

Abstract
Recent advances in the development of exchange and correlation functionals to be employed in density functional theory calculations combined with the availability of ever more powerful high-performance computing facilities, let predictive computational materials science become reality. In order to assess the quality of calculated material properties, Jacob's ladder provides an informal classification, where exchange and correlation functionals of similar capabilities are placed at the same rung of this ladder, while improved and more accurate ones are placed at higher rungs. Climbing Jacob's ladder, i.e. employing more accurate exchange and correlation functionals, increases the quality of the results and the computational demands, and provides some guidance as to what accuracies and computational costs to expect from specific calculations. This is particular important for materials whose electronic ground state properties are incorrectly described, e.g. small band gap semiconductors, and materials, where system sizes for subsequent investigations, like defect properties or band offsets in heterostructures, become prohibitively large for more accurate exchange and correlation functionals.
Here, we provide a systematic density functional theory study on the ground state properties of Ag2ZnSnSe4 and Cu2ZnSnSe4 for the lowest four rungs of Jacob's ladder. Cu2ZnSnSe4, and in particular its alloys with Ag, is a promising candidate material for future thin-film solar cell absorber layers. In the present work, the obtained material properties are compared to available experimental data, allowing to benchmark the accuracy of the employed exchange and correlation functionals. We also provide a comparative study for subsequent quasiparticle calculations. Therein, the influence of differently obtained eigenvalues and orbitals as starting points are critically assessed with respect to available experimental data. Our results show that, structural properties based on the SCAN functional show overall best agreement with available experimental data, whereas additional hybrid functional calculations are necessary for satisfying results on electronic and optical properties.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Novel photovoltaic materials based on the kesterite crystal structure are ingredients for third generation thin-film solar cells based only on earth-abundant and non-toxic elements. Prominent examples include Cu2ZnSnS4( CZTS), Cu2ZnSnSe4( CZTSe), and their solid solution Cu2ZnSn(Sx Se1 − x )4( CZTSSe), where the latter one allows to continuously tune the electronic band gap between around 1.0 eV for CZTSe [1, 2] and 1.53–1.67 eV for CZTS [3], respectively, thereby including the optimum band gap of 1.34 eV for single junction photovoltaic cells [4]. Subsequently, the overall potential of CZTSSe based solar cell devices has been demonstrated with a reported power conversion efficiency of 12.6% [5]. Following up on the question posed very early on, of why kesterite solar cells are not 20% efficient [6], the last years have seen a lot of experimental investigations into the structural, electronic, and optical properties of kesterite based solar cell materials, as well as theoretical investigations based on first-principles calculations employing density functional theory (DFT) for a comprehensive atomic understanding.
These combined experimental and theoretical efforts have identified the most likely cause for limiting further advances as a large defect density of Cu-Zn antisites [7], either intrinsic Cu or Zn
or Zn antisite defects or Cu
antisite defects or Cu –Zn
–Zn defect complexes observed for the Cu–Zn disorder [8]. Looking at the final device geometry, i.e. a CZTSSe/CdS interface for a p–n junction, these defects are accumulating at the interface region, thereby pinning the Fermi level in the middle of the band gap [7]. Subsequently this leads to only a small band bending into the absorber layer and a large deficit in the open-circuit voltage (
defect complexes observed for the Cu–Zn disorder [8]. Looking at the final device geometry, i.e. a CZTSSe/CdS interface for a p–n junction, these defects are accumulating at the interface region, thereby pinning the Fermi level in the middle of the band gap [7]. Subsequently this leads to only a small band bending into the absorber layer and a large deficit in the open-circuit voltage ( ) [9]. Experimentally, this can be verified by the difference in the band gap determined from internal quantum efficiency and photoluminescence measurements [10].
) [9]. Experimentally, this can be verified by the difference in the band gap determined from internal quantum efficiency and photoluminescence measurements [10].
In order to significantly reduce the Cu–Zn antisite defect density, a partial replacement of Cu with larger Ag cations has been proposed [11, 12], based on promising results on kesterite type Ag2ZnSnSe4( AZTSe) [13, 14]. DFT calculations indicate an increase in the formation energy of I–II antisite defects, leading to a significant reduction in I-II antisite defect density by an order of magnitude and subsequently a reduced band tailing [9, 15].
From a first-principles perspective, kesterite type materials pose a very challenging task. First of all, they are quaternary materials with a complicated interplay of interactions among the atoms. Secondly, as potential absorber layers in solar cell devices, they have band gaps in the range of 1.0–1.5 eV, which are notoriously difficult to treat accurately by DFT methods due to an underestimation of the band gap in simpler exchange and correlation functionals. One way to circumvent this particular band gap problem is to employ more accurate exchange and correlation functionals, i.e. hybrid functionals, which incorporate some fraction of Hartree–Fock exact exchange and have been shown to yield better electronic properties of semiconducting materials. Another way would be to perform additional quasiparticle investigations based on many-body perturbation theory, i.e. subsequent GW calculations. While those two approaches can routinely be applied to the bulk properties of kesterite type materials, an increased demand of computational resources prohibits their use for subsequent material properties investigations, e.g. formation energies of defects and defect complexes or the calculation of band offsets in modelled device heterostructures.
To this end, a well tested and proven theoretical approach would be highly desirable, providing a guide towards a useful combination of available exchange and correlation functionals together with their overall accuracy and their demands on computational resources. Here, we provide a detailed first-principles investigation on the structural, electronic, and optical properties of AZTSe and CZTSe in both, the kesterite and stannite crystal structure, and benchmark the results obtained from exchange and correlation functionals of increasing complexity with respect to available experimental data.
The paper is organised as follows. Section 2 provides information on the structural polymorphs of AZTSe and CZTSe, as well as the necessary theoretical background for first-principles calculations, the employed exchange and correlation functionals, and important computational details. In a first step, in section 3 the obtained results on the structural, electronic, and optical properties are compared with and benchmarked against available experimental data. Results from subsequent quasiparticle calculations based on the GW approximation will be discussed as well. In a second step, the overall performance of the employed exchange and correlation functionals will be critically discussed, leading to suggestions for material properties investigation, such as the calculation of defects and defect complexes or band offsets in modelled heterostructures. Finally, section 4 provides a short summary and conclusion, and gives an outlook into future investigations.
