range 1 mV) are hardly visible. Middle trace shows a 0.05 Hz low-pass filtering of the upper trace, full range 0.1 mV. Two SPCs are readily visible, but so are other deflections due to baseline noise. Lower trace shows the time-integrated signal (full range 3 mVs). Compared to the low-pass filtered trace, abrupt baseline noise is much less conspicuous. The initial electronegative part of the two SPCs is more pronounced due to the integrating process that reverses the differentiating effect of the AC-filtering during recording.
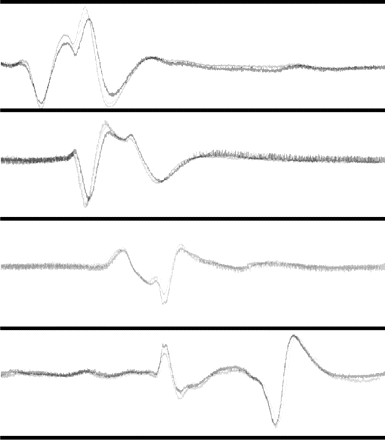
Three sets of 22-min recordings of integrated ECoG signal from three consecutive episodes of CSD (Patient no. 4). The figure illustrates that the ECoG signals for the same patient were stereotyped as indicated in this figure by superimposing traces from three different CSD episodes.
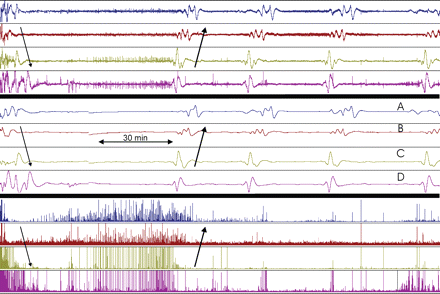
Three hour recording of ECoG from Channels A to D (Patient no. 9). The three sets of traces represent the same period (same setting as in Fig. 2): upper four traces show the unfiltered signal (full scale 3 mV). Middle four traces show the integrated signal (full scale 100 mVs). Lower four traces show the power of the 0.5–70 Hz band of the signal (full scale: 0.05 mV2). Baseline ECoG activity showed burst-suppression pattern: 2 s bursts and 10–30 s suppressions, amplitude 300–1000 μV. Initially, a CSD spreading from Channel A to D depressed this ECoG activity for 30–40 min. The CSD was accompanied by SPCs and spread from Channels B to D at a velocity of 2–3 mm/min (thin arrows). After ∼1 h another CSD accompanied by SPCs spread from D to A (thick arrows). This time, the ECoG activity did not recover. After 29 min, SPCs spread from channel D to A with exactly the same time sequence and shape as detected during the last CSD. The ECoG remained depressed indicating compromised metabolism. The event was therefore classified as a PID. Two stereotyped PIDs followed after intervals of 32 and 39 min. Just before the last of these PIDs slight recovery of ECoG in Channel D was noticed (lower right). During 5 h a total of 21 stereotyped PIDs were recorded. These PIDs were either similar to those in the figure or spread in the direction A to D in a second stereotyped pattern.
Criteria for ECoG events
| ECoG | SPC | Comment | |
|---|---|---|---|
| Synchronous ECoG depressions | Simultaneous loss of power—ECoG amplitude of ≥50%, in two or more channels | No accompanying SPC | Must be distinguished from abrupt loss of amplitude due to movement artefacts |
| Spreading episodes, general definitions | Depressions: Loss of power—ECoG amplitude of 50%, gradually developing over 30–60 s | Bi- or tri-phasic change in potential of 2–5 min' duration and 0.1–4 mV amplitude | Depressions may develop in two steps separated by several minutes |
| CSD, fast recovery | Depressions lasting for 2–10 min in at least one channel | Accompanying stereotyped and spreading SPC in at least the same and the neighbouring channels | Normal recovery time when compared with CSDs in the intact rodent brain |
| CSD, prolonged recovery | Depressions lasting for more than 10 min in the fastest recovering channel | Latency for spreading between channels within 0.5–25 min | The prolonged recovery suggests early or partial energy failure |
| PID | No locally generated ECoG in any of the channels that display a spreading SPC | Stereotyped and spreading SPCs in at least two channels. Latency for spreading between channels within 0.5–25 min | The occurrence of repeated SPCs in otherwise electrically silent tissue points to severe but incomplete energy failure |
| ECoG | SPC | Comment | |
|---|---|---|---|
| Synchronous ECoG depressions | Simultaneous loss of power—ECoG amplitude of ≥50%, in two or more channels | No accompanying SPC | Must be distinguished from abrupt loss of amplitude due to movement artefacts |
| Spreading episodes, general definitions | Depressions: Loss of power—ECoG amplitude of 50%, gradually developing over 30–60 s | Bi- or tri-phasic change in potential of 2–5 min' duration and 0.1–4 mV amplitude | Depressions may develop in two steps separated by several minutes |
| CSD, fast recovery | Depressions lasting for 2–10 min in at least one channel | Accompanying stereotyped and spreading SPC in at least the same and the neighbouring channels | Normal recovery time when compared with CSDs in the intact rodent brain |
| CSD, prolonged recovery | Depressions lasting for more than 10 min in the fastest recovering channel | Latency for spreading between channels within 0.5–25 min | The prolonged recovery suggests early or partial energy failure |
| PID | No locally generated ECoG in any of the channels that display a spreading SPC | Stereotyped and spreading SPCs in at least two channels. Latency for spreading between channels within 0.5–25 min | The occurrence of repeated SPCs in otherwise electrically silent tissue points to severe but incomplete energy failure |
CSD = cortical spreading depression; PID = peri-infarct depolarisation; ECoG = electrocorticogram; SPC = slow potential changes of ECoG
Criteria for ECoG events
| ECoG | SPC | Comment | |
|---|---|---|---|
| Synchronous ECoG depressions | Simultaneous loss of power—ECoG amplitude of ≥50%, in two or more channels | No accompanying SPC | Must be distinguished from abrupt loss of amplitude due to movement artefacts |
| Spreading episodes, general definitions | Depressions: Loss of power—ECoG amplitude of 50%, gradually developing over 30–60 s | Bi- or tri-phasic change in potential of 2–5 min' duration and 0.1–4 mV amplitude | Depressions may develop in two steps separated by several minutes |
| CSD, fast recovery | Depressions lasting for 2–10 min in at least one channel | Accompanying stereotyped and spreading SPC in at least the same and the neighbouring channels | Normal recovery time when compared with CSDs in the intact rodent brain |
| CSD, prolonged recovery | Depressions lasting for more than 10 min in the fastest recovering channel | Latency for spreading between channels within 0.5–25 min | The prolonged recovery suggests early or partial energy failure |
