

Abstract
Lithium salts govern important characteristics of lithium-ion batteries, including their efficiency (ion conductivity), operating voltage (potential window), and thermal stability. Herein, a series of lithium borates (lithium difluoro(perfluoropinacolato)borate (PFP-F2), lithium difluoro(2-hydroxy-3,3,3,3ʹ,3ʹ,3ʹ-hexafluoroisobutirato)borate (HHIB-F2), lithium (perfluoropinacolato)(oxalato)borate (PFP-Ox), lithium bis(2-hydroxy-3,3,3,3ʹ,3ʹ,3ʹ-hexafluoroisobutirato)borate (HHIB2), and lithium (2-hydroxy-3,3,3,3ʹ,3ʹ,3ʹ-hexafluoroisobutirato)(oxalato)borate) containing CF3 and C=O groups were developed as potential electrolytes for lithium-ion batteries. The proposed lithium borates were synthesized in good purity from lithium tetrafluoroborate and lithium difluoro(oxalato)borate using trimethylchlorosilane and three types of bidentate ligands in ethylmethylcarbonate. The applicability of the novel lithium borates as electrolytes for lithium-ion batteries was demonstrated based on thermal and electrochemical stability evaluations. In addition, lithium borates with >4 CF3 groups (PFP-F2, PFP-Ox, and HHIB2) exhibited outstanding stability against hydrolysis (water contamination). HHIB-F2 showed the best ionic conductivity owing to the balanced incorporation of increased Li+ dissociation and mobility by introducing the CF3 group and reducing the anion size. HHIB-F2 and HHIB2-containing electrolytes showed better cycle performance than their conventional BF4 counterparts. This study suggests that the new lithium borates, HHIB2, and HHIB-F2, are promising lithium salts for lithium-ion batteries, providing a new direction for the lithium salt molecular design.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Lithium-ion batteries are commonly used as power sources for mobile PCs and smartphones because of their lightweight and high-capacity characteristics [1, 2]. Furthermore, the electrification of automobiles in recent years has been aggressively pursued to reduce greenhouse gas emissions. In this context, it is critical to develop Li-ion batteries suitable for automobiles, necessitating considerable input and output characteristics (especially at low temperatures), durability at high temperatures and improved safety [3–5]. Most electrolytes used in commercial Li-ion batteries contain lithium hexafluorophosphate (LiPF6) as a lithium salt [5–8] due to its high ionic conductivity [9, 10], wide potential window [9, 11], and low cost [12]. However, LiPF6 has several issues, including low thermal stability (<70 °C) [13] and hydrogen fluoride (HF) formation via hydrolysis [14]. Therefore, the development of an alternative lithium salt remains an important and challenging task.
Lithium tetrafluoroborate (LiBF4) is a Li salt with high thermal stability [15, 16] and better hydrolysis resistance compared to LiPF6 [17], but its low ionic conductivity hinders its widespread use. To overcome those disadvantages, the chelate complexes and borate esters as anion receptors [18, 19] were first proposed, and it has been expanded to polymer electrolyte systems [20]. Furthermore, a recent publication focused on the potential use of lithium borates as electrolyte additives [7, 21–24]. For solution-based electrolytes, previous studies have suggested that a promoter of ionic conductivity is the negative charge delocalization on the central boron atom [18, 25, 26] and various electron-withdrawing groups, i.e. CF3 or C=O, have been introduced into the anions to tune this delocalization. Although anions including lithium bis(perfluoropinacolato)borate (LiBPFPB) [27, 28], lithium bis(2-hydroxy-3,3,3,3ʹ,3ʹ,3ʹ-hexafluoroisobutirato) borate (HHIB2) [29], lithium bis(oxalato)borate (LiBOB) [30, 31], and lithium bis(malonato)borate (LiBMB) [31] have been proposed, the ionic conductivity of LiBOB, which is the highest among the above-mentioned salts, was still comparable to that of LiBF4. The negligible improvement of ionic conductivity for the listed anions likely originates from the increased molecular weight of the Li salt caused by the introduction of electron-withdrawing group [32]. Larger molecular weights increase the solution viscosity and thereby reduce Li+ diffusivity. Therefore, to improve the ionic conductivity, it is necessary to limit the molecular weight increase while retaining the effects of the electron-withdrawing group to reduce electron density on the central boron.
To fulfill the battery electrolyte requirements, lithium difluoro(oxalato)borate (LiDFOB) [33] with a chelated oxalate moiety and two fluorine atoms was introduced. LiDFOB showed higher thermal stability than LiBF4 and exhibited the highest ionic conductivity among previously reported lithium borates [15, 34, 35], mainly due to its high stability that originated from the chelating effect and electron-withdrawing group addition without significant molecular weight gain. The concept of such an asymmetric complex was successfully applied to LiBMB, yielding lithium difluoro-2-methyl-2-fluoromalonatoborate (LiDFMFMB) [36], wherein the chelated malonate moiety was halved. LiDFMFMB showed greatly improved solubility in carbonate solvent and conductivity, which are known disadvantages of LiBMB. Although these borates feature the electron-withdrawing properties of the carbonyl group, the introduction of a CF3 group with a more significant electron-withdrawing effect can be more effective for the delocalization of the anion on the boron.
The mass (Mcal) was calculated considering the borate anion moiety without lithium cations. The experimentally obtained mass (Mexp) was measured using high-resolution mass spectra-electrospray ionization (HRMS-ESI) (negative ionization mode). NMR spectra were obtained for each lithium borate dissolved in deuterated acetonitrile (CD3CN).