2. Background and computational details
2.1. Structural polymorphs
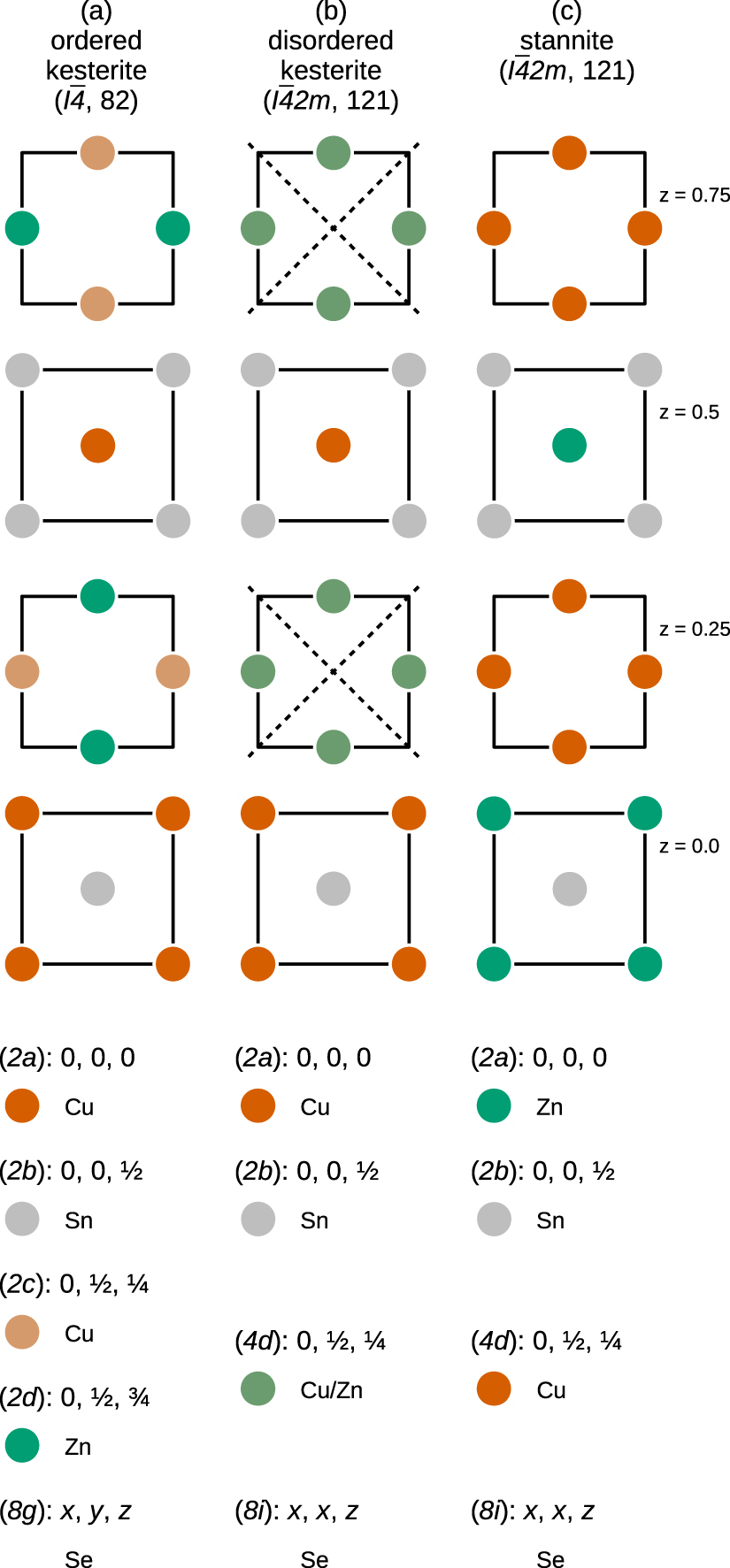
At ambient condition AZTSe and CZTSe crystallise in the kesterite crystal structure (space group  , no. 82), as shown in figure 1(a). The kesterite crystal structure can be most easily understood as originating from a binary II–VI zinc-blende crystal structure via pairwise cation substitutions, with the restriction that the bonding to adjacent cations has to fulfil the so-called octet rule, i.e. the valence shell of each atom comprises eight electrons. Starting from a II–VI zinc-blende material, doubling the unit cell along the c axis, and additionally replacing the group-II element by a pair of group-I and group-III elements, can lead to ternary I–III–VI2 compounds of the chalcopyrite and the CuAu-like structure, respectively. A further replacement of the group-III elements by pairs of group-II and group-IV elements in the chalcopyrite structure leads to the I2–II–IV–VI4 kesterite crystal structure. However, performing the same replacement starting from the CuAu-like structure can lead to the I2–II–IV–VI4 stannite crystal structure (space group
, no. 82), as shown in figure 1(a). The kesterite crystal structure can be most easily understood as originating from a binary II–VI zinc-blende crystal structure via pairwise cation substitutions, with the restriction that the bonding to adjacent cations has to fulfil the so-called octet rule, i.e. the valence shell of each atom comprises eight electrons. Starting from a II–VI zinc-blende material, doubling the unit cell along the c axis, and additionally replacing the group-II element by a pair of group-I and group-III elements, can lead to ternary I–III–VI2 compounds of the chalcopyrite and the CuAu-like structure, respectively. A further replacement of the group-III elements by pairs of group-II and group-IV elements in the chalcopyrite structure leads to the I2–II–IV–VI4 kesterite crystal structure. However, performing the same replacement starting from the CuAu-like structure can lead to the I2–II–IV–VI4 stannite crystal structure (space group  , no. 121), which is shown in figure 1(c).
, no. 121), which is shown in figure 1(c).
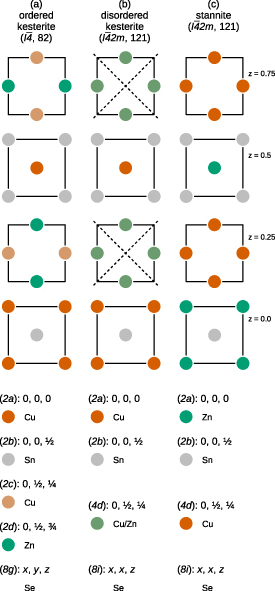
Figure 1. Cu, Zn, and Sn cation planes with z = 0.0, 0.25, 0.5 and 0.75 in the (a) ordered kesterite (space group  , no. 82), (b) disordered kesterite (space group
, no. 82), (b) disordered kesterite (space group  , no. 121), and (c) stannite (space group
, no. 121), and (c) stannite (space group  , no. 121) crystal structures, respectively. The dashed lines in the disordered kesterite structure (b) denote the additional symmetry elements due to the totally random distribution of Cu and Zn cations in the z = 0.25 and z = 0.75 planes. The lower parts list the colour-coded Wyckoff positions for each of the structural polymorphs.
, no. 121) crystal structures, respectively. The dashed lines in the disordered kesterite structure (b) denote the additional symmetry elements due to the totally random distribution of Cu and Zn cations in the z = 0.25 and z = 0.75 planes. The lower parts list the colour-coded Wyckoff positions for each of the structural polymorphs.
Download figure:
Standard image High-resolution imageTo make matters worse, the ionic radii of Cu and Zn are nearly identical, giving rise to a high probability of cation exchange in the Cu–Zn planes perpendicular to the crystallographic c axis, located at z = 0.25 and z = 0.75. While partial exchange of Cu and Zn cations leads to an increase in the Cu and Zn
and Zn defect density, a totally random distribution of Cu and Zn cations within the Cu–Zn planes leads to the introduction of additional symmetry elements and has therefore been named disordered kesterite structure, shown in figure 1(b). In terms of structural investigations this disordered kesterite structure crystallises in the same space group as the stannite crystal structure, and has hindered the correct crystal structure determination of possible solar absorber materials for some time.
defect density, a totally random distribution of Cu and Zn cations within the Cu–Zn planes leads to the introduction of additional symmetry elements and has therefore been named disordered kesterite structure, shown in figure 1(b). In terms of structural investigations this disordered kesterite structure crystallises in the same space group as the stannite crystal structure, and has hindered the correct crystal structure determination of possible solar absorber materials for some time.
2.2. Theoretical background
In recent years, first-principles calculations based on DFT have become a very powerful tool for materials science investigations, not only due to the development of ever more accurate exchange and correlation functionals, but also due to the wide-spread availability of local, regional, and national high-performance computing (HPC) facilities, providing the necessary computational resources for investigations of increasing complexities. With the constant development of improved exchange and correlation functionals to be employed in first-principles investigations, for somebody not that familiar with the topic it became more and more difficult to judge the accuracy and reliability of the reported results. In order to allow for some guidance, the so-called Jacob's ladder can provide a first hint [16], as adopted in figure 2.