| PID | No locally generated ECoG in any of the channels that display a spreading SPC | Stereotyped and spreading SPCs in at least two channels. Latency for spreading between channels within 0.5–25 min | The occurrence of repeated SPCs in otherwise electrically silent tissue points to severe but incomplete energy failure |
| ECoG | SPC | Comment | |
|---|---|---|---|
| Synchronous ECoG depressions | Simultaneous loss of power—ECoG amplitude of ≥50%, in two or more channels | No accompanying SPC | Must be distinguished from abrupt loss of amplitude due to movement artefacts |
| Spreading episodes, general definitions | Depressions: Loss of power—ECoG amplitude of 50%, gradually developing over 30–60 s | Bi- or tri-phasic change in potential of 2–5 min' duration and 0.1–4 mV amplitude | Depressions may develop in two steps separated by several minutes |
| CSD, fast recovery | Depressions lasting for 2–10 min in at least one channel | Accompanying stereotyped and spreading SPC in at least the same and the neighbouring channels | Normal recovery time when compared with CSDs in the intact rodent brain |
| CSD, prolonged recovery | Depressions lasting for more than 10 min in the fastest recovering channel | Latency for spreading between channels within 0.5–25 min | The prolonged recovery suggests early or partial energy failure |
| PID | No locally generated ECoG in any of the channels that display a spreading SPC | Stereotyped and spreading SPCs in at least two channels. Latency for spreading between channels within 0.5–25 min | The occurrence of repeated SPCs in otherwise electrically silent tissue points to severe but incomplete energy failure |
CSD = cortical spreading depression; PID = peri-infarct depolarisation; ECoG = electrocorticogram; SPC = slow potential changes of ECoG
Power-analysis and criteria for spreading events
For enhancement of the ECoG signal (AC) variations in the conventional EEG-frequency bands, the signal was filtered (0.5–70 Hz) and squared to obtain the power. This improved our ability to detect periods of ECoG depression by simple visual detection (Fig. 2). For example, a reduction of the raw signal of ∼30% was represented as a 50% reduction of the power signal. Since these moderate depressions were usually accompanied by spreading stereotyped SPCs, the criteria for spreading events were modified accordingly (Table 1).
Results
Data for the 12 patients are summarized in Table 2. Mean age was 48 years for females (n = 4) and 41 years for males (n = 8). All traumatic brain injury patients were males (n = 5). Trauma mechanisms were assault (n = 3), fall (n = 1) or road traffic accident (n = 1). Of the non-traumatic cases, two patients had subarachnoid haemorrhage due to intracranial aneurysms, two had intracerebral haematomas due to an arterio-venous malformation and three had spontaneous haemorrhages, possibly due to hypertension and atherosclerosis. Injuries were located in the frontal, parietal and/or temporal lobes (n = 8, 4, and 4). Initial preoperative Glasgow coma scale score (GCS) was in the range 6–15. Six-month recovery ranged between fatal outcome and upper good recovery.
Summary of clinical electrocorticogram data in 12 patients
| Pt | M/F | Age | Pre-surgery GCS | eGOS at 6 months | Principal lesion site | Cause | Number of ECoG depressions | Number of PIDs | Velocity range mm/min | Hours from injury to | Hours from injury to | Hours of effective ECoG | Spreading events per hour | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Synchronous | CSD fast recovery | CSD prolonged recovery | first record | first spreading event | recording | ||||||||||||
| 1 | M | 22 | 9 | 4 | L front-par. contusion, depressed skull fracture | Assault | 1 | 9a | 1–5 | 42.5 | 46.7 | 35.8 | 0.28 | ||||
| 2 | F | 39 | 15 | 7 | SAH---R MCA | aneurysm | 41 | ||||||||||
| 3 | F | 42 | 14 | 5 | R frontal ICH | Spontan. | 65 | ||||||||||
| 4 | M | 48 | 6 | 0 | L parieto-temp.ICH | RTA | 8 | 1 | 1.66–10 | 12.5 | 16.5 | 44 | 0.20 | ||||
| 5 | M | 24 | 12 | 6 | L front-parietal ICH | AVM | 11b | 0.41–4 | 18.6 | 21.7 | 29.9 | 0.37 | |||||
| 6 | F | 61 | 15 | 5 | SAH---L MCA | aneurysm | 1 | 22.6 | |||||||||
| 7 | M | 21 | 15 | 5 | R front. ICH | AVM | 34 | 2–10 | 72.7 | 72.9 | 43 | 0.79 | |||||
| 8 | F | 51 | 12 | 7 | R temporo-parietal ICH | Spontan. | 59.6 | ||||||||||
| 9 | M | 67 | 3c | 0 | R fronto-temp ASDH | Fall | 2 | 21 | 3–5 | 15.4 | 19.3 | 53.8 | 0.43 | ||||
| 10 | M | 40 | 7 | 5 | L front.contusion/ICH | Assault | 129 | ||||||||||
| 11 | M | 57 | 7 | d | R frontal ICH + SAH | Spontan. | 70.6 | ||||||||||
| 12 | M | 48 | 11 | 5 | L temp R front cont | Assault | 3 | 5 | 4 | 0.7–10 | 21.2 | 23.6 | 56.3 | 0.21 | |||
| Pt | M/F | Age | Pre-surgery GCS | eGOS at 6 months | Principal lesion site | Cause | Number of ECoG depressions | Number of PIDs | Velocity range mm/min | Hours from injury to | Hours from injury to | Hours of effective ECoG | Spreading events per hour | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Synchronous | CSD fast recovery | CSD prolonged recovery | first record | first spreading event | recording | ||||||||||||
| 1 | M | 22 | 9 | 4 | L front-par. contusion, depressed skull fracture | Assault | 1 | 9a | 1–5 | 42.5 | 46.7 | 35.8 | 0.28 | ||||
| 2 | F | 39 | 15 | 7 | SAH---R MCA | aneurysm | 41 | ||||||||||
| 3 | F | 42 | 14 | 5 | R frontal ICH | Spontan. | 65 | ||||||||||
| 4 | M | 48 | 6 | 0 | L parieto-temp.ICH | RTA | 8 | 1 | 1.66–10 | 12.5 | 16.5 | 44 | 0.20 | ||||
| 5 | M | 24 | 12 | 6 | L front-parietal ICH | AVM | 11b | 0.41–4 | 18.6 | 21.7 | 29.9 | 0.37 | |||||
| 6 | F | 61 | 15 | 5 | SAH---L MCA | aneurysm | 1 | 22.6 | |||||||||
| 7 | M | 21 | 15 | 5 | R front. ICH | AVM | 34 | 2–10 | 72.7 | 72.9 | 43 | 0.79 | |||||
| 8 | F | 51 | 12 | 7 | R temporo-parietal ICH | Spontan. | 59.6 | ||||||||||
| 9 | M | 67 | 3c | 0 | R fronto-temp ASDH | Fall | 2 | 21 | 3–5 | 15.4 | 19.3 | 53.8 | 0.43 | ||||
| 10 | M | 40 | 7 | 5 | L front.contusion/ICH | Assault | 129 | ||||||||||
| 11 | M | 57 | 7 | d | R frontal ICH + SAH | Spontan. | 70.6 | ||||||||||
| 12 | M | 48 | 11 | 5 | L temp R front cont | Assault | 3 | 5 | 4 | 0.7–10 | 21.2 | 23.6 | 56.3 | 0.21 | |||
GCS = Glasgow coma scale; eGOS = extended Glasgow outcome scale at 6 months; ECoG = electrocorticogram; SAH = subarachnoid haemmorrhage; MCA = middle cerebral artery; ICH = Intracerebral haematoma, AVM = arterio-venous malformation; ASDH = acute subdural haematoma. RTA = road traffic accident.