Herein, we synthesize a series of lithium borates to clarify the effect of the number of CF3 and C=O groups on ionic conductivity as well as their applicability as lithium-ion battery electrolytes. Lithium difluoro(perfluoropinacolato)borate (PFP-F2), lithium difluoro(2-hydroxy-3,3,3,3ʹ,3ʹ,3ʹ-hexafluoro isobutirato)borate (HHIB-F2), lithium (perfluoropinacolato)(oxalato)borate (PFP-Ox), lithium bis(2hydroxyy-3,3,3,3ʹ,3ʹ,3ʹ-hexafluoroisobutirato)borate (HHIB2), and lithium (2-hydroxy-3,3,3,3ʹ,3ʹ,3ʹ-hexafluoroisobutirato)(oxalato)borate (HHIB-Ox) were synthesized from trimethylchlorosilane, bidentate ligands (PFP, HHIB, and Ox), and boron sources (LiBF4 and LiDFOB). The prepared borates were thermally stable up to 140 °C, and the symmetric bis-chelated borate, HHIB2, showed the best thermal stability up to 350 °C. Although the greatest disadvantage of the conversion from a bis-chelate to mono-chelate for molecular size reduction is the decreased thermal stability, the thermal stability of both bis- and mono-chelate was sufficient for lithium-ion battery electrolyte. The solubility of the salt was significantly improved by introducing the CF3 group into the LiBOB structure. PFP-F2 and HHIB2 showed improved hydrolysis resistance with negligible degradation after 2 day in an aqueous solution. The electrochemical window of electrolytes containing PFP-F2, PFP-Ox, HHIB2, HHIB-F2, and HHIB-Ox is comparable or wider than that of BF4-containing electrolytes. HHIB-F2 showed the best ionic conductivity of 7.1 mS cm−1 by incorporating a good balance of anion size reduction (increased Li+ mobility) and CF3 group introduction (increased Li+ dissociation). Furthermore, all five lithium borates showed superior ionic conductivity compared to conventional BF4 anions. The lithium-ion batteries composed of HHIB-F2 and HHIB2-containing electrolytes exhibited larger 1st cycle capacities and superior cycling stabilities compared to LiBF4 under accelerated test conditions (60 °C; 3C), confirming the applicability of these lithium borates for lithium-ion batteries applications. The incorporation of 2–4 CF3 groups into the molecule provided the best balance, imparting moderate electrochemical stability while maintaining ionic conductivity as an electrolyte.
2. Results and discussion
2.1. Characterizations of lithium borates
The structures of the synthesized lithium borates were confirmed by high-resolution mass spectrometry and 11 B, 19F, 13C, and 7Li nuclear magnetic resonance (NMR) spectroscopy (table 1). The experimentally obtained mass (Mexp) agreed with the calculated mass (Mcal). The mass difference (|Mexp− Mcal|/Mexp × 106) for each lithium borate (PFP-F2, PFP-Ox, HHIB2, HHIB-F2, and HHIB-Ox) was 1.39, 4.83, 3.74, 2.95, and 1.89 ppm, respectively, confirming the successful synthesis of the lithium borates. The 11B, 19F, 13C, and 7Li NMR spectra further confirmed the structure and high purity of the synthesized lithium borates (figures S2–S5). The 1H NMR showed no signals from impurities or by-products, except for solvated ethylmethylcarbonate (EMC) and the residual protonated acetonitrile-d3 methyl group (CD3CN; figure S1). The 13C NMR spectra showed characteristic peaks from C=O (140–147 ppm), CF3 (112–113 ppm), and (CF3)2 C (79–84 ppm), as well as peaks from the solvated EMC (figure S2). The (CF3)2 C peaks for the HHIB group (HHIB2, HHIB-F2, and HHIB-Ox) and PFP group (PFP2, PFP-F2, and PFP-Ox) were observed at ca. 79 and 84 ppm, respectively. The (CF3)2 C peak chemical shift was affected by the C=O within the structure. The CF3 peak chemical shifts corresponded well with the number of electron-withdrawing CF3 groups attached to the beta carbon: PFP-group (PFP2, PFP-F2, and PFP-Ox) > HHIB-group (HHIB2, HHIB-F2, and HHIB-Ox). The C=O peaks from the solvated EMC were detected for most of the lithium borates, except for HHIB-F2, Ox2, and Ox-F2. The C=O (ca. 140 ppm) chemical shifts for the solvated EMC were slightly shifted, while the CH3, CH3CH2, and CH3 CH2 chemical shifts for the solvated EMC were similar to those observed in the EMC solution. The C=O peak shift suggests a strong interaction between the EMC and Li+ via the carbonyl moiety, making it difficult to remove EMC from lithium borates. An additional C=O feature was detected for PFP-Ox, HHIB2, HHIB-F2, HHIB-Ox, Ox2, and Ox-F2 at 141–147 ppm. The C=O peaks from the HHIB group can be seen at the lowest magnetic field (ca. 146 ppm), indicating that they are strongly affected by the alpha carbon CF3 group.
Table 1. Structure-related parameters obtained from NMR and high-resolution mass spectrometry.