Figure 2. Jacob's ladder, placing exchange and correlation functionals of similar capabilities at the same rung [16].
Download figure:
Standard image High-resolution imageHere, exchange and correlation functionals of similar capabilities are grouped together at the same rung of this ladder, while improved and more accurate ones are placed at a higher rung. Functionals of the local density approximation (LDA) can be found at the lowest rung and are based solely on the electron density, the more common examples include the parametrisations after Perdew and Zunger [17] and Perdew and Wang [18]. The next rung depicts the generalised gradient approximation (GGA) functionals, which additionally take into account the gradient of the electron density. Examples include the parametrisations after Perdew, Burke, and Ernzerhof (PBE) [19], the PBE parametrisation revised for solids (PBEsol) [20], and the parametrisation after Armiento and Mattsson (AM05) [21], respectively. Additional inclusion of the second derivative of the electron density (the Laplacian) and the orbital kinetic energy density leads to the rung of the so-called meta-GGAs, with the SCAN functional [22] being a prominent example. This SCAN functional satisfies all known possible exact constraints for the exact density functional, and has been claimed to match or improve on the accuracy of computationally more demanding hybrid functionals [23].
The mentioned hybrid functionals replace a pre-defined fraction of the underlying exchange and correlation energy by a Hartree–Fock exact exchange term. Prominent examples include the parametrisations after Heyd, Scuseria, and Ernzerhof [24], or the PBE0 functional [25]. Recent years witnessed a surge in investigations trying to improve hybrid functional investigations by adjusting the pre-defined fraction of Hartree–Fock exact exchange, ultimately being determined in a fully self-consistent manner [26, 27].
On the one hand, by climbing Jacob's ladder and employing ever more sophisticated exchange and correlation functionals, we expect to obtain improved accuracy from our first-principles calculations, as indicated by the right arrow in figure 2. On the other hand, this is accompanied by an increase in computational resources, as depicted by the left arrow in figure 2. However, with the more wide-spread availability of HPC facilities, hybrid functional investigations became more and more common in recent years and led to a better description of structural, electronic, and optical properties of a range of materials [28–31].
Apart from the calculation of electronic and optical properties by means of exchange and correlation functionals of four distinct rungs of Jacob's ladder as described above, we also performed quasiparticle calculations of those material properties based on the GW approximation introduced by Hedin [32]. While the GW approximation remains the most accurate method for quasiparticle calculations available, its numerical requirements and perturbative nature make approximations unavoidable [33–37].
From a numerical requirements' point of view several flavours of practical GW calculations have been established over the years. The most simple one is the single-shot  method, with G0 being the noninteracting Green's function of the system and W0 its screened Coulomb interaction. Extensions iterate the eigenvalues to self-consistency in the Green's function alone (GW0), or also in the screened interaction W( GW). Methodical extensions see the inclusion of vertex corrections as well [38], however, this is beyond the scope of the present work.
method, with G0 being the noninteracting Green's function of the system and W0 its screened Coulomb interaction. Extensions iterate the eigenvalues to self-consistency in the Green's function alone (GW0), or also in the screened interaction W( GW). Methodical extensions see the inclusion of vertex corrections as well [38], however, this is beyond the scope of the present work.
From a perturbative nature's point of view, all mentioned flavours of GW calculations depend on the starting one-electron energies and orbitals, usually taken as Kohn-Sham eigenenergies and orbitals from preceding DFT calculations. This immediately implies that the accuracy of GW calculations will be influenced by the accuracy of the starting one-electron energies and orbitals, as will be discussed later in the results section.
2.3. Computational details
All the calculations of the present work have been performed using the Vienna ab initio Simulations package (VASP, 5.4.4) [39–41] together with the projector-augmented wave (PAW) method [42, 43]. Structural relaxations employed the recommended PAW potentials supplied by VASP that provided 17, 17, 12, 14, and 6 valence electrons for the Cu, Ag, Zn, Sn, and Se atoms, respectively. The electronic band structures, the optical properties, and the many-body perturbation theory calculations based on the GW approximation made use of the PAW potentials recommended for GW calculations, and provided 19, 19, 20, 14, and 6 valence electrons for the Cu, Ag, Zn, Sn, and Se atoms instead.
In order to judge the accuracy and computational efficiency of several rungs of Jacob's ladder, we employed the following exchange-correlation potentials: for the LDA functional we chose the Perdew and Zunger parametrisation [17, 44], for the GGA functional we chose the Perdew, Burke, and Ernzerhof implementation revised for solids (PBEsol) [20], as meta-GGA functional we chose the relatively new SCAN functional [22], and finally as hybrid functional we chose the range-separated HSE06 functional [24], respectively.
Structural relaxations of the kesterite and stannite crystal structures have been performed for the standard primitive unit cells, containing one formula unit (f.u.) for both structural polymorphs. The ground state structures have been optimised by analysing the total energy curves, which have been obtained for several volumes around the experimentally known ground state volume. Keeping these volumes fixed, the respective lattice constants and all internal coordinates have been allowed to relax until the forces on all atoms were below 0.001 eV Å–1. While the ground state volume V0 has been obtained by a spline fit to the total energy curves, a subsequent analysis via Murnaghan's equation of state [45, 46]

additionally yielded the bulk modulus B0 and its pressure derivative  , respectively. A final relaxation of internal coordinates has been performed for the ground state volumes, yielding the overall ground states that have been further analysed using the AFLOW [47] and FINDSYM [48, 49] packages.
, respectively. A final relaxation of internal coordinates has been performed for the ground state volumes, yielding the overall ground states that have been further analysed using the AFLOW [47] and FINDSYM [48, 49] packages.
The obtained relaxed ground state structures served as a starting point for subsequent calculations of the electronic band structures and the real and imaginary parts of the dielectric functions, which have been obtained by summing over empty states using Fermi's Golden Rule, transition matrix elements, and applying a Kramers-Kronig transformation [50]. In order to ensure converged results the number of empty bands in the calculations of the optical properties have been increased by a factor of four. The final real and imaginary parts of the dielectric functions have been obtained by diagonalising the dielectric tensors for every energy point and averaging over the resulting main diagonal elements, as applied before to non-cubic oxide [51] and amorphous materials [52, 53].
Together with the other technical parameters, k-point grid of 6 × 6 × 6, cut-off energy of the plane-wave expansion of 500 eV, and a convergence criteria for the total energy of 10−6 eV, this ensured well-converged results. Due to the increased numerical demand the k-point grid has been reduced to 4 × 4 × 4 for the GW calculations.