In a1 and b7 of these episodes, ECoG depression occurred in one channel only, while the spread of SPC was evidenced in three and two channels, respectively.
GCS was 10 a few hours before surgery.
Lost to follow up. 4 weeks post-op, partial recovery of left-sided motor deficit only and residual cognitive deficit.
Summary of clinical electrocorticogram data in 12 patients
| Pt | M/F | Age | Pre-surgery GCS | eGOS at 6 months | Principal lesion site | Cause | Number of ECoG depressions | Number of PIDs | Velocity range mm/min | Hours from injury to | Hours from injury to | Hours of effective ECoG | Spreading events per hour | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Synchronous | CSD fast recovery | CSD prolonged recovery | first record | first spreading event | recording | ||||||||||||
| 1 | M | 22 | 9 | 4 | L front-par. contusion, depressed skull fracture | Assault | 1 | 9a | 1–5 | 42.5 | 46.7 | 35.8 | 0.28 | ||||
| 2 | F | 39 | 15 | 7 | SAH---R MCA | aneurysm | 41 | ||||||||||
| 3 | F | 42 | 14 | 5 | R frontal ICH | Spontan. | 65 | ||||||||||
| 4 | M | 48 | 6 | 0 | L parieto-temp.ICH | RTA | 8 | 1 | 1.66–10 | 12.5 | 16.5 | 44 | 0.20 | ||||
| 5 | M | 24 | 12 | 6 | L front-parietal ICH | AVM | 11b | 0.41–4 | 18.6 | 21.7 | 29.9 | 0.37 | |||||
| 6 | F | 61 | 15 | 5 | SAH---L MCA | aneurysm | 1 | 22.6 | |||||||||
| 7 | M | 21 | 15 | 5 | R front. ICH | AVM | 34 | 2–10 | 72.7 | 72.9 | 43 | 0.79 | |||||
| 8 | F | 51 | 12 | 7 | R temporo-parietal ICH | Spontan. | 59.6 | ||||||||||
| 9 | M | 67 | 3c | 0 | R fronto-temp ASDH | Fall | 2 | 21 | 3–5 | 15.4 | 19.3 | 53.8 | 0.43 | ||||
| 10 | M | 40 | 7 | 5 | L front.contusion/ICH | Assault | 129 | ||||||||||
| 11 | M | 57 | 7 | d | R frontal ICH + SAH | Spontan. | 70.6 | ||||||||||
| 12 | M | 48 | 11 | 5 | L temp R front cont | Assault | 3 | 5 | 4 | 0.7–10 | 21.2 | 23.6 | 56.3 | 0.21 | |||
| Pt | M/F | Age | Pre-surgery GCS | eGOS at 6 months | Principal lesion site | Cause | Number of ECoG depressions | Number of PIDs | Velocity range mm/min | Hours from injury to | Hours from injury to | Hours of effective ECoG | Spreading events per hour | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Synchronous | CSD fast recovery | CSD prolonged recovery | first record | first spreading event | recording | ||||||||||||
| 1 | M | 22 | 9 | 4 | L front-par. contusion, depressed skull fracture | Assault | 1 | 9a | 1–5 | 42.5 | 46.7 | 35.8 | 0.28 | ||||
| 2 | F | 39 | 15 | 7 | SAH---R MCA | aneurysm | 41 | ||||||||||
| 3 | F | 42 | 14 | 5 | R frontal ICH | Spontan. | 65 | ||||||||||
| 4 | M | 48 | 6 | 0 | L parieto-temp.ICH | RTA | 8 | 1 | 1.66–10 | 12.5 | 16.5 | 44 | 0.20 | ||||
| 5 | M | 24 | 12 | 6 | L front-parietal ICH | AVM | 11b | 0.41–4 | 18.6 | 21.7 | 29.9 | 0.37 | |||||
| 6 | F | 61 | 15 | 5 | SAH---L MCA | aneurysm | 1 | 22.6 | |||||||||
| 7 | M | 21 | 15 | 5 | R front. ICH | AVM | 34 | 2–10 | 72.7 | 72.9 | 43 | 0.79 | |||||
| 8 | F | 51 | 12 | 7 | R temporo-parietal ICH | Spontan. | 59.6 | ||||||||||
| 9 | M | 67 | 3c | 0 | R fronto-temp ASDH | Fall | 2 | 21 | 3–5 | 15.4 | 19.3 | 53.8 | 0.43 | ||||
| 10 | M | 40 | 7 | 5 | L front.contusion/ICH | Assault | 129 | ||||||||||
| 11 | M | 57 | 7 | d | R frontal ICH + SAH | Spontan. | 70.6 | ||||||||||
| 12 | M | 48 | 11 | 5 | L temp R front cont | Assault | 3 | 5 | 4 | 0.7–10 | 21.2 | 23.6 | 56.3 | 0.21 | |||
GCS = Glasgow coma scale; eGOS = extended Glasgow outcome scale at 6 months; ECoG = electrocorticogram; SAH = subarachnoid haemmorrhage; MCA = middle cerebral artery; ICH = Intracerebral haematoma, AVM = arterio-venous malformation; ASDH = acute subdural haematoma. RTA = road traffic accident.