| Lithium borate | 19F NMR [ppm] | 11B NMR [ppm] | 13C NMR [ppm] | 7Li NMR [ppm] | Mexp Mcal| |Mexp− Mcal|/Mexp × 106 | |
|---|---|---|---|---|---|---|
| Structure | Abbreviation | |||||
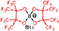
| PFP2 (LiBPFPB) | δ 94.1 | δ 12.7 | δ 112.5 δ 83.5 | δ 0.3 | — — — |
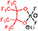
| PFP-F2 | δ 94.1 δ 16.6 | δ 6.4 | δ 112.7 δ 83.9 | δ 0.2 | 380.9768 380.9773 1.39 |
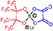
| PFP-Ox | δ 94.2 | δ 10.7 | δ 142.7 δ 112.2 δ 84.0 | δ 0.2 | 430.9581 430.9602 4.83 |
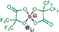
| HHIB2 | δ 88.2 | δ 11.4 | δ 146.3 δ 111.8 δ 79.1 | δ 0.3 | 430.9586 430.9602 3.74 |
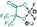
| HHIB-F2 | δ 88.1 δ 15.4 | δ 5.7 | δ 147.3 δ 112.1 δ 79.1 | δ 0.2 | 258.9811 258.9818 2.95 |
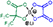
| HHIB-Ox | δ 88.2 | δ 10.1 | δ 146.1 δ 142.3 δ 111.6 δ 79.1 | δ 0.2 | 308.9641 308.9647 1.89 |
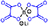
| Ox2 (LiBOB) | — | δ 8.8 | δ 141.8 | δ 0.2 | — — — |

| Ox-F2 (LiDFOB) | δ 10.3 | δ 4.4 | δ 143.0 | δ 0.1 | — — — |
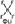
| LiBF4 | δ 10.3 | δ 0.0 | — | δ 0.0 | — — — |
NMR analysis suggests that the effect of the electron density reduction (shift to a lower magnetic field) caused by the two CF3 groups is stronger than that of the carbonyl group. The 19F NMR shows the CF3 group within the PFP-group and HHIB-group at 94.1 and 88.2 ppm, respectively. The PFP CF3 group signal is located at a lower field than that in HHIB, likely because C(CF3)2, which has greater electron-withdrawing properties than carbonyl, is attached to the β-position of the CF3 group. The result indicates that the CF3 group peak position varies significantly depending on the chelate ring substituent. A similar trend was observed for the F atom peak that is directly attached to the boron center. The F peak position was significantly affected by the opposite substituent (PFP, HHIB, oxalate, or F), specifically 16 ppm for PFP-F2 and HHIB-F2 and 10 ppm for Ox-F2 and LiBF4 (figure S3). However, the peaks arising from the CF3 groups within PFP2, PFP-F2, and PFP-Ox were similar, indicating negligible induction effects of PFP, F, and Ox bound to boron on the opposite CF3 group in the PFP chelate ring. The same trend was observed for the CF3 groups within the borate-containing HHIB group (HHIB2, HHIB-F2, and HHIB-Ox). In the 11B NMR spectra, the boron peaks were downshifted with an increasing number of CF3 groups, indicating that the electron-withdrawing CF3 group delocalized the negative charge on the boron center (figures S4(a) and (b)). In contrast, the 7Li NMR suggested almost no Li peak position dependence on the structure of the lithium borates or electron density of the boron center, probably because of the dominant effect of the solvated deuterated acetonitrile around Li+ (figure S5).
2.2. Fundamental properties of lithium borates
2.2.1. Thermal stability
Thermogravimetry-differential thermal analysis (TG-DTA) measurements revealed that the inductive effect, resonance stabilization effect of the substituent group, and chelate effect determine the thermal stability of lithium borates. HHIB2, Ox2, and PFP2 showed significantly higher thermal stabilities than that of LiBF4 (table 2).
Table 2. Stability data for each of the newly prepared lithium borates.
| Lithium borate | Decomposition | Decomposition |
|---|---|---|
| PFP2 | 220 | <0.1 |
| PFP-F2 | 140 | <0.1 |
| PFP-Ox | 170 | 4.5 |
| HHIB2 | 350 | <0.1 |
| HHIB-F2 | 180 | 92.8 |
| HHIB-Ox | 170 | 86.7 |
| Ox2 | 310 | 68.7 |
| Ox-F2 | 230 | 92.8 |
| LiBF4 | 190 | 17.7 |
a Estimated from TG-DTA analysis at 10 °C min−1 under N2 atmosphere. b The decomposition rate was evaluated by 11B-NMR analysis of a 0.8 M aqueous solution after 2 day at 25. c Values measured using DSC are reported [27].
Table 2 lists the decomposition temperatures of the prepared lithium borates estimated by TG-DTA measurements (TG-DTA curves shown in figure S6). The lithium borates solvated by EMC (PFP-F2, PFP-Ox, HHIB2, and HHIB-Ox) showed a mass decrease from EMC volatilization at 100 °C–150 °C. The DTA curve showed that PFP2 solvated by EMC melts at 105 °C, consistent with the melting temperature of 120 °C for pure borate PFP2 [27]. Furthermore, the TG curve of PFP2 showed a seamless decrease in the mass from 110 to 220 °C, indicating continuous volatilization of EMC and PFP2. HHIB2 offered the highest thermal stability (350 °C), followed by Ox2 (310 °C) and PFP2 (220 °C).
Bis-chelated borates were more stable than their corresponding mono-chelated borates with the same ligand (PFP2 > PFP-F2, HHIB2 > HHIB-F2, Ox2 > Ox-F2), consistent with the general trend derived from the chelate effect [37, 38]. It should be noted that the asymmetric bis-chelated borates (PFP-Ox, HHIB-Ox) were significantly destabilized compared to the symmetric bis-chelated borates (PFP2, HHIB2, Ox2), likely due to the lowering of the complex symmetry. Among the three symmetric bis-chelated borates, HHIB2 with two HHIB ligands showed the best thermal stability, due to the strong inductive effect of the CF3 group [39] and the resonance stabilization effect of the carbonyl group [26]. The better thermal stability of Ox2 compared to PFP2 can also be explained by the resonance effect of the carbonyl group. LiBF4 showed lower thermal stability than HHIB2, Ox2, and PFP2, partly because of the lower bonding energy of B–F (757 kJ mol−1) [40] compared to that of B–O (803 kJ mol−1) [41]. The same argument is consistent with the lower thermal stability of HHIB-F2, Ox-F2, and PFP-F2 compared to the corresponding bis-chelated lithium borates. Thermal stability was improved when the two fluorine atoms attached to boron were replaced by the Ox ligand (PFP-F2 < PFP-Ox). Although the B–F bond in HHIB-F2 was replaced by a more stable B–O bond (HHIB-Ox), HHIB-Ox was more thermally unstable than HHIB-F2 because of the distorted HHIB-Ox structure.
2.2.2. Hydrolytic stability
The hydrolytic stabilities of PFP2, PFP-F2, PFP-Ox, and HHIB2 were significantly improved compared to that of LiBF4. The result suggests the effective inhibition of nucleophilic attack from water to the boron atom at the center of borate, by the steric hindrance of the CF3 group (table 2).
Table 2 shows the decomposition rates of each borate in water estimated from the 11B NMR analysis (figure S7). The decomposition rates of PFP2, PFP-F2, PFP-Ox, and HHIB2 were <1%, suggesting that they were particularly resistant to hydrolysis. The sterically hindered CF3 groups prevented water molecules from accessing the reactive site, namely, the boron center and C=O group, effectively suppressing hydrolysis. Although HHIB-F2 and HHIB-Ox contain the CF3 group, hydrolysis was not completely suppressed because water molecules can approach the boron center from the opposite side (F and Ox sides) of the HHIB moiety.