3. Results and discussion
3.1. Structural properties
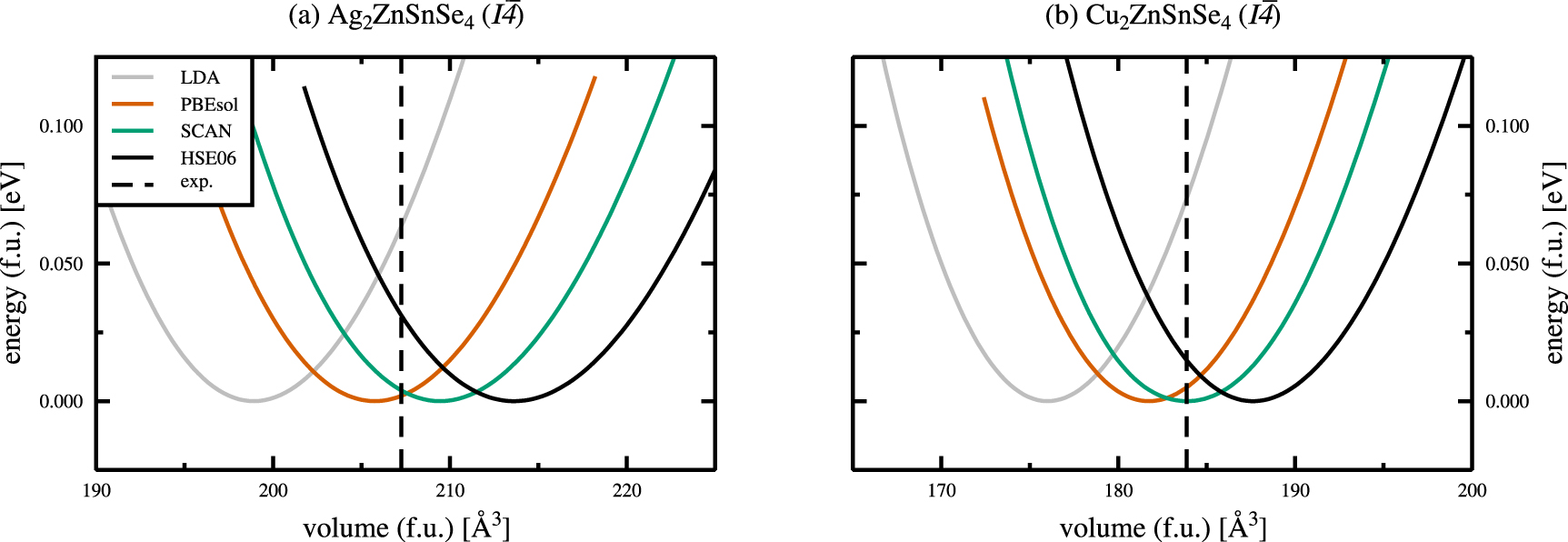
The total energy curves for various employed exchange and correlation functionals, normalised to one functional unit (f.u.) and rescaled to zero energy, are shown in figure 3 for the kesterite crystal structures of AZTSe (a) and CZTSe (b), respectively. The dashed vertical lines indicate the experimental ground state volumes taken from a combined x-ray and neutron powder diffraction study of Gurieva et al[ 2]. The respective total energy curves for the stannite crystal structures of AZTSe and CZTSe can be found in the appendix (figure A1).

Figure 3. Total energy curves for the kesterite crystal structures of Ag2ZnSnSe4( a) and Cu2ZnSnSe4( b), calculated using various exchange and correlation functionals. The dashed lines (- - - -) indicate the experimental values of Gurieva et al[ 2], and all energies are normalised to one functional unit (f.u.) and rescaled to zero energy, respectively.
Download figure:
Standard image High-resolution image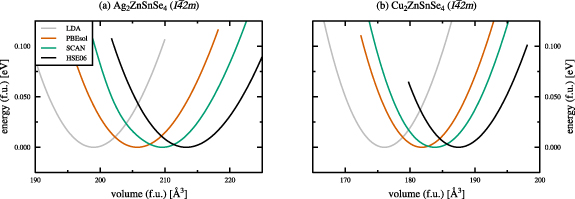
Figure A1. Total energy curves for the stannite crystal structures of Ag2ZnSnSe4( a) and Cu2ZnSnSe4( b), calculated using various exchange and correlation functionals. All energies are normalised to one functional unit (f.u.) and rescaled to zero energy, respectively.
Download figure:
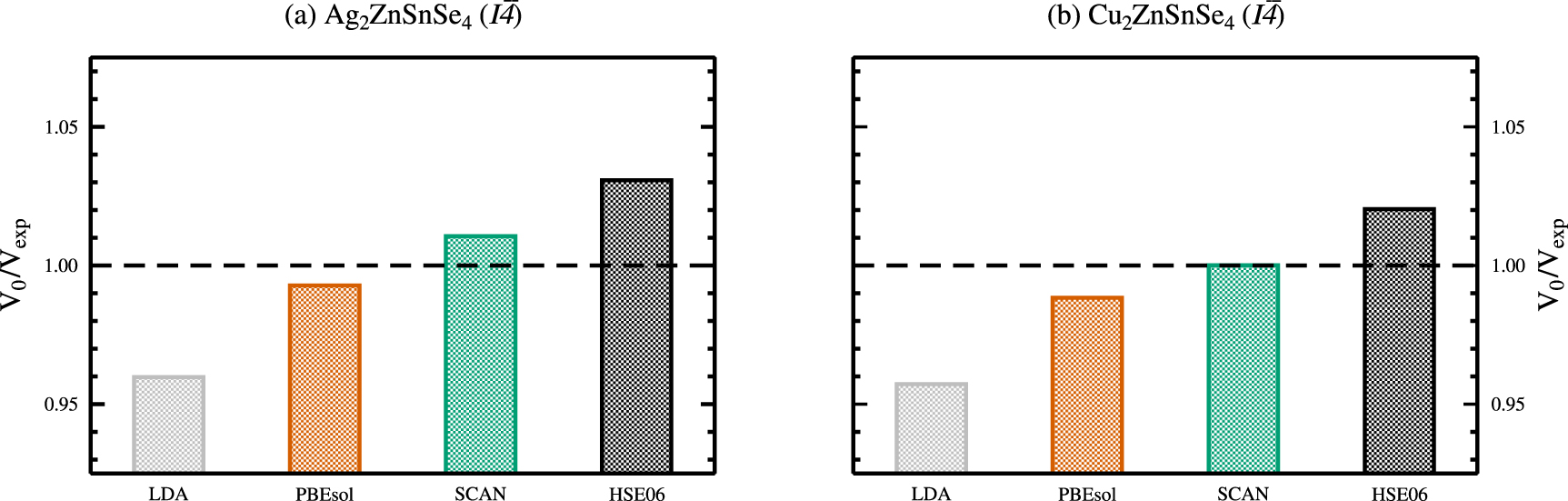
Standard image High-resolution imageThe obtained ground state structural properties of the kesterite and stannite crystal structures of AZTSe and CZTSe are given in tables 1 and 2 for various exchange and correlation functionals in comparison to experimental data, respectively. For both, kesterite-type AZTSe and CZTSe, the LDA calculated ground state volume is severely underestimated by approximately 4%, as shown in figures 3 and 4. Deviations from the experimental ground state volumes of about 1% are obtained for the PBEsol and the SCAN functionals, with a much better agreement of the SCAN functional in case of kesterite-type CZTSe. The hybrid functional HSE06 overestimates the experimental ground state volumes by 2%–3%, respectively.
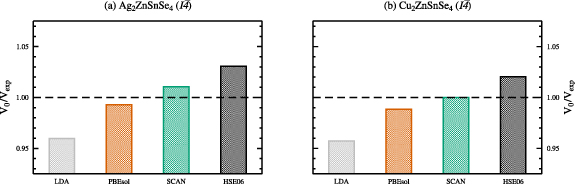
Figure 4. Ground state unit cell volumes V0 with respect to the experimental volume  of Gurieva et al[ 2] for the kesterite crystal structures of Ag2ZnSnSe4( a) and Cu2ZnSnSe4( b), calculated using various exchange and correlation functionals. The experimental volumes correspond to
of Gurieva et al[ 2] for the kesterite crystal structures of Ag2ZnSnSe4( a) and Cu2ZnSnSe4( b), calculated using various exchange and correlation functionals. The experimental volumes correspond to  (dashed horizontal line).
(dashed horizontal line).