In a1 and b7 of these episodes, ECoG depression occurred in one channel only, while the spread of SPC was evidenced in three and two channels, respectively.
GCS was 10 a few hours before surgery.
Lost to follow up. 4 weeks post-op, partial recovery of left-sided motor deficit only and residual cognitive deficit.
ECoG background activity
Monitoring was initiated either in the final phase of surgery or soon afterwards. The ECoG pattern when monitoring started often showed a burst-suppression pattern, but within hours, a continuous, but irregular 1–3 Hz (delta) activity was established in most patients. Repeated CSD episodes changed the background ECoG activity from delta waves to a burst-suppression pattern which ceased when the cerebral cortex no longer produced CSD. Focal seizure activity was detected in two patients (Nos 1 and 7). The relationship of repeated seizure activity with CSD is described in a separate publication.
CSDs and PIDs
Six patients had a total of 73 episodes of CSD. Every episode of ECoG activity depression was accompanied by stereotyped SPCs. The spreading episodes radiated from the exposed injury site (43%) or had the opposite direction (57%). In 3 out of the 6 patients we encountered both directions of spread in various episodes. In two patients (Nos 9 and 12) (Fig. 5), local ECoG activity failed to recover for several hours after a few episodes of CSD. In this period a total of 25 SPCs continued to spread between the channels. The shape, duration and amplitude of the SPCs were similar to the previous recordings taken at a time when background ECoG activity was still obtained. Therefore, these episodes represent spreading depolarization in otherwise electrically silent tissue, i.e. PIDs. The medication of the patients was unchanged immediately before and during these periods. Mean age for the CSD/PID group (n = 6) was 38.3 versus 48.3 years for the other six patients (not significant, P = 0.27 by t-test). The overall age distribution was bimodal: all three patients <25 years had CSD, while the remaining 9 patients (age 39–69) had an incidence of 33%. The initial evaluation of the data using our original criteria (Strong et al., 2002) showed 49 episodes of CSD as opposed to a total of 98 episodes of CSD/PID using the present analysis.
Duration and timing of events
The duration of the period of depressed ECoG activity showed a bimodal distribution (Fig. 6). In three patients (Nos 4, 5, and 7) the CSD episodes (n = 54) had a short recovery time of 6 min (2.5–11) (median, range). This compares well to the recovery time for experimental CSD in the normally perfused brain (Leao, 1944b; Hansen and Zeuthen, 1981; Martins-Ferreira et al., 2000). In three patients (Nos 1, 9 and 12) the recovery time was 8–22 min for the first CSD episode, but became prolonged later. In one of these patients (No. 1) the recovery time gradually increased up to 86 min with eventual good recovery. In 2 out of 3 patients (Nos 9 and 12) the first CSD episodes were followed by persistently depressed ECoG activity for 3.7 and 8 h, respectively, while recurrent PIDs were recorded. At the end of this period, a burst-suppression pattern gradually reappeared.
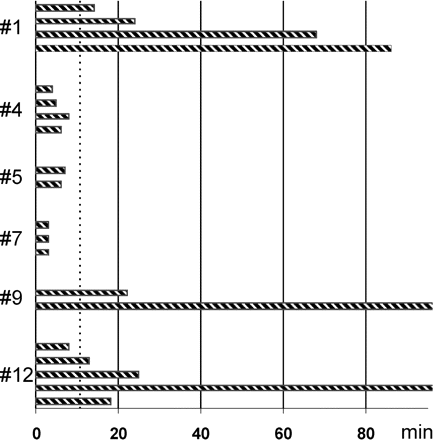
The recovery time of ECoG activity varied between and within patients. The time to return of locally generated ECoG activity for 20 different episodes of CSD in six patients is shown. Bars represent the period of suppressed ECoG activity in Channel A for separate episodes of CSD in a top–down time sequence. Patient identities (Table 2) are indicated along the vertical axis. For simplicity, only the first CSD episode within each 12-h interval of recording was included except for Patient no. 12, where the suppression time was markedly prolonged episode by episode. Recovery time varied between 2.5 and 86 min. In 2 CSD episodes (Patients no. 9 and 12) ECoG activity remained depressed for several hours, while recurrent PIDs were recorded. A recovery of the ECoG within10 min, as indicated by the dotted line, was considered fast while a depression lasting for >10 min indicated prolonged recovery. The difference in recovery time between CSD episodes in a single patient represents variation over time in the capacity of the tissue to regenerate transmembrane ion gradients, i.e. variation in the match between energy demand and availability. The figure also illustrates variability between patients.
The time delay between the onset of monitoring and the first depolarizing episode was 0.2–4.2 h. Most ECoG depressions occurred at regular intervals of ∼30 min. A few series of CSDs/PIDs showed repetition intervals of 8–12 min.
SPC
Every episode of transient ECoG depression was accompanied by a marked and stereotyped slow baseline change, i.e. an SPC (Fig. 2). The profile of the SPC varied between patients, but remained largely constant within the same patient given that the CSD propagated in the same direction (Fig. 4). The SPCs lasted for 2–3 min for the larger monophasic component, and 3–5 min for the biphasic steep components (Fig. 4). The amplitude from peak to peak was 0.06–3 mV for the raw signal (Fig. 5), and 1.3–68 mVs for the integrated signal (Figs 2 and 4). The well-defined shape of the SPCs allowed the latency between onsets at two neighbouring electrodes to be measured more accurately than the onset of ECoG depressions. The latency varied between 1 and 24 min corresponding to propagating velocities of 3.3 (0.41–10) mm/min (median, range)—comparing favourably with the results reported previously (Strong et al., 2002). The wide range of velocities may be explained by variations in the orientation of the propagating wave front with respect to the electrode strip (Strong et al., 2002).
When the first part of the SPC appeared, the ECoG background activity of that channel usually showed a minor reduction of amplitude. The marked depression of ECoG background activity usually coincided with the onset of the late component of the SPC which reflects depression at both electrodes recorded by that channel. Several recordings showed an SPC spreading from channels showing an ECoG depression into a neighbouring channel where ECoG background activity persisted. Probably, the CSD wave stopped at the first electrode, while a sulcus prevented the wave reaching the second electrode recorded by the latter channel. Thus, the SPCs enabled us to demonstrate the spreading nature of the events when ECoG depressions were only significant in one channel.