To clarify the hydrolysis mechanism, the hydrolyzed compounds were characterized by further analysis of the 11B NMR spectra. The major hydrolysis products of Ox-F2 were BF4, BF3(OH) [34], and B(Ox)(OH)2 [42]. Similarly, the main hydrolysis products of HHIB-F2 were BF4, BF3(OH), and B(HHIB)(OH)2. This indicates that the fluorine anion generated by the hydrolysis of the B–F bond nucleophilically attacks boron and reforms the B–F bond to produce BF4 and BF3(OH). The low decomposition rate of BF4 in water can be attributed to the re-formation of B–F bonds even after their hydrolysis.
2.2.3. Electrochemical stability
The electrochemical stability of each lithium borate was evaluated via linear sweep voltammetry (LSV) with a glassy carbon electrode (GCE) in an EMC- ethylene carbonate (EC) 2:1 (vol/vol) solution. The electrochemical windows of the electrolytes containing PFP-F2, PFP-Ox, HHIB2, HHIB-F2, and HHIB-Ox were comparable to or even wider than those of Ox2, Ox-F2, and BF4-containing electrolytes. The result confirmed the applicability of the newly prepared lithium borates as electrolytes for lithium-ion batteries (figure 1).

Figure 1. Electrochemical stability of lithium borates. Linear sweep voltammograms of PFP2, PFP-F2, PFP-Ox, HHIB2, HHIB-F2, HHIB-Ox, Ox2, Ox-F2, and LiBF4 in EC/EMC (1/2 v) from 3.0 to 0.0 VLi at (a) 1st and (b) 2nd cycle and from 3.0 to 6.0 VLi at (c) 1st and (d) 2nd cycle at a scan rate of 5 mV s−1. A glassy carbon electrode was used as a working electrode, and lithium metal was used as the counter and reference electrodes. Introducing the CF3 group into the molecule increases the solubility (wt.%) (table S1). The comparison of ionic conductivity was made at 0.8 M since the maximum solubility of Ox2 in the EC-EMC (1:2 vol/vol) solution was 0.8 M.
Download figure:
Standard image High-resolution imageA large reductive current was observed below 0.5 VLi, which corresponds to the EC reduction (figure 1(a)). During the initial potential sweep, a small reductive current was observed at 0.5–2.0 VLi for most of the salts except for LiBF4. The onset potential of the reduction peak is in the following order; HHIB2 < PFP-F2 < HHIB-F2 ∼ PFP2 < Ox-F2 < Ox2 ∼ PFP-Ox < HHIB-Ox. To further clarify the origin of the reduction peak, we performed LSV in borate-containing 1,2-dimethoxyethane (DME) solvent, with higher reduction stability compared to EC (figures S8(a) and (b)). The LSV showed a similar small reduction peak, and the onset potential trend was independent of the solvent. In order to confirm the origin of this small reductive peak, LSV was performed in an EMC-EC 2:1 (vol/vol) solution containing potential impurities, such as PFP, HHIB, and Ox (figure S8(c)). Although a reduction current appeared at ca. 1.3VLi for PFP, Ox, and HHIB-containing EMC-EC electrolytes, no reductive peak corresponding to those in figure 1(a) was observed, suggesting a negligible impurity contribution to the reductive peaks. Therefore, we conclude that the reductive peak observed in figure 1(a) was originated from partial salt decomposition. Although the strong electron-withdrawing nature of the CF3 group generally decreases the LUMO level [43], the presence of the CF3 group did not decrease the reduction stability of PFP2, PFP-F2, HHIB2, or HHIB-F2. Furthermore, borates containing Ox ligands, including Ox2 and OxF2, showed relatively low reduction stability (high onset potential) among the salt tested, which is in agreement with the previous studies suggesting Ox-contained borates easily decompose via reduction reactions and generate solid electrolyte interphase (SEI) on the anode surface [15, 20, 44–46]. The energy level calculations of highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) for each borate (figure S9) suggest that the LUMO level is in the order of PFP2 > PFP-F2 ≫ PFP-Ox and HHIB2 > HHIB-F2 ≫ HHIB-Ox, which gave the following general trend in the LUMO level; borates with CF3 group > B–F bond > Ox. Although the presence of solvents significantly affects the redox potentials of the molecule [47], the observed LUMO-level trend is partially reflected in the LSV results, where borates with Ox ligands have relatively low reduction stability.
We here propose that the partial decomposition of the borates form an SEI layer on the GCE surface, inhibiting the further decomposition of the borates and solvent. Our hypothesis was supported by the absence of reductive current at 0.5–2.0 VLi during the second potential sweep in both EC/EMC and DME system, indicating the inhibition of the further borate reduction by the SEI formed during the initial cycle (figures 1(b) and S8(b)). Furthermore, the onset potential of the EC and DME reduction reaction shifted slightly lower in the second cycle compared to that of the initial cycle, suggesting that the SEI can also prevent EC and DME decomposition.
Oxidation stability was evaluated for all prepared lithium borates, confirming that all borates were electrochemically stable up to ca. 4.8 VLi. This range covered the usual operating voltage of the lithium-ion battery (up to ca. 4.2 VLi [4, 5, 7]; figures 1(c) and (d)). The oxidation stability of bis- and mono-chelated lithium borates depends on the ligands; the oxidation stability followed the order: oxalate, HHIB, and PFP. The strong electron-withdrawing nature of the CF3 group lowered the HOMO level and improved oxidation resistance [25]. With the exception of Ox-F2, the oxidation current decreased in the second cycle, suggesting that oxidative decomposition products deactivate the GCE. Despite the presence of easily oxidized oxalate groups, PFP-Ox and HHIB-Ox showed excellent oxidation resistance. Thus, the observed trend indicates that CF3-and/or oxalate-containing oxidized products can effectively prevent further decomposition of the borates.
