

Abstract
The dynamics of biologically relevant molecules exposed to ionizing radiation contains many facets and spans several orders of magnitude in time and energy. In the extreme ultraviolet (XUV) spectral range, multi-electronic phenomena and bands of correlated states with inner-valence holes must be accounted for in addition to a plethora of vibrational modes and available dissociation channels. The ability to track changes in charge density and bond length during ultrafast reactions is an important endeavor toward more general abilities to simulate and control photochemical processes, possibly inspired by those that have evolved biologically. By using attosecond XUV pulses extending up to 35 eV and few-femtosecond near-infrared pulses, we have previously time-resolved correlated electronic dynamics and charge migration occurring in the biologically relevant molecule adenine after XUV-induced sudden ionization. Here, using additional experimental data, we comprehensively report on both electronic and vibrational dynamics of this nucleobase in an energy range little explored to date with high temporal resolution. The time-dependent yields of parent and fragment ions in the mass spectra are analyzed to extract exponential time constants and oscillation periods. Together with time-dependent density functional theory and ab-initio Green's function methods, we identify different vibrational and electronic processes. Beyond providing further insights into the XUV-induced dynamics of an important nucleobase, our work demonstrates that yields of specific dissociation outcomes can be influenced by sufficiently well-timed ultrashort pulses, therefore providing a new route for the control of the multi-electronic and dissociative dynamics of a DNA building block.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
The localization and transport of charge is essential for many photoreactions in biomolecules and functional materials. Just as femtosecond laser technology led to the femtochemistry paradigm, where coherent vibrational wavepackets can be excited and the nuclear motion of molecules be tracked in real time [1, 2], attosecond technology is making it possible to time-resolve electronic dynamics, including multi-electronic transitions with core- or inner-valence holes [3–6] and charge migration across a molecule [7–9]. There is a great interest in utilizing such pulses of light to understand if and how the speed and efficiency of reactions can be improved by better utilizing charge migration and other electronic dynamics, a research field that can be called 'attochemistry' [10–13].
Attosecond pulses are generated via a high-harmonic generation process [14, 15], where a near-infrared (NIR) laser pulse is focused to tunnel-ionize a medium and accelerate electrons away from and then back to their parent ion. The excess kinetic energy of each recombining electron becomes emitted as a photon, which may extend into the extreme ultraviolet (XUV) or soft x-ray regime, depending on the driving laser and medium. Crucially, coherence is preserved from the driving NIR laser to and within the generated attosecond pulse spectrum, allowing pump–probe experiments with sub-femtosecond precision. Attosecond methods based on photoelectron spectroscopy can measure pulse durations [16–18] or time-delays in XUV photoionization processes [19–26] and in the subsequent NIR-probing [27]. Spectroscopy on the initial photoelectron is, however, not sensitive to later dynamics within the molecule, for which transient absorption spectroscopy [28, 29] and pump–probe mass spectrometry are often used [30–33].
Charge migration is defined as a net change in electron or hole probability density within a molecule without any accompanying motion of nuclei (which would be called charge transfer instead) [34, 35]. In the mid-1990s it was suggested that this occurred in oligopeptides (short chains of amino acids) after site-selective photoionization [36], based on observations of which charged fragments were obtained. Theoretical work for di- and tetrapeptides showed that a hole could transfer already in 1–5 fs [37, 38]. Such a redistribution of charge within a molecule can in some cases be achieved through a quantum beat of coherently excited orbitals of different energy, while in other cases it must be described as an intrinsic correlation-driven electronic relaxation that lets the occupation of electronic states evolve over time. Experimentally, the use of an isolated attosecond pulse and a few-femtosecond NIR probing pulse has allowed approximately 4 fs periodic charge migration to be confirmed within the single amino acids phenylalanine [7] and tryptophan [39–41].
A related class of biomolecules are DNA-building blocks, such as nucleosides and nucleobases. Attosecond charge migration from one nucleobase to its complementary nucleobase was recently examined in simulations after removing an oxygen-1s electron [42]. Such processes could have direct consequences for the probability of DNA-damage, for which improved control or biological targeting could have applications in radiation therapy of tumors. In general, ionizing radiation is more likely to damage DNA indirectly via radicals and low-energy secondary electrons created when the primary radiation interacts with the much more abundant water molecules in tissue [43–45]. Although the relaxation of DNA-building blocks after 5 eV ultraviolet photoexcitation [46] has been studied extensively with pump–probe cross-correlations of 80–160 fs (features down to 50 fs fitted) [47–51], we are not aware of any time-resolved studies in the 10–50 eV energy range relevant for DNA strand-breakage [43, 52]. The time resolution achieved in the ultraviolet would be insufficient to examine initial inner-valence multi-electronic dynamics as well as any coherently excited vibrational wavepackets and their implications for the structural integrity of the molecule. We have previously reported on the dissociation dynamics of the nucleobase thymine and its nucleoside thymidine ionized by XUV attosecond pulses and probed by few-femtosecond NIR-pulses [53].