Download figure:
Standard image High-resolution imageTable 1. Ground state structural properties (ground
state volume V0, lattice constants a0 and c0, fractional coordinates x, y, and z of the anion Wyckoff position 8g, and bulk modulus B0 and its pressure derivative  ) obtained by various exchange and correlation functionals in comparison to experimental data for the kesterite crystal structures of Ag2ZnSnSe4 and Cu2ZnSnSe4.
) obtained by various exchange and correlation functionals in comparison to experimental data for the kesterite crystal structures of Ag2ZnSnSe4 and Cu2ZnSnSe4.
| Ag2ZnSnSe4 | V0( Å3) | a0( Å) | c0( Å) | x | y | z | B0( GPa) |

|
|---|---|---|---|---|---|---|---|---|
| LDA | 198.912 | 5.916 | 11.365 | 0.262 | 0.241 | 0.131 | 62.8 | 5.0 |
| PBEsol | 205.756 | 5.987 | 11.479 | 0.264 | 0.242 | 0.130 | 56.1 | 4.9 |
| SCAN | 209.448 | 6.072 | 11.362 | 0.269 | 0.243 | 0.130 | 53.5 | 5.0 |
| HSE06 | 213.628 | 6.096 | 11.496 | 0.272 | 0.246 | 0.128 | 49.3 | 5.0 |
| exp. [2] | 207.262 | 6.046 | 11.340 | |||||
| exp. [11] | 205.866 | 6.036 | 11.301 | 0.251 | 0.231 | 0.128 | ||
| Cu2ZnSnSe4 | ||||||||
| LDA | 176.011 | 5.605 | 11.207 | 0.228 | 0.239 | 0.131 | 72.9 | 4.8 |
| PBEsol | 181.736 | 5.665 | 11.327 | 0.229 | 0.239 | 0.131 | 66.1 | 5.0 |
| SCAN | 183.887 | 5.695 | 11.340 | 0.232 | 0.240 | 0.130 | 64.3 | 5.2 |
| HSE06 | 187.615 | 5.732 | 11.422 | 0.239 | 0.244 | 0.129 | 59.6 | 5.0 |
| exp. [1] | 183.976 | 5.695 | 11.345 | |||||
| exp. [11] | 183.702 | 5.692 | 11.340 | 0.251 | 0.240 | 0.128 | ||
| exp. [2] | 183.880 | 5.693 | 11.347 |
Table 2. Ground state structural properties (ground state volume V0, lattice constants a0 and c0, fractional coordinates x and z of the anion Wyckoff position 8i, and bulk modulus B0 and its pressure derivative
 ) obtained by various exchange and correlation functionals for the stannite crystal structures of Ag2ZnSnSe4 and Cu2ZnSnSe4.
) obtained by various exchange and correlation functionals for the stannite crystal structures of Ag2ZnSnSe4 and Cu2ZnSnSe4.
| Ag2ZnSnSe4 | V0( Å3) | a0( Å) | c0( Å) | x | z | B0( GPa) |

|
|---|---|---|---|---|---|---|---|
| LDA | 199.001 | 5.724 | 12.149 | 0.759 | 0.879 | 62.4 | 4.6 |
| PBEsol | 205.720 | 5.776 | 12.334 | 0.758 | 0.880 | 55.7 | 5.0 |
| SCAN | 209.603 | 5.768 | 12.599 | 0.758 | 0.882 | 53.6 | 3.3 |
| HSE06 | 213.282 | 5.800 | 12.681 | 0.754 | 0.884 | 49.6 | 4.9 |
| Cu2ZnSnSe4 | |||||||
| LDA | 176.051 | 5.607 | 11.198 | 0.761 | 0.863 | 72.8 | 4.7 |
| PBEsol | 181.793 | 5.667 | 11.320 | 0.760 | 0.864 | 66.0 | 4.9 |
| SCAN | 183.838 | 5.677 | 11.407 | 0.760 | 0.865 | 64.7 | 5.0 |
| HSE06 | 187.463 | 5.706 | 11.514 | 0.756 | 0.868 | 60.1 | 5.0 |
The obtained ground state volumes increase with the increasing sophistication of the employed exchange and correlation functionals (LDA, PBEsol, SCAN, and HSE06). This trend is equally observed for the lattice parameters a and c for both materials, AZTSe and CZTSe, and for both crystal structures, given for kesterite in table 1 and for stannite in table 2, respectively. The bulk modulus B0 shows the same decreasing trend for both materials and crystal structures, and its pressure derivative is mostly around 5.
The fractional coordinates of the anion Wyckoff positions show a complementary trend, i.e. increasing (decreasing) for the kesterite-type 8g fractional coordinates x and y( z), and decreasing (increasing) for the stannite-type 8i fractional coordinates x( z), respectively. The performance of various exchange and correlation functionals in describing fractional coordinates will become important in comparison to precise experimental local structure investigations, and has been shown to substantially influence the electronic band gaps in CZTS and CZTSe [54].
3.2. Electronic and optical properties
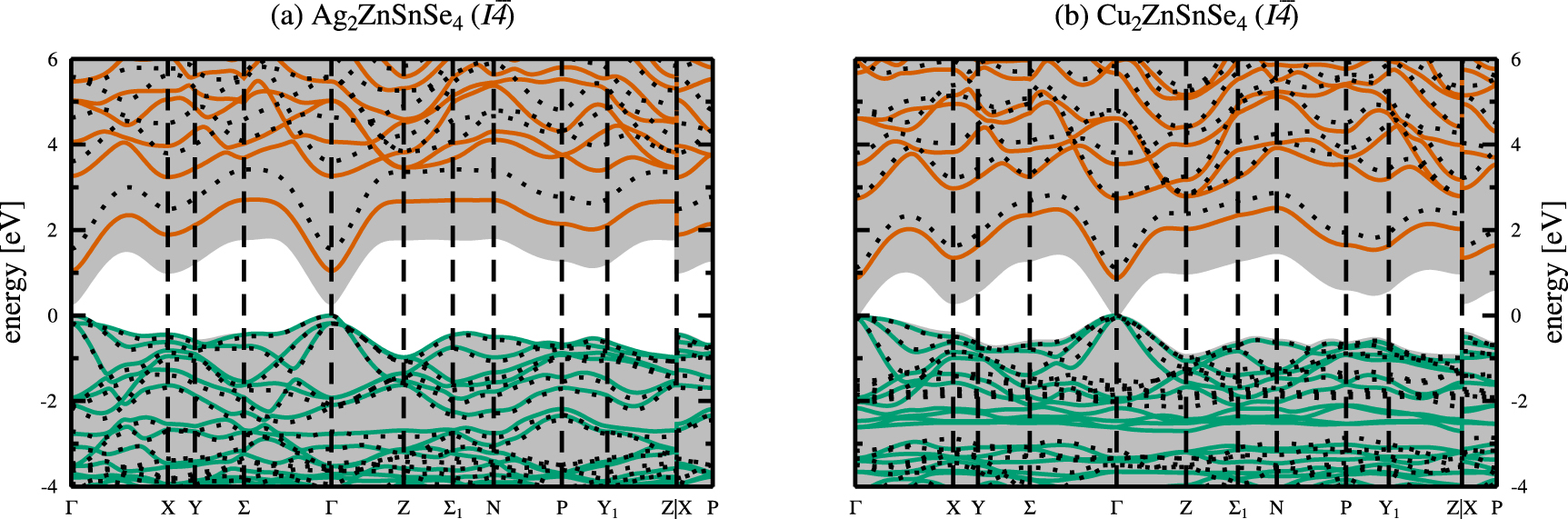
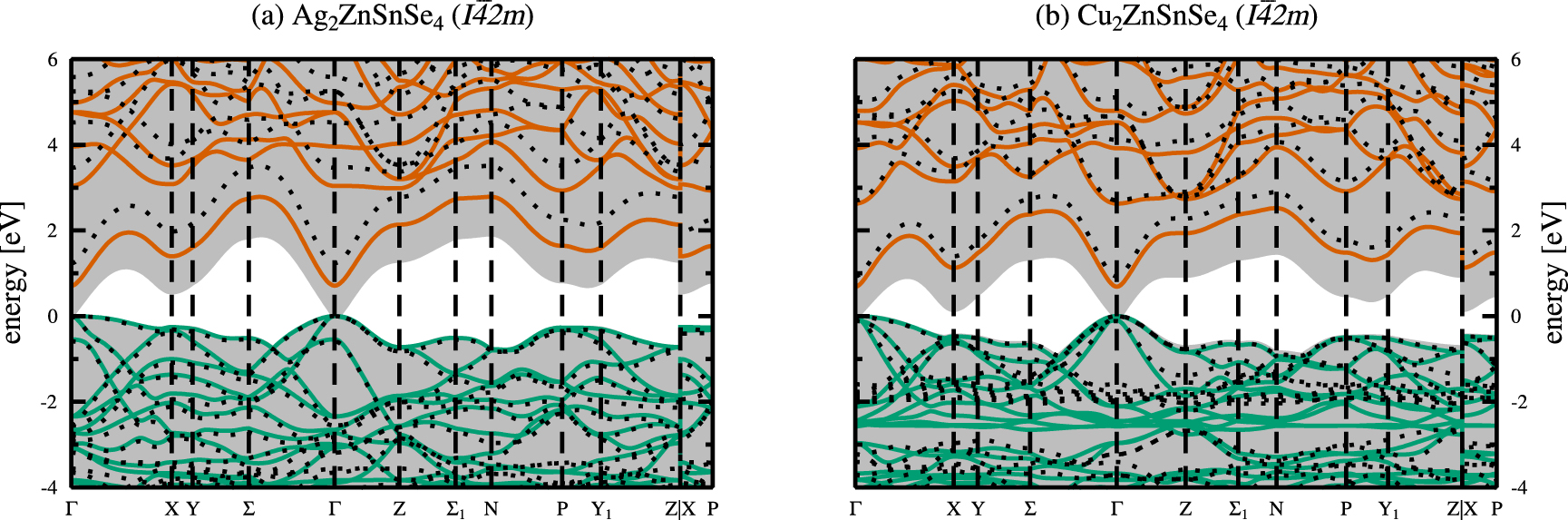