2.3. Relationship between borate structure and ionic conductivity
The ionic conductivity for novel lithium borate electrolyte can be tuned by introducing an electron-withdrawing CF3 group to the anion structure (improved Li+ dissociation) and by reducing the molecular weight and anion size (improved Li+ diffusion). HHIB-F2, containing 2 CF3 groups together with small and light F atoms, optimally balanced the above-mentioned design requirements, resulting in the highest ionic conductivity among the newly prepared lithium borates (table 3).
Table 3. Ionic conductivity in the 0.8 M EC-EMC (1:2 v/v) solution.
| Lithium borate | Ionic conductivity at 30 °C (mS cm−1) |
|---|---|
| PFP2 | 4.2 |
| PFP-F2 | 5.9 |
| PFP-Ox | 4.3 |
| HHIB2 | 5.8 |
| HHIB-F2 | 7.1 |
| HHIB-Ox | 6.0 |
| Ox2 | 6.0 |
| Ox-F2 | 6.6 |
| LiBF4 | 3.6 |
The ionic conductivities of 0.8 M lithium borate in EC-EMC (1:2 vol/vol) solution at 30 °C are listed in table 3. All newly synthesized lithium borates showed better ionic conductivity than conventional LiBF4. The conductivity followed the order: HHIB-F2 > Ox-F2 > HHIB-Ox > Ox2 > PFP-F2 > HHIB2 > PFP-Ox > PFP2, suggesting that the anion size and electron-withdrawing nature of the ligand played significant roles in determining ionic conductivity.
Lithium borates with large PFP ligands (PFP2, PFP-Ox, and PFP-F2) and HHIB2, which contains two relatively large HHIB ligands, showed low ionic conductivity. However, the introduction of an electron-withdrawing group (CF3 or F) improved the ionic conductivity (replacing the carbonyl group with the CF3 group: HHIB-Ox > Ox2, HHIB-F2 > Ox-F2, replacing the Ox group with F: HHIB-F2 > HHIB-Ox, Ox-F2 > Ox2). The ionic conductivity can be determined by the dissociation degree of the salt and/or the mobility of anions and cations [6, 48, 49]. To clarify the effect of the dissociation degree on the ionic conductivity, 11B NMR and Raman spectroscopy were performed. Although the degree of Li dissociation was dictated by the boron atom electron density, no clear relationship between Li dissociation and ionic conductivity was confirmed (figure 2).
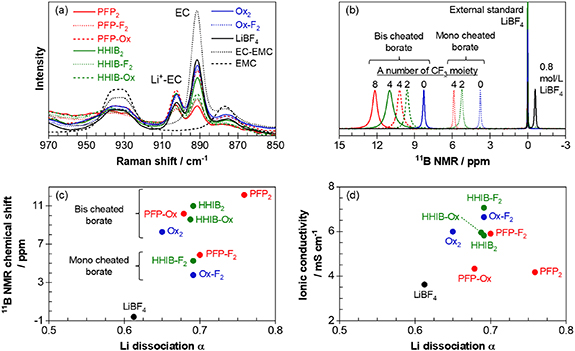
Figure 2. Relationships among the borate structure, Li dissociability, and ionic conductivity. (a) Raman spectra of 0.8 M EC-EMC (1/2 v) solution of each lithium borate in the 850–960 cm−1 region. (b) 11B NMR spectra of 0.8 M EC-EMC (1/2 v/v) solution of each lithium borate. LiBF4 dissolved in CD3CN was used as an external standard (0 ppm). (c) 11B NMR chemical shift of 0.8 M EC-EMC (1/2 v/v) solution versus the Li dissociation α (calculated from table S1). (d) Ionic conductivities of 0.8 M EC-EMC (1/2 v/v) solution versus the Li dissociation α. The ionic conductivity was measured at 30 °C, while 11B NMR and Raman spectra were measured at 25 °C.
Download figure:
Standard image High-resolution imageThe Li dissociation degree (α) was estimated from the Raman intensity ratio of the EC-related peaks (Ibound), considering the difference in the EC molar number (table S2). The Raman spectra of 0.8 M lithium borate in EC-EMC (1:2) solution at 850–960 cm−1 are shown in figure 2(a). The peak at approximately 880–900 cm−1 was assigned to C–O symmetric stretching of the non-solvated EC (denoted as free-EC) [50–52]. The small peak at approximately 905 cm−1 corresponds to the C–O symmetric stretching vibration from EC interacting with the Li cation (denoted as Li+-EC). The Li dissociation degree (α) was estimated using the following method: the peak intensities of free-EC and Li+-EC (denoted as IEC and ILi+-EC, respectively) were calculated by integrating the deconvoluted free-EC and Li+ -EC peak areas [50, 52]. From the ratio of IEC to ILi+-EC and the amount of EC used to prepare the 0.8 M solution, the molar number of EC interacting with Li+ was calculated. A solvation number of 4 was assumed for Li+ [52–54] (EC to Li+ is 3, and EMC to Li+ is 1), allowing for the determination of the molar number of Li+ solvated by EC. Considering that the solvated Li+ is dissociated, it can be divided by the total molar number of Li+ in the solution to obtain an estimated α. The lithium borates showed an estimated α of 0.65–0.76, slightly higher than that of LiBF4. The Li+ dissociation degree was in accordance with the number of electron-withdrawing CF3 groups within the molecule: PFP2 > HHIB2 > PFP-Ox > HHIB-Ox > Ox2 for bis-chelated lithium borates and PFP-F2 > HHIB-F2 > Ox-F2 for the mono-chelated lithium borates.