Non-spreading episodes
In 6 out of the 12 patients no CSD episodes were recorded. One of these six patients (No. 6) showed a single non-spreading episode without accompanying SPC. The episode occurred during operation, when the middle cerebral artery was clipped temporarily to bring the blood flow in the middle cerebral artery to a transient hold. The ECoG depression lasted for 6 min followed by a burst-suppression activity. In Patient no. 1, a similar episode was recorded at 15 min after the recordings commenced in the ICU. The two episodes were not accompanied by an SPC, and were therefore not explained by depolarization of the tissue. Thus, one possible mechanism for the non-spreading events is temporary hypoperfusion causing transient silencing of the ECoG activity. This question may be addressed in future studies that include local CBF measurement. In our previous paper (Strong et al., 2002), non-spreading episodes were encountered more frequently. This difference is difficult to define, but could relate to improvement of recording conditions in this study.
Case reports
Patient no. 4
This 48-year-old male suffered multiple traumas in a road traffic accident. CT-scan showed a left parietal and temporal intracerebral haematoma and intraventricular haemorrhage. Upon evacuation of the haematomas at 7.5 h after injury, the electrode strip was placed over the left middle frontal gyrus. Re-operation was performed at 16.5 h after injury to remove a newly formed, extradural haematoma. Thrombocytopenia was noted and corrected during the re-operation. During the period of monitoring, the ICP remained >25 mmHg, steadily climbing, but re-intervention was deemed inappropriate. The patient developed sepsis and died after 11 days.
ECoG was monitored from 12.5 to 88.5 h after the accident (Fig. 2). Initially, a pattern of burst-suppression activity was detected. At 15 h after the accident, three episodes of CSD were detected at 24–28 min interval. No further events were detected for the following 38 h while the background ECoG changed into a stable delta-activity. From 54 to 80 h after the injury, six CSD episodes were recorded, accompanied by remarkably stereotyped SPCs (Fig. 4). This was followed by the reappearance of a burst-suppression pattern.
Patient no. 5
This 24-year-old male was previously healthy but woke up confused and vomiting. CT-scan showed a left frontoparietal intracerebral haematoma, 3 × 5 cm2, with midline shift. Angiography revealed an arterio–venous malformation arising from suprasylvian middle cerebral artery branches, draining via a single vein to the superior sagittal sinus. At admission the GCS was 12, but dropped to 7 prior to craniotomy, evacuation of the haematoma and resection of the arterio–venous malformation. ECoG monitoring over the left middle frontal gyrus was started at 18.6 h post ictus. From 21.7 to 34 h post-ictus, 11 episodes of CSD were recorded spreading from Channel A. During this period, the ECoG background activity of Channel A turned into a burst-suppression pattern, while continuous delta-activity persisted in the other channels. Channel A gradually recovered over the first 3 h following the last CSD, and for the next 10 h ECoG was stable. At this time, right-sided arm and leg weakness were noted for the first time. Recovery was slow; at 6 months the patient was dysphasic and hemiparetic: power in the right arm was MRC 3/5, and in the leg, 4/5.
Patient no. 9
This 67-year-old male was admitted after a fall. Initial GCS was 13, but dropped to 10 after a few hours. CT-scan revealed an extensive right hemisphere acute subdural haematoma, with midline shift and right frontal lobe contusion. GCS dropped further to three, and the pupils became non-reactive bilaterally, before surgical evacuation of the subdural haematoma. ECoG monitoring over the partially contused right middle frontal gyrus started at 17.5 h after injury. After 40 min of monitoring, we recorded 2 consecutive episodes of depression of ECoG activity, accompanied by stereotyped spreading SPCs (Fig. 5). Following the first episode, ECoG background activity returned, but after the second episode the ECoG activity remained depressed in all channels. Over the next 5 h, 21 stereotyped episodes of spreading SPCs were detected. ECoG remained depressed for a total of 8 h, after which a burst-suppression pattern developed and then gradually recovered. Arterial oxygen saturation remained >90% and no extra sedative was administered to the patient in this period to explain the lack of cortical activity. The pupils remained fixed; on the second day high blood pressure and low pulse were noted, and deterioration continued until death 2 days later.
Patient no. 12
This 48-year-old man had a history of non-migrainous headaches, and his mother suffered from migraine with aura. The patient was assaulted. GCS at the scene was 11. At admission, he had generalized tonic-clonic seizures which were controlled by diazepam, phenytoin loading and maintenance. The patient was maintained on insulin in the ICU. CT revealed a left temporal lobe contusion and haematoma (burst lobe), left petrous temporal fracture, right frontal contusion and right orbital fracture. The left temporal contusion was evacuated, and an ECoG strip was placed over the left superior temporal gyrus, close to the contused tissue. Monitoring started at 21.2 h after injury. Initial ECoG showed no evidence of cortical activity, but after 1.7 h a burst-suppression pattern emerged. After another 40 min, six CSDs were observed at 17–38 min intervals. The recovery time increased from 8 to 25 min, presenting a sparse burst-suppression pattern (40–50 s suppressions). After the last CSD, ECoG background activity remained depressed for 3.7 h. During the first 80 min of this period four PIDs were observed at intervals of 5–10 min. After another 2.5 h, the ECoG background activity gradually recovered. At 19 and 23 h after recovery another two CSD episodes were observed with recovery times of 6–18 min. No further events were observed for the following 35 h while the ECoG gradually changed into an almost continuous 1.5–2 Hz activity of low amplitude (150 μV). After 2 weeks GCS was 14 (dysphasic). The patient had lower moderate disability at 6-month follow-up.
Discussion
General
The data presented here provide definite evidence that the spreading depression of ECoG background activity, which can be detected in cortical tissue in ∼50% of patients with acute brain injury, is identical to Leao's cortical spreading depression (Leao, 1944b). We observed that every spreading ECoG depression was accompanied by a stereotyped SPC, which represents the depolarization of neurons and glia that is an integral part of CSD. The profile, duration and amplitude of the SPCs were consistent with the characteristics of the DC potential changes observed during CSD in animal experiments (Leao, 1951). The SPCs and the depression of the ECoG background activity occurred at the same time and propagated together in the cortical tissue that was monitored by the electrode strip. The speed of spread of the SPCs was similar to that of Leao's CSD. Taken together this provides definite evidence that the transient, spreading ECoG depressions we have observed in human neocortex adjacent to a brain lesion are CSDs. SPCs were also observed in two patients with long-term depression of ECoG. This represents the first electrophysiological recordings in humans of PID, characteristic of the ischaemic penumbra (Nedergaard and Hansen, 1993). Repeated depolarizations of brain tissue, spreading through cortical regions adjacent to cortical lesions are common in acutely injured human cortex. Whether PID/CSD contributes to the expansion of the focal brain damage following the initial insult will need to be addressed in a future study.