Based on the obtained ground state structures we calculated the electronic band structures as outlined in section 2.3. The electronic band structures are shown in figure 5 for the kesterite crystal structures of AZTSe (a) and CZTSe (b), respectively. The respective electronic band structures for the stannite crystal structures of AZTSe and CZTSe can be found in the appendix (figure A2). In all electronic band structure figures, green and red lines depict the valence and conduction bands calculated with the hybrid HSE06 functional, whereas the shaded grey background indicates the SCAN calculated valence and conduction bands. The dotted black lines are the results of many-body perturbation theory calculations by means of the single-shot  approximation with the HSE06 eigenvalues and orbitals as starting points.
approximation with the HSE06 eigenvalues and orbitals as starting points.

Figure 5. Electronic band structures for the kesterite crystal structures of Ag2ZnSnSe4( a) and Cu2ZnSnSe4( b), with zero energy at the top of the valence bands. Shown are the valence (green) and conduction bands (red), calculated using the hybrid HSE06 functional [24]. The dotted lines ( ) and the shaded grey backgrounds show the results from the
) and the shaded grey backgrounds show the results from the  and SCAN calculations, respectively.
and SCAN calculations, respectively.
Download figure:
Standard image High-resolution image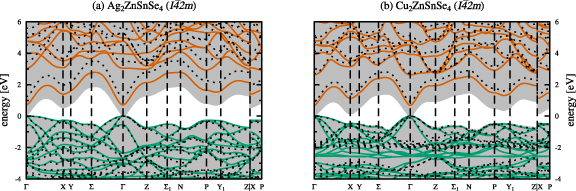
Figure A2. Electronic band structures for the stannite crystal structures of Ag2ZnSnSe4( a) and Cu2ZnSnSe4( b), with zero energy at the top of the valence bands. Shown are the valence (green) and conduction bands (red), calculated using the hybrid HSE06 functional [24]. The dotted lines ( ) and the shaded grey backgrounds show the results from the
) and the shaded grey backgrounds show the results from the  and SCAN calculations, respectively.
and SCAN calculations, respectively.
Download figure:
Standard image High-resolution imageAs a first approximation to the electronic band gaps of the materials, we look at the Kohn-Sham eigenvalue differences between the valence and conduction bands for the LDA, PBEsol, SCAN, and HSE06 functionals, and the quasiparticle energy differences in case of  calculations, respectively. For kesterite-type AZTSe the electronic band gaps amount to 0.16, 0.12, 0.30, and 1.04 eV for the LDA, PBEsol, SCAN, and hybrid HSE06 functionals. The closest agreement to the experimental band gap of 1.31 eV [2] is obtained for the hybrid HSE06 calculation. Similarly, for kesterite-type CZTSe the electronic band gaps amount to 0.01, 0.02, and 0.03 eV, i.e. close to a metallic solution, and 0.87 eV for the LDA, PBEsol, SCAN, and hybrid HSE06 functionals. Again, the hybrid HSE06 calculation yields closest agreement with the experimental band gap ranging between 0.94 eV [2] and 1.0 eV [1]. The closer agreement of the calculated band gap energies with the experimental values in case of CZTSe is possibly due to the better agreement of the obtained fractional coordinates of the anion Wyckoff position with respect to the experimental values, as given in table 1.
calculations, respectively. For kesterite-type AZTSe the electronic band gaps amount to 0.16, 0.12, 0.30, and 1.04 eV for the LDA, PBEsol, SCAN, and hybrid HSE06 functionals. The closest agreement to the experimental band gap of 1.31 eV [2] is obtained for the hybrid HSE06 calculation. Similarly, for kesterite-type CZTSe the electronic band gaps amount to 0.01, 0.02, and 0.03 eV, i.e. close to a metallic solution, and 0.87 eV for the LDA, PBEsol, SCAN, and hybrid HSE06 functionals. Again, the hybrid HSE06 calculation yields closest agreement with the experimental band gap ranging between 0.94 eV [2] and 1.0 eV [1]. The closer agreement of the calculated band gap energies with the experimental values in case of CZTSe is possibly due to the better agreement of the obtained fractional coordinates of the anion Wyckoff position with respect to the experimental values, as given in table 1.
From the band gap values and the electronic band structures in Figure 5 it becomes apparent, that the simpler non-hybrid exchange and correlation functionals show the known band gap underestimation with respect to the experimental values. This severe underestimation of the electronic band gaps obtained by non-hybrid functionals is shown in figure 6.