Herein, we propose that the electron density of the boron atom varies with the number of electron-withdrawing groups, which also dictates the Li dissociation degree. This is supported by the chemical shift analysis of the 11B NMR spectra, which can be used to estimate the boron atom electron density (figure 2(b)). For the bis-chelated lithium borates (PFP2, HHIB2, PFP-Ox, HHIB-Ox, and Ox2), the chemical shifts of borate were large towards a low magnetic field (PFP2 > HHIB2 > PFP-Ox > HHIB-Ox > Ox2) in accordance with the number of CF3 groups, indicating lower boron atom electron density in the same order. The same trend was observed for the mono-chelated lithium borates (PFP-F2 > HHIB-F2 > Ox-F2). The difference in the boron electron density for the bis- and mono-chelated lithium borates (bis-chelated < mono-chelated) can be explained by considering the resonance effect, which delocalizes the negative charge depending on the ligand structure. Furthermore, the cyclic structures of the bis- and mono-chelated borates benefited from the resonance effect compared to the simple BF4 ligand.
A good correlation between the 11B NMR chemical shifts and α values was confirmed in both the mono- and bis-chelated borates (figure 2(c)). This suggests that the boron atom electron density, which was affected by the number of electron-withdrawing groups and resonance effect from the ligand structure, dictated the degree of Li dissociation. Thus, the results support our hypothesis, and the 11B NMR chemical shifts can be used as a descriptor of the Li dissociation degree. However, the Li dissociation degree did not show a clear correlation with ionic conductivity (figure 2(d)), suggesting that the ionic conductivity was influenced by different parameters such as ion mobility.
The relationship between ionic conductivity and Li-ion mobility (self-diffusion coefficient of Li+, DLi) was studied by calculating the self-diffusion coefficient (D) of lithium cations using the 7Li signals from PFG NMR [55]. The ionic conductivity was tied to DLi, and was significantly affected by the solution viscosity, which was determined by the anion molecular weight and size (figure 3).
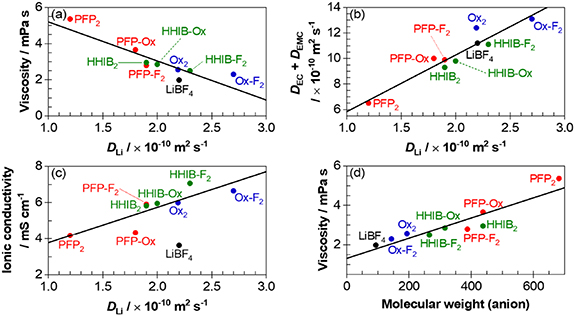
Figure 3. Relationships among the borate structure, Li mobility, and ionic conductivity. (a) Viscosity of 0.8 M EC-EMC (1/2 v/v) solution versus the self-diffusion coefficients of the lithium cation. (b) Self-diffusion coefficients of the solvent (EC + EMC) versus that of the lithium cation. (c) Ionic conductivities of 0.8 M EC-EMC (1/2 v/v) solution versus the self-diffusion coefficients of the lithium cation. (d) Molecular weight of the borate anion versus the viscosity and mass fraction (anion) of the 0.8 M EC-EMC (1/2 v/v) solution. The self-diffusion coefficient, viscosity, and ionic conductivity were measured at 30 °C.
Download figure:
Standard image High-resolution imageTo estimate the mobility of the ionic species and solvents, the self-diffusion coefficient (D) of borate anions, solvents, and lithium cations were calculated from 19F (except for non-fluorinated BOB, for which 11B was used), 1H, and 7Li signals of PFG NMR, respectively (table S3). The validity of the obtained self-diffusion coefficient (D) was confirmed by the inverse proportionality of the Li+ self-diffusion coefficient (DLi) to the viscosity (figure 3(a)), in agreement with the Stokes–Einstein equation expressed as follows:
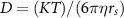
where κ is the Boltzmann constant, η is the viscosity, and rs is the Stokes radius of the diffused species. The observed linear relationship between DLi and the self-diffusion coefficient of the solvents (DEC, DEMC) provides further support (figure 3(b)) and is consistent with the literature [56].
Figure 3(c) reveals that the ionic conductivity correlates well with the Li+ self-diffusion coefficients (D Li ), suggesting that D Li can be used as a descriptor for ionic conductivity. The large deviation in LiBF4 likely originates from the significantly lower degree of Li+ dissociation. Based on the above equation, D Li can be tuned by the electrolyte viscosity and Stokes radius of Li+. The anion viscosity and molecular weight exhibited a linear relationship (figure 3(d)), suggesting that the molecular weight determines the solution viscosity. For example, BF4, which has the lowest molecular weight, showed the lowest viscosity, whereas the highest molecular weight, PFP2, showed the highest viscosity. Here, the difference in the complex structure (bis- or mono-chelated) had a negligible effect on the correlation between viscosity and molecular weight. It was also found that reducing the anion size (PFP2 vs. HHIB2, HHIB-F2) decreased the viscosity and effectively improved ionic conductivity.
2.4. Applicability of lithium borates to lithium-ion batteries
Based on the above-described results, HHIB2 and HHIB-F2 were chosen as electrolytes and their charge–discharge performances were evaluated using a LiNi0.33Mn0.33Co0.33O2 cathode and artificial graphite anode. After stabilizing the cells by pre-charge/discharge at a 0.2 C rate at 25 °C (figure S10), we conducted cycle tests at a 3 C rate at 60 °C to confirm the performance of each electrolyte under severe conditions. The newly synthesized lithium borates showed notably larger initial capacity and better cycling stability compared to conventional LiBF4, suggesting their applicability as electrolytes in lithium-ion batteries (figure 4).

Figure 4. Charge/discharge test for lithium-ion batteries prepared with the newly developed borates. (a)–(c) Voltage profiles and (d) coulombic efficiency from the 1st to 100th cycles of LiNi0.33Mn0.33Co0.33O2/artificial graphite cells at 3C rate and 60 °C. The cutoff potential was set to 4.3 V. 1.0 M of HHIB2, HHIB-F2, and LiBF4 (EC-EMC = ½ v/v) solutions were used as electrolytes. The initial charge/discharge cycle (pre-cycling) was performed at 25 °C and 0.2 C before the cycle test (see figure S10).