Long-term monitoring and slow potential changes
In pursuit of a therapeutic strategy, the main technical problem is to secure stable and sensitive recordings that may be interpreted online, to allow selection of patients who might benefit from drug intervention. The present paper describes two important means of enhancing changes compatible with CSD/PID during monitoring. Changes of signal amplitude are more readily detected, and repeated stereotyped patterns of these changes better appreciated, when the EEG-filtered signal is displayed as a compressed power trend. The time-integrated curves show SPCs during CSD/PID that are remarkably stereotyped, and often easy to recognize. The combined monitoring of the raw ECoG signal, power and integrated curves in a 90-min compressed screen frame is a very efficient way to monitor these patients (Fig. 2). With the use of this analytical framework, and provided there are stable recording conditions in the ICU, it is unlikely that a CSD or PID event will be overlooked if it spreads below the monitoring strip. The location of the strip is important for the sensitivity, but the CSD may reach the strip from two or more different sources (Fig. 1). We observed that in half of the patients the depolarization waves propagated along the strip in either direction. This might suggest that CSD propagates over large areas of the same cerebral lobe. Spread of PIDs between gyri is common after middle cerebral artery occlusion in cats (gyrencephalic brain) (Strong et al., 2000). On the other hand, experience in some experiments in animals with gyrencephalic brains suggests that sulci may represent barriers to CSD (Bowyer et al., 1999a, 1999b), while major cytoarchitectonic changes may block CSD in rodents, lisencephalic squirrel monkey cortex and larger gyrencephalic brains (Leao, 1944b; Rebert, 1970; Bures et al., 1974). In all six patients displaying CSD, the first CSD was observed within 4.2 h of start of recording. In one patient three CSD episodes were observed at 16–18 h after injury followed by a CSD-free period, while at 54 h new CSD episodes were observed. This biphasic pattern of CSD episodes is similar to the time distribution of CSD/PID in experimental animals under conditions of focal brain damage that induces transient PID/CSDs within the first 2 h post-injury, while a second CSD peak is observed at 12 h (Hartings et al., 2003). The 36-h CSD-free interval in this patient suggests that a long observation period is needed for studies of CSD in the acutely injured human cerebral cortex.
Electrophysiological evidence of CSD in human brain
Bures and co-workers obtained the first direct evidence for a CSD in human grey matter in vivo (Sramka et al., 1977). They showed that microinjections of potassium chloride into the caudate nucleus or the hippocampus elicited spreading depressions. A large negative shift of DC potential was observed in the hippocampus spreading at a rate of 3.2 mm/min, consistent with the propagation rate of CSD in rodents. They obtained similar recordings in the caudate nucleus. Notably, there was no suppression of the spontaneous electrical activity in the caudate nucleus recorded locally while the CSD propagated. This was due to technical limitations of the method: the electrodes were not positioned along the path of propagation and the recordings were not bipolar, but monopolar (Sramka et al., 1977). Another issue is the pick-up area of the electrodes. The ECoG depression is confined to an ∼30-mm wide band, which is surrounded by much larger volumes of tissue with preserved ECoG activity. This may explain the preserved spontaneous activity due to volume conduction of activity from remote generators. Nevertheless, the DC potential changes were unequivocal and were taken as definite evidence of the existence of spreading depression in human brain. The recording conditions that we used in the present, and in our previous study (Strong et al., 2002), took Bures' considerations into account.
A second line of evidence comes from in vitro studies of excised temporal cortical tissue from humans undergoing a brain operation for epilepsy, or other intracranial disorders. In one such study, Avoli et al. (1991) described DC changes characteristic for CSD by exposing brain tissue to artificial CSF with a reduced concentration of Mg2+. The CSD episodes alternated with periods of seizure activity, and both pathological reactions were blocked by NMDA-receptor antagonists (Avoli et al., 1991). Speckmann's group has successfully elicited seizure activity and CSD in human brain slices as well, and has demonstrated important interaction between the two events (Gorji, 2001; Gorji and Speckmann, 2004). Thus, the in vitro evidence is strong and provides direct evidence that human brain cortex can and does support CSD.
The third line of evidence comes from the work of Mayevsky who observed repetitive episodes of CSDs in a head injured patient in vivo (i.e. in only 1 out of the 14 studied) starting at ∼30 h after start of the monitoring (Mayevsky et al., 1996). The recordings showed characteristic transients of potassium, cerebral blood flow, cerebral blood volume, oxidation of the cortex and ECoG suppression consistent with CSD. Measurements were performed at a single point, therefore the putative spreading nature of the events could not be demonstrated. The recording equipment was positioned over the hemisphere contralateral to the lesion for practical reasons (Mayevsky et al., 1996), which may explain the low incidence of CSD/PID episodes reported.
In comparison, we positioned the electrode strip immediately adjacent to the lesion, radiating into apparently normal brain tissue. Most likely, this may explain why we found evidence of CSD/PID in 50% of our patients with acutely injured human brain cortex. It is likely that some CSD/PID episodes escaped detection by our electrodes, because the depolarization waves failed to propagate to the recording site. However, we reasoned that the close proximity between lesion and electrodes would minimize this shortcoming of the methodology, and facilitate the detection of at least some of CSD/PID episodes in patients in whom the reaction occurred. In rodents, CSD spreads in all directions from the point of stimulation, and in pigs CSDs have been found to propagate into and out of sulci as well, even though the velocity of spread is slower in the sulcus than on the crest of the gyrus (Bowyer et al., 1999b). We observed slow velocities of spread between some electrodes (0.4 mm/min) corresponding to a latency of 25 min between two neighbouring electrodes, while a ‘normal’ propagation velocity in rodents is ∼2–3 mm/min. The slower velocity of spread may correspond to a ‘true’ travel length of ∼5 cm which may indicate spread into a sulcus and up again. Additional studies using more electrodes than in the present study would be needed to describe in more detail the propagation characteristics of CSD in the human brain, which are likely to be more complex than in rodents.
PIDs and the ischaemic penumbra
The observations in two patients of a few initial CSDs followed by repeated episodes of PIDs show that depolarizing episodes may be linked to the expansion of the penumbra and thereby potentially of the core infarct zone. In both patients a period of repeated SPCs, in otherwise electrically silenced cortex, was followed by 2.4–3 h of complete ECoG silence followed by slow recovery of the ECoG activity. The reason for the prolonged silencing of the ECoG after the depolarizations could be a reduced blood flow in the penumbra region of the traumatized tissue (Astrup et al., 1977). The fact that the tissue were able to produce repeated SPC episodes suggests that perfusion and in turn the substrate supply for brain energy metabolism were sufficient for the re-establishing of the ionic gradients across the cell membranes. Eventually, the ECoG background activity resumed, which suggests that the tissue was indeed viable. We postulate that this is the first electrophysiological evidence of a penumbra in human brain tissue, and that the spreading SPCs which we have recorded represent the human correlate of the PIDs recorded in experimental focal brain injury.