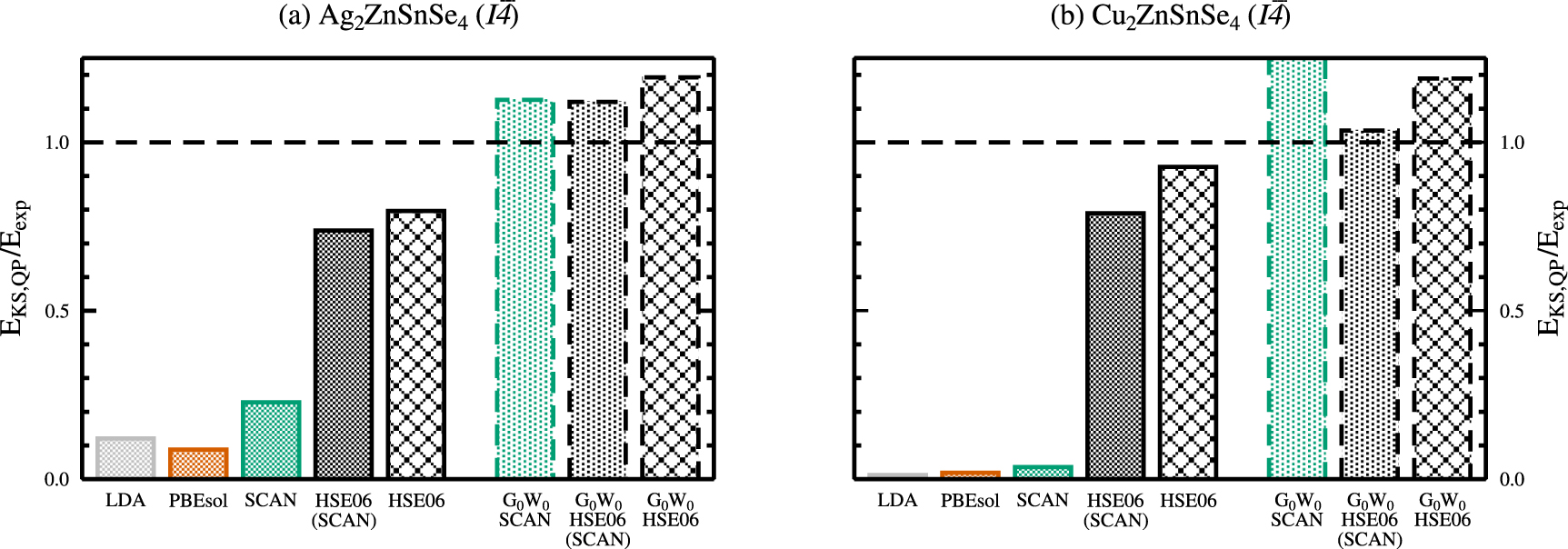
Figure 6. Kohn-Sham eigenvalue differences  between the valence and conduction bands for the LDA, PBEsol, SCAN, and HSE06 functionals and quasiparticle energy differences
between the valence and conduction bands for the LDA, PBEsol, SCAN, and HSE06 functionals and quasiparticle energy differences  in case of
in case of  calculations with respect to the experimental band gap
calculations with respect to the experimental band gap  for the kesterite crystal structures of Ag2ZnSnSe4( a) and Cu2ZnSnSe4( b). The experimental band gaps correspond to
for the kesterite crystal structures of Ag2ZnSnSe4( a) and Cu2ZnSnSe4( b). The experimental band gaps correspond to  (dashed horizontal line).
(dashed horizontal line).
Download figure:
Standard image High-resolution imageOne important objective of the present work is to benchmark different computational approaches against the agreement with experimental structural and electronic properties. As for the structural properties, overall best agreement with available experimental data has been obtained by the SCAN functional, and the PBEsol functional being very close. The electronic properties, however, show best agreement for the hybrid HSE06 functional.
While the present investigation deals with relatively small unit cells, where even a full structural relaxation employing the hybrid HSE06 functional is feasible, for many other possible investigations structural relaxations based on hybrid functional become prohibitively expensive in terms of computational resources. This includes all types of investigations requiring larger supercells, e.g. formation energies of defects and defect complexes or the calculation of band offsets in modelled device heterostructures. For these types of investigations it is indispensable to estimate the deviations introduced by less accurate exchange and correlation functionals.
Over the last years it became more and more popular to perform structural relaxations of larger unit cells employing computationally cheaper functionals, and obtain the electronic properties by means of hybrid functional calculations without further relaxing the structure, so-called single-shot or one-shot hybrid calculations. The HSE06 calculated electronic band gaps based on the SCAN optimised structures amount to 0.97 and 0.74 eV for AZTSe and CZTSe, being somewhat lower that the HSE06 values obtained for the HSE06 optimised structures of 1.04 and 0.87 eV, respectively. Compared to the full hybrid HSE06 investigation, this approach yields the second best agreement with respect to the experimental band gaps, however, for larger unit cells it is presently the only approach available for restricted computational resources.
In addition, we performed quasiparticle calculations based on the single-shot  method. For kesterite-type AZTSe and depending on the starting eigenvalues and orbitals, the
method. For kesterite-type AZTSe and depending on the starting eigenvalues and orbitals, the  calculations yield a quasiparticle gap of 1.48, 1.47, and 1.56 eV for the plain SCAN functional, the HSE06 calculations on top of the SCAN optimised structures, and the hybrid HSE06 functional, respectively. The respective
calculations yield a quasiparticle gap of 1.48, 1.47, and 1.56 eV for the plain SCAN functional, the HSE06 calculations on top of the SCAN optimised structures, and the hybrid HSE06 functional, respectively. The respective  calculations for kesterite-type CZTSe yield quasiparticle gaps of 1.43, 0.97, and 1.12 eV, with starting eigenvalues and orbitals from the plain SCAN functional, the HSE06 calculations on top of the SCAN optimised structures, and the hybrid HSE06 functional, respectively. It can be seen, that in all cases, the
calculations for kesterite-type CZTSe yield quasiparticle gaps of 1.43, 0.97, and 1.12 eV, with starting eigenvalues and orbitals from the plain SCAN functional, the HSE06 calculations on top of the SCAN optimised structures, and the hybrid HSE06 functional, respectively. It can be seen, that in all cases, the  quasiparticle gaps are overestimated with respect to the experimental band gaps; however, the agreement is best for the starting eigenvalues and orbitals taken from a single-shot hybrid HSE06 calculation on top of the SCAN optimised structures (figure 6). Various test calculations to iterate the Green's functions to self-consistency, i.e. GW0 calculations, showed only negligible influence on the quasiparticle gaps.
quasiparticle gaps are overestimated with respect to the experimental band gaps; however, the agreement is best for the starting eigenvalues and orbitals taken from a single-shot hybrid HSE06 calculation on top of the SCAN optimised structures (figure 6). Various test calculations to iterate the Green's functions to self-consistency, i.e. GW0 calculations, showed only negligible influence on the quasiparticle gaps.
In terms of a direct comparison between different theoretical approaches, the  quasiparticle gaps are enlarged by approximately 0.5 eV (0.25 eV) for kesterite-type AZTSe (CZTSe) for starting eigenvalues and orbitals taken from both, the HSE06 calculations on top of the SCAN optimised structures, and the hybrid HSE06 functional, respectively. The same trend is observed for the stannite-type structures. The total overestimation of the quasiparticle gap of 1.43 eV with starting eigenvalues and orbitals from a SCAN calculation for kesterite-type CZTSe highlights the perturbative character of the GW method in general, and that the near metallic solution of the SCAN calculation is inadequate to provide starting conditions for subsequent quasiparticle calculations.
quasiparticle gaps are enlarged by approximately 0.5 eV (0.25 eV) for kesterite-type AZTSe (CZTSe) for starting eigenvalues and orbitals taken from both, the HSE06 calculations on top of the SCAN optimised structures, and the hybrid HSE06 functional, respectively. The same trend is observed for the stannite-type structures. The total overestimation of the quasiparticle gap of 1.43 eV with starting eigenvalues and orbitals from a SCAN calculation for kesterite-type CZTSe highlights the perturbative character of the GW method in general, and that the near metallic solution of the SCAN calculation is inadequate to provide starting conditions for subsequent quasiparticle calculations.