Download figure:
Standard image High-resolution imageFigures 4(a)–(c) shows the charge–discharge profiles of the 1st, 50th, and 100th cycles at 60 °C. It should be noted that the charge–discharge profile was obtained after SEI formation via pre-cycling treatment (see figures S10(a)–(c)). The capacities of the 1st cycle for cells prepared with HHIB2 (155.8 mAh g−1) and HHIB-F2 (154.8 mAh g−1) were notably larger than the cell with the LiBF4 electrolyte (138.0 mAh g−1). The difference in the 1st cycle capacity is largely due to undesirable side reactions (solvent and electrolyte decomposition), which consume Li+ and decrease capacity. Therefore, the notably lower 1st cycle capacity of LiBF4 compared to HHIB2 and HHIB-F2 is due to the progressive loss of Li+ through side reactions, in agreement with the understanding that LiBF4 cannot form a good SEI to inhibit side reactions [57–59].
Further support comes from the cycling stability test, where the capacity fades from the 1st to 100th cycle was as follows: HHIB2 (19.6%) < HHIB-F2 (23.7%) < LiBF4 (28.6%). Relatively high cycling stability was observed for HHIB2 due to the high anionic thermal stability. Although HHIB-F2 and LiBF4 have similar thermal stability, there is a large difference in cycle capacity. Figure 4(d) shows that LiBF4 has low coulombic efficiency up to 30 cycles, suggesting the side reaction is not sufficiently suppressed. In contrast, HHIB-F2 showed a coulombic efficiency of >99% after the 10th cycle, confirming the formation of the SEI by HHIB-F2, which can effectively suppress side reactions. The effect of SEI from borates was further confirmed by the AC impedance spectroscopy using coin cells (figure S10(d)). HHIB2 shows the largest semi-circle, indicating high resistance to SEI due to reductive decomposition products. Conversely, HHIB-F2 exhibited the smallest semi-circle, indicating the SEI component that increases resistance has been reduced and/or the Li+ conductivity of SEI has been improved by HHIB-F2. To confirm the cause of the difference in resistance, x-ray photoelectron spectroscopy (XPS) was performed to analyze the surface of the negative electrode after the pre-charge/discharge (figure S11). HHIB-F2-derived SEI contains a trace amount of CF3 and B-F, while HHIB2-derived SEI mainly consists of Li2O. Although the details are still not clear, this difference in the composition may be responsible for the resistance of SEI, which may affect the battery performance.
3. Conclusions
Five lithium borates (PFP-F2, PFP-Ox, HHIB2, HHIB-F2, and HHIB-Ox) were synthesized from trimethylchlorosilane, three bidentate ligands (PFP, HHIB, Ox), and boron sources (LiBF4, LiDFOB) as promising electrolytes for lithium-ion batteries. These borates were thermally stable up to 140 °C, and symmetric bis-chelated borates, HHIB2, with two HHIB ligands, showed the best thermal stability up to 350 °C because of the strong inductive effect of the CF3 group and resonance stabilization effect of the carbonyl group. The hydrolysis resistance was also improved by CF3 group introduction; in particular, PFP-F2 and HHIB2 showed negligible degradation after 2 day in an aqueous solution. The electrochemical windows of the PFP-F2, PFP-Ox, HHIB2, HHIB-F2, and HHIB-Ox electrolytes are comparable to or even wider than that of BF4-containing electrolytes, confirming the applicability of lithium borates as lithium-ion battery electrolytes.
The 11B NMR and Raman spectroscopic measurements confirmed that the electron density at the boron center correlate well with Li+ dissociability. Li+ mobility is related to the solution viscosity, which is determined by the anion molecular weight and size. HHIB-F2 showed the best ionic conductivity of 7.1 mS cm−1, nearly a two-fold increase compared to conventional BF4 anions, by incorporating a good balance of anion size reduction (increased Li+ mobility) and CF3 group contribution (increased Li+ dissociation).
Finally, the charge/discharge properties of lithium-ion batteries with HHIB-F2 and HHIB2, which showed the highest ionic conductivity among the salts tested, and higher ionic conductivity than LiBF4 and higher hydrolysis resistance than LiBF4, respectively. The lithium-ion batteries with HHIB-F2 and HHIB2-containing electrolytes exhibited a larger 1st cycle capacity and superior cycling stability compared to LiBF4 under accelerated test conditions (60 °C, 3C). This study clarifies the influence of the complex structure (number of CF3 groups, presence or absence of C=O or B–F bonds) on the electrochemical and physical properties of the electrolyte, providing a reliable and promising approach for high-performance electrolyte development.
4. Experimental
4.1. Lithium borate syntheses
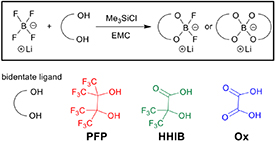
Both mono- (BF2 type; PFP-F2, HHIB-F2, Ox-F2) and bis-chelated lithium borates (PFP2, HHIB2) were synthesized using LiBF4 (KISHIDA CHEMICAL, battery grade) and corresponding bidentate ligands (perfluoropinacol, 2-hydroxy-3,3,3,3ʹ,3ʹ,3ʹ-hexafluoroisobutiric acid, oxalic acid) as starting materials. Perfluoropinacol (>98% purity) was purchased from TOKYO CHEMICAL INDUSTRY and oxalic acid was obtained by drying oxalic acid dihydrate (>99%), purchased from FUJIFILM Wako Pure Chemical, at 90 °C under reduced pressure. In addition, 2-hydoxy-3,3,3,3ʹ,3ʹ,3ʹ-hexafluoroisobutiric acid was synthesized according to a literature procedure [60]. Anhydrous alkyl carbonates such as EMC or DMC (KISHIDA CHEMICAL, battery grade) were selected as reaction solvents. Trimethylchlorosilane (Me3SiCl, TOKYO CHEMICAL INDUSTRY, >98%) was used as a defluorination reagent to minimize residual chlorine. The molar ratio between Me3SiCl and the ligand dictates the final product structure; bis-chelated lithium borates were mainly obtained using four equivalents of Me3SiCl and two equivalents of ligand, while mono-chelated lithium borates were obtained using two equivalents of Me3SiCl and one equivalent of ligand. Asymmetric bis-chelated lithium borates (PFP-Ox, HHIB-Ox) were synthesized by treating Me3SiCl with Ox-F2 and the corresponding ligand (perfluoropinacol, 2-hydoxy-3,3,3,3ʹ,3ʹ,3ʹ-hexafluoroisobutiric acid). The general synthetic method for lithium borate is shown in Scheme 1. The individual synthesis steps are shown in the SI, and all manipulations were performed under nitrogen atmosphere with a dew point of ⩽−50 °C. The lithium borate Ox2 (LiBOB) was not synthesized and was purchased from Albemarle with a purity of ⩾99%.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Scheme 1. General synthetic method.