In animals, a CSD/PID leads to a massive leaking of ions over the cell membranes along the ion gradients, and the subsequent re-establishment of the concentration gradients of, e.g. sodium, potassium and calcium, is a heavy metabolic task for the cells. In normally perfused tissue, local blood flow increases ∼100% for more than a minute during CSD (Leao 1944a; Fabricius et al., 1995). The local oxygen tension may transiently decrease or increase depending on the blood flow reaction (Lehmenkuhler et al., 1976; Back et al., 1994; Wolf et al., 1996), while the tissue lactate concentration increases more than 100% for several minutes in the normally perfused brain due to activation of glycolytic pathways (Lauritzen et al., 1990; Scheller et al., 1992).
The hemodynamic and metabolic changes associated with PIDs are different. Importantly, transient ‘reduction’ in tissue pO2 is characteristic of PID (Back et al., 1994), and reductions in perfusion of varying duration propagate across the cerebral cortex coupled with the depolarization (Strong et al., 2003). A microdialysis study of changes in extracellular glucose and lactate in the penumbra indicated transient reductions in glucose coupled with increases in lactate; the transient events appear to be superimposed on a gradual decline in dialysate glucose, suggesting that the supply of glucose for energy consumption is compromized under these conditions (Hopwood et al., 2005). This may explain in part the capacity of PIDs to increase infarct size (Gill et al., 1992; Mies et al., 1993; Back et al., 1996; Busch et al., 1996; Takano et al., 1996)
Clinical perspective in relation to treatment
CSD is efficiently blocked by NMDA antagonists in animals (Lauritzen and Hansen, 1992) and man (Avoli et al., 1991; Gorji, 2001). However, several intervention studies applying NMDA-antagonists in largely unselected human brain trauma patients have been unsuccessful (Heiss et al., 1999; Lees et al., 2000; Sacco et al., 2001). We suggest that identification of those patients that display CSD/PID may provide a means of selecting those patients who are likely to benefit from treatment with NMDA-receptor antagonists. The CSD/PID-prone patients reported here displayed multiple episodes (nine or more) and intervention could in principle be added after the first or second episode. Propofol is known to block gap junctions in vitro and might therefore be supposed to inhibit CSD events, which in part depend on preserved function of gap junctions (Nedergaard et al., 1995). It is worthy of note that all six patients displaying CSD/PIDs were treated with propofol. The dosage of propofol, however, was in the lower range, and could have been insufficient to block gap junctions. Nevertheless, it points to an important requirement for future intervention studies: any anti-CSD treatment must be monitored for effect until a dose–response relationship has been established.
The occurrence of CSD/PID showed a possible dependence on age. When 12 of the patients from our previous publication (Strong et al., 2002) were included (excluding 2 patients who were monitored for <12 h), the age-dependent incidence of patients with CSD/PID was virtually the same as in the present material: 21–24 years: 83% (n = 6); 39–69 years: 33% (n = 18). Incidence of trauma as the primary injury for the young group was 66 versus 56% for older patients. Therefore, a different mechanism of injury is not an obvious explanation for this trend. Among the 24 patients, all the 6 young patients were males. In the older group there were 9 males and 9 females, and incidence of CSD/PIDs in both of these gender groups was 33%. It is quite possible that CSD may be a more common finding in young patients, who have a better prognosis after brain injury. The finding needs to be confirmed in a larger series, but may point to a target group for an intervention study.
Conclusions
We present direct and unequivocal electrophysiological evidence for the existence of CSD and PID in human cerebral cortex. We used the unfiltered ECoG, simple expressions as power and ECoG integration to obtain this result, which doubled the sensitivity of the ECoG for these types of events, and made detection of PIDs possible. The method is robust and may become a standard monitoring device in the ICU in the future. For acutely injured patients, non-invasive methods of CSD detection, preferably applied over periods of days, are needed. Whatever methods emerge are likely to be less specific than the ECoG of the exposed cortex, and for this reason any one method will best be supported by simultaneous use of an additional method. Eventually, this will determine to what extent the large body of knowledge about CSD and focal ischaemia in animals will be applicable to acute pathology in the human brain. The clinical impact of pharmacological blockade of CSD/PID events may become the subject of a future intervention study, but in the first place it is necessary to establish the impact of these events on outcome.
Note: We have initiated an international research group: COSBID (CoOperative Study of Brain Injury Depolarisations). The primary goal of this group is to register ECoG in a cohort of brain injury patients, and compare the occurrence of spreading events (CSD/PID) with outcome. Further information is available at www.cosbid.org.
We thank the patients and their relatives for their assistance with the study, and the medical and nursing staff of the Intensive Care Unit of King's College Hospital, London, for their support and collaboration. We are grateful to The Wellcome Trust, GlaxoSmithKline, HeadFirst and the Rosetrees Charitable Trust for financial support.
References
Astrup J, Symon L, Branston NM, Lassen NA. Cortical evoked potential and extracellular K+ and H+ at critical levels of brain ischemia.
Avoli M, Drapeau C, Louvel J, Pumain R, Olivier A, Villemure JG. Epileptiform activity induced by low extracellular magnesium in the human cortex maintained in vitro.
Back T, Kohno K, Hossmann KA. Cortical negative DC deflections following middle cerebral artery occlusion and KCl-induced spreading depression—effect on blood flow, tissue oxygenation, and electroencephalogram.
Back T, Ginsberg MD, Dietrich WD, Watson BD. Induction of spreading depression in the ischemic hemisphere following experimental middle cerebral artery occlusion: effect on infarct morphology.
Bowyer SM, Okada YC, Papuashvili N, Moran JE, Barkley GL, Welch KM, et al. Analysis of MEG signals of spreading cortical depression with propagation constrained to a rectangular cortical strip. I. Lissencephalic rabbit model.
Bowyer SM, Tepley N, Papuashvili N, Kato S, Barkley GL, Welch KM, et al. Analysis of MEG signals of spreading cortical depression with propagation constrained to a rectangular cortical strip. II. Gyrencephalic swine model.
Bures J, Buresova O, Krivanek J. The mechanism and applications of Leao's spreading depression of electroencephalographic activity. Prague: Academia,
Busch E, Gyngell ML, Eis M, Hoehn Berlage M, Hossmann KA. Potassium-induced cortical spreading depressions during focal cerebral ischemia in rats: contribution to lesion growth assessed by diffusion-weighted NMR and biochemical imaging.