Based on the overall best agreement with the experimental band gaps for the hybrid HSE06 calculations, we also calculated the optical properties. The real (red) and imaginary (green) parts of the dielectric function are shown in figure 7 for the kesterite crystal structures of AZTSe (a) and CZTSe (b), respectively. In case of CZTSe, the dashed lines present experimental results of Leon et al[ 55], obtained via spectroscopic ellipsometry on bulk crystals. Although the overall structure of the theoretical results is much more pronounced, the important peak positions agree well with each other.  calculations of the dielectric functions basically show the same peak structure, and are only shifted to reflect the change in the band gap energies. The respective dielectric functions for the stannite crystal structures of AZTSe and CZTSe can be found in the appendix (figure A3).
calculations of the dielectric functions basically show the same peak structure, and are only shifted to reflect the change in the band gap energies. The respective dielectric functions for the stannite crystal structures of AZTSe and CZTSe can be found in the appendix (figure A3).

Figure 7. Real (red) and imaginary (green) parts of the dielectric functions for the kesterite crystal structures of Ag2ZnSnSe4( a) and Cu2ZnSnSe4( b), obtained by hybrid HSE06 calculations. The dashed lines in (b) present experimental results of León et al[ 55], obtained via spectroscopic ellipsometry on bulk crystals.
Download figure:
Standard image High-resolution image
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure A3. Real (red) and imaginary (green) parts of the dielectric functions for the stannite crystal structures of Ag2ZnSnSe4( a) and Cu2ZnSnSe4( b).
Download figure:
Standard image High-resolution image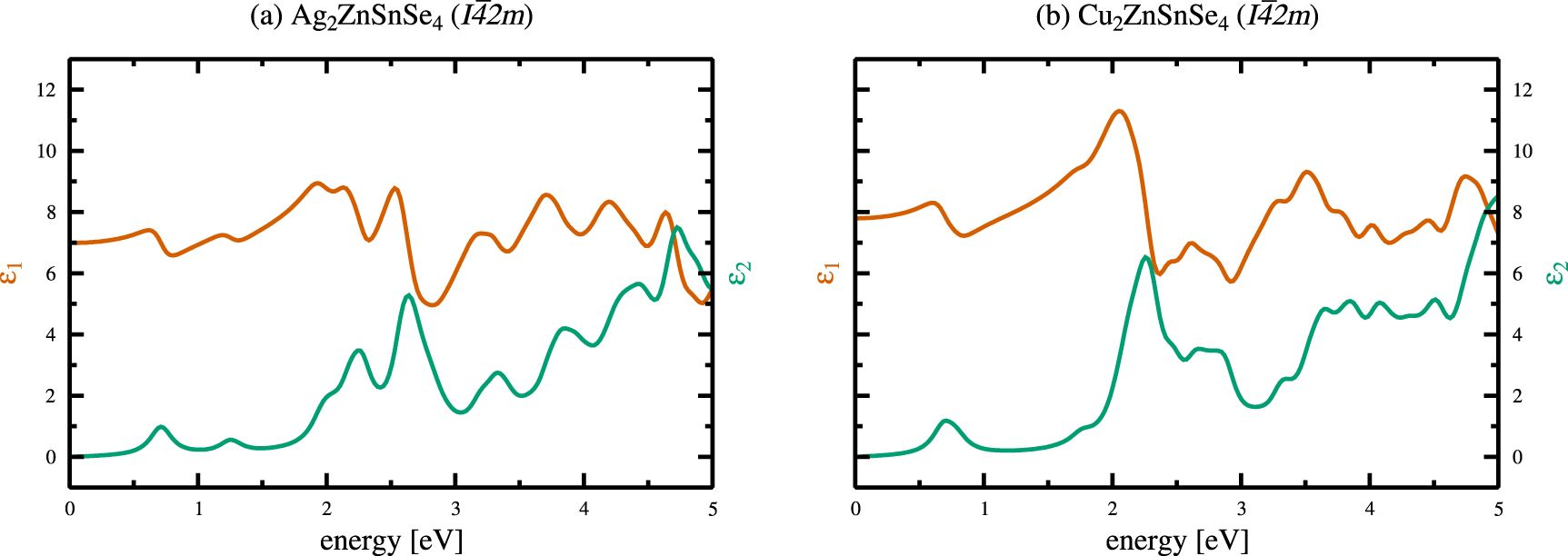{kind=link}
4. Conclusions
In terms of a methodical overview, the present work provides a detailed first-principles investigation of the ground state structural, electronic, and optical properties of the kesterite and stannite crystal structures of AZTSe and CZTSe. The DFT calculations employed exchange and correlation functionals covering the lowest four rungs of Jacob's ladder, thereby including the LDA parametrised by Perdew and Zunger [17], the GGA with the PBE parametrisation revised for solids [20], the meta-GGA with the SCAN functional [22], and the hybrid HSE06 functional [24], respectively. It has been shown, that overall best agreement for the ground state structural properties is obtained by the SCAN functional, while the electronic and optical properties are overall best described by the hybrid HSE06 functional. Performing a single-shot HSE06 calculation on top of the SCAN functional's optimised structures still provided acceptable agreement with respect to experimental data. Subsequent quasiparticle calculations based on the GW approximation revealed a strong influence of the starting eigenvalues and orbitals, and tended to open the gaps for all considered cases. Overall best agreement has been obtained for the starting eigenvalues and orbitals stemming from a single-shot HSE06 calculation on top of the SCAN functional's optimised structures.
While the presented full HSE06 structural optimisations might be suitable for the unit cells employed in the present work, they become prohibitively expensive in terms of computational time for subsequent material property investigations, e.g. defects and defect complexes or the calculation of band offsets in heterostructures. In those cases, performing a structural relaxation based on the SCAN functional followed by a single-shot HSE06 calculation for an accurate description of the electronic properties, provides a reasonable compromise of accuracy and required computational time. This approach seems to be more widely applicable and has recently been suggested for oxide perovskites [56]. Moreover, the eigenvalues and orbitals provided by single-shot HSE06 calculations on top of the SCAN functional's optimised structures, turn out to be an excellent starting point for subsequent quasiparticle calculations based on the GW approximation.
In terms of the material properties, the present work provides a case study into the accuracy and expected agreement with available experimental results for the kesterite and stannite crystal structures of AZTSe and CZTSe. The identified methodical recipes will be beneficial for subsequent investigations requiring larger unit cells, e.g. defects and defect complexes or band offsets. They are equally applicable to investigations of solid solutions based on the two materials, AZTSe and CZTSe, or to get a detailed atomic scale insight into the intrinsic disorder effects. Lastly, the identified numerical recipes are expected to be transferable to similar kesterite and stannite materials as well, thereby widening the impact of the present results considerably.
Conflicts of interest
There are no conflicts to declare.
Acknowledgments
We are grateful to S Levcenko for providing the spectroscopic ellipsometry data for Cu2ZnSnSe4. This work made use of computational resources provided by the North-German Supercomputing Alliance (HLRN, www.hlrn.de), and the ZEDAT and DIRAC high-performance computing facilities of the Freie Universität Berlin and the Helmholtz-Zentrum Berlin, respectively.