Download figure:
Standard image High-resolution image{kind=link}
4.2. Analytical methods
The 1H, 7Li, 11B, 13C, and 19F NMR spectra used for characterization were recorded using a JEOL JNM-ECZ400S spectrometer in deuterated acetonitrile (CD3CN) at ambient temperature. HRMS were recorded using an LTQ Orbitrap Discovery (Thermo Fisher Scientific) mass spectrometer in ESI mode. TG and DTA were performed with equipment from Rigaku (Thermo plus TG8120) using 10 mg samples at a heating rate of 10 °C min−1 under a nitrogen atmosphere. LSV data were collected at a scan rate of 5 mV s−1 with an electrochemical analyzer (ALS-604E, BAS). Glassy carbon was used as the working electrode and lithium metal was used as both the counter and reference electrodes. The ionic conductivity was measured using a conductivity meter with a submersible-type cell (HORIBA, ES-51/3552-10D) at 30 °C. The cell constant was calibrated before use using a potassium chloride standard solution (TAKEMURADENKISEISAKUSHO, 1,413 μS cm−1). Raman spectra were collected using equipment from RENISHAW (inVia Raman microscope WiRE5) equipped with an argon laser emitting a 514.5 nm line. The samples were placed in quartz cells and self-diffusivity data were measured using the pulsed field gradient spin-echo diffusion method [48, 61, 62]. PGSE NMR measurements were performed using a JEOL JNM-ECZ400S spectrometer. The diffusion coefficients of the anions, lithium, and solvents were measured at 30 °C using 19F, 7Li, and 1H NMR, respectively. The interval between the gradient pulses (Δ) was 50 ms and the maximum magnetic field gradient (g) was 0.3 T m−1. Tubes were purchased from SIGEMI (BMS-005J) and used to seal the samples. 11B NMR was used to measure the lithium bis(oxalato)borate anions. However, the echo signal did not decay under these conditions and attempts to obtain the self-diffusivity value were unsuccessful. Therefore, only the bis(oxalato)borate anion was measured using a JEOL JNM-ECA400 spectrometer with Δ = 25 ms and maximum g = 12.0 T m−1. A rheometer MCR102 (Anton Paar) was used for viscosity measurements, and calibration was performed before use using a standard calibration solution (Nippon Grease, JS2.5, and JS10). The HOMO and LUMO energy levels were determined using density functional theory with the functional B3LYP with the 6–31Gdp basis set in Gaussian 09 program. XPS (PHI 5000 VersaProbe II, ULVAC-PHI, Japan) measurements were carried out using Al Kα radiation (1486.6 eV) under ultrahigh vacuum. The samples were transferred from the Ar-filled glove-box to the XPS chamber using a transfer vessel to minimize exposure to air. A charge neutralizer was employed for all measurements, and the graphite peak at 284.3 eV was used as a reference for energy scale adjustment. The obtained spectra were analyzed by Multipack software (v. 9.6.0.15). Line syntheses of elemental spectra were conducted using Gaussian–Lorentzian (80:20) curve fitting with Shirley background subtraction.
4.3. Coin cell assembly and electrochemical tests
Battery-grade EC was purchased from KISHIDA CHEMICAL. The electrolyte consisted of carbonate solutions EC/EMC (1/2 vol/vol) and electrolytes (PFP-F2, HHIB-F2, HHIB2, and LiBF4). The electrolyte concentration was 1.0 M, and the composite NMC111 cathode consisted of 90.2 wt% LiNi0.33Mn0.33Co0.33O2 active material (MX-6; Umicore), 3.8 wt% conductive carbon (HS-100; Denka), and 6.0 wt% polyvinylidene difluoride (PVDF) binder (L#7208; KUREHA). The composite graphite anode contained 90.0 wt% graphite (MAG-D; Hitachi Chemical) along with a 10.0 wt% PVDF binder (L#9130; KUREHA). A cellulose separator (TF40-30) was purchased from NIPPON KODOSHI. The active mass loading of the cathodes and anodes was 12.2 and 6.7 mg cm−2, respectively. Graphite/NMC111 full 2032-type coin cells were built with a cathode (d = 10.0 mm), anode (d = 12.0 mm), separator (d = 16.0 mm), and 40 μl of electrolyte solution in each cell prepared in a glove box filled with argon (content of H2O below 1 ppm) for electrochemical performance measurements. The assembled graphite/NMC111 cells were charged at a constant current 0.2 C to 4.3 V (CC–CV mode) at 25 °C. After resting for 1 h, the charged cells were discharged in CC mode (0.2 C) to 3.0 V at 25 °C. This process is called the pre-charge/discharge. The cycling tests were conducted under the following conditions. The cells were charged to 4.3 V in CC–CV mode (3C) at 60 °C, and after resting for 1 min at 60 °C, the charged cells were discharged to 3.0 V in CC mode (3 C) at 60 °C. The charge/discharge cycle was repeated 100 times. After the pre-charge/discharge (i.e. before cycling), the cells were probed by electrochemical impedance spectroscopy (EIS; ALS-660 C electrochemical analyzer, BAS, Japan) at 25 °C and at a state of charge of 100% (25 °C, 0.2 C charging) using an amplitude of 10 mV and a frequency range of 100 kHz to 10 mHz.
Acknowledgments
The authors would like to thank Katsumasa Mori and Hayato Hesaka for technical assistance with the experiments.
Data availability statement
The data that support the findings of this study are available upon reasonable request from the authors.
Conflict of interest
There are no conflicts to declare.
Supplementary data (2.1 MB PDF)