Fabricius M, Akgoren N, Lauritzen M. Arginine–nitric oxide pathway and cerebrovascular regulation in cortical spreading depression.
Gill R, Andine P, Hillered L, Persson L, Hagberg H. The effect of MK-801 on cortical spreading depression in the penumbral zone following focal ischaemia in the rat.
Gorji A, Speckmann EJ. Spreading depression enhances the spontaneous epileptiform activity in human neocortical tissues.
Hansen AJ, Zeuthen T. Extracellular ion concentrations during spreading depression and ischemia in the rat brain cortex.
Hartings JA, Rolli ML, Lu XCM, Tortella FC. Delayed secondary phase of peri-infarct depolarizations after focal cerebral ischemia: relation to infarct growth and neuroprotection.
Heiss WD, Thiel A, Grond M, Graf R. Which targets are relevant for therapy of acute ischemic stroke?
Hopwood SE, Parkin MC, Bezzina EL, Boutelle MG, Strong AJ. Transient changes in cortical glucose and lactate levels associated with peri-infarct depolarisations, studied with rapid-sampling microdialysis.
Hossmann KA. Viability thresholds and the penumbra of focal ischemia [see comments].
Koroleva VI, Bures J. The use of spreading depression waves for acute and long-term monitoring of the penumbra zone of focal ischemic damage in rats.
Kraig RP, Nicholson C. Extracellular ionic variations during spreading depression.
Lauritzen M, Hansen AJ. The effect of glutamate receptor blockade on anoxic depolarization and cortical spreading depression.
Lauritzen M, Hansen AJ, Kronborg D, Wieloch T. Cortical spreading depression is associated with arachidonic acid accumulation and preservation of energy charge.
Leao AAP. Pial circulation and spreading depression activity in the cerebral cortex.
Leao AAP. The slow voltage variation of cortical spreading depression of activity.
Lees KR, Asplund K, Carolei A, Davis SM, Diener HC, Kaste M, et al. Glycine antagonist (gavestinel) in neuroprotection (GAIN International) in patients with acute stroke: a randomised controlled trial. GAIN International Investigators 10.
Lehmenkuhler A, Speckmann EJ, Caspers H. Cortical spreading depression in relation to potassium activity, oxygen tension, local flow and carbon dioxide tension. In: Kessler M, Clark LC, Lübbers DW, Silver IA, Simon W, editors. Ion and enzyme electrodes in biology and medicine. München, Germany: Urban and Scwerzenberg;
Marrannes R, Willems R, De Prins E, Wauquier A. Evidence for a role of the N-methyl-d-aspartate (NMDA) receptor in cortical spreading depression in the rat.
Martins-Ferreira H, Nedergaard M, Nicholson C. Perspectives on spreading depression.
Mayevsky A, Doron A, Manor T, Meilin S, Zarchin N, Ouaknine GE. Cortical spreading depression recorded from the human brain using a multiparametric monitoring system.
Mies G, Iijima T, Hossmann KA. Correlation between peri-infarct DC shifts and ischaemic neuronal damage in rat.
Nedergaard M, Hansen AJ. Spreading depression is not associated with neuronal injury in the normal brain.
Nedergaard M, Hansen AJ. Characterization of cortical depolarizations evoked in focal cerebral ischemia.
Nedergaard M, Cooper AJ, Goldman SA. Gap junctions are required for the propagation of spreading depression.
Nilsson P, Hillered L, Olsson Y, Sheardown MJ, Hansen AJ. Regional changes in interstitial K+ and Ca2+ levels following cortical compression contusion trauma in rats.
Ohta K, Graf R, Rosner G, Heiss WD. Calcium ion transients in peri-infarct depolarizations may deteriorate ion homeostasis and expand infarction in focal cerebral ischemia in cats.
Park CK, Nehls DG, Graham DI, Teasdale GM, McCulloch J. The glutamate antagonist MK-801 reduces focal ischemic brain damage in the rat.
Parkin M, Hopwood S, Jones DA, Hashemi P, Landolt H, Fabricius M, et al. Dynamic changes in brain glucose and lactate in pericontusional areas of the human cerebral cortex, monitored with rapid sampling on-line microdialysis: relationship with depolarisation-like events.
Rebert CS. Spreading depression in squirrel monkey lissencephalic cortex.
Sacco RL, DeRosa JT, Haley EC Jr, Levin B, Ordronneau P, Phillips SJ, et al. Glycine antagonist in neuroprotection for patients with acute stroke: GAIN Americas: a randomized controlled trial.
Scheller D, Kolb J, Tegtmeier F. Lactate and pH change in close correlation in the extracellular space of the rat brain during cortical spreading depression.
Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression- like depolarization.
Sramka M, Brozek G, Bures J, Nadvornik P. Functional ablation by spreading depression: possible use in human stereotactic neurosurgery.
Strong AJ, Harland SP, Meldrum BS, Whittington DJ. The use of in vivo fluorescence image sequences to indicate the occurrence and propagation of transient focal depolarizations in cerebral ischemia.
Strong AJ, Smith SE, Whittington DJ, Meldrum BS, Parsons AA, Krupinski J, et al. Factors influencing the frequency of fluorescence transients as markers of peri-infarct depolarizations in focal cerebral ischemia.
Strong AJ, Fabricius M, Boutelle MG, Hibbins SJ, Hopwood SE, Jones R, et al. Spreading and synchronous depressions of cortical activity in acutely injured human brain.
Strong AJ, Hopwood SE, Boutelle MG, Parkin MC, Bezzina EL, Su Y, et al. Measuring dynamic changes in perfusion in the penumbra with high spatial and temporal resolution using laser speckle contrast imaging: comparison with indicator clearance.
Takano K, Latour LL, Formato JE, Carano RA, Helmer KG, Hasegawa Y, et al. The role of spreading depression in focal ischemia evaluated by diffusion mapping.
Wolf T, Lindauer U, Obrig H, Dreier J, Back T, Villringer A, et al. Systemic nitric oxide synthase inhibition does not affect brain oxygenation during cortical spreading depression in rats: a noninvasive near-infrared spectroscopy and laser-Doppler flowmetry study.
Author notes
1Department of Clinical Neurophysiology, Glostrup Hospital, University of Copenhagen, Denmark, 2Department of Clinical Neurosciences, King's College London and 3Department of Bioengineering, Imperial College London, University of London, London, UK


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}