In this work, we describe the time-resolved electronic and vibrational dynamics of the nucleobase adenine, ionized by 17–35 eV photons. In the first part, we discuss how XUV-induced charge migration in the adenine cation enables the production of intact dications. We use additional experimental data beyond our recent report on this phenomenon [6], which to our knowledge constituted the first photoionization study of adenine above the 23.5 eV dication appearance energy [54]. In the second part, we instead examine the fragment ions and discuss signatures of different types of vibrational dynamics. Together, the two parts provide complementary information about the femtosecond dynamics in photoionized adenine.
2. Experimental set-up
Carrier–phase stabilized 25 fs pulses from a titanium:sapphire laser are sent through a hollow-core fiber with a pressure gradient of helium for self-phase modulation, and compressed to 4 fs with a central wavelength of about 700 nm (1.77 eV) by chirped mirrors [55, 56]. A beam splitter gives two replicas of this NIR pulse, one of which is sent through waveplates for polarization gating [57–59] and focused in a pulsed jet of krypton to generate [14, 15] the XUV isolated attosecond pulse. An aluminum filter is then used to block the residual NIR while transmitting most of the XUV [60]. The resulting attosecond pulse has a duration below 300 as and a continuous XUV spectrum from 17 to 35 eV, with the highest spectral intensity near 27 eV [6]. The other part of the NIR-beam is used as a probe at an estimated intensity of 1–1.4 × 1013 W cm−2, recombined collinearly using an annular mirror after a piezoelectric delay-stage to achieve a Mach–Zehnder-like interferometer. To reduce most of the thermal drift, an active stabilization system for the delay is locked onto the spatial fringes of a helium:neon laser that is co-propagating through most of the interferometer (giving an in-loop residual RMS error of 20 as).
Adenine was sublimated at 190 ∘C with helium as carrier and buffer gas, entering the electrostatic time-of-flight mass-spectrometer through a 1 mm skimmer. To avoid detecting the comparably high amount of helium, the microchannel plate and phosphor screen detector voltages were gated. Because this gives a detector efficiency that gradually drops off for the lightest ions, we choose to present only ions with a mass/charge ratio of 25 u or greater.
3. Results and discussion
Results concerning three topics will be presented after an overview of adenine's main fragmentation channels in section 3.1. In section 3.2, we summarize our recent discovery of electronic dynamics that redistributes the charge and allows a stable dication to be produced. Thereafter, we examine the onset and decay times of several fragmentation channels in section 3.3 and finally the oscillations in two of them in section 3.4.
3.1. Pump–probe mass spectra
An overview of the mass spectra obtained when adenine (135 u, 8.20 eV ionization energy [63]) is photoionized by an XUV attosecond pulse, and probed by a NIR pulse is shown in figure 1. The ionic fragment with the lowest appearance energy is 108 u (11.3 to 12.3 eV [54, 63, 64]), via the loss of neutral 27 u as depicted in figure 2(a) [61–63, 65]. Adenine furthermore exhibits sequential ejection of neutral 27 u CHN-groups, indicated by solid arrows in figure 1. This gives rise to the fragments Cn
Hn
N (n = 5 is the parent cation) with appearance energies increasing gradually to 14 ± 0.6 eV [54] for n = 1 (a charged 27 u CHN
(n = 5 is the parent cation) with appearance energies increasing gradually to 14 ± 0.6 eV [54] for n = 1 (a charged 27 u CHN fragment). CHN should be seen as a sum formula, referring to either of the HCN or HNC isomers, which are usually not distinguished. As exemplified in figures 2(a) and (b), some hydrogen migration is necessary for 81 u and 27 u ions, while (d) shows a shortcut to 54 u by ejecting C2H2N2 in one step.
fragment). CHN should be seen as a sum formula, referring to either of the HCN or HNC isomers, which are usually not distinguished. As exemplified in figures 2(a) and (b), some hydrogen migration is necessary for 81 u and 27 u ions, while (d) shows a shortcut to 54 u by ejecting C2H2N2 in one step.

Figure 1. Mass spectra of photoionized adenine. The top panel shows the results of sending the NIR pulse after or before the XUV pulse, and the bottom panel makes the small but meaningful difference between them visible. With the NIR just after the XUV, the adenine dication appears and the yield is increased for small fragments and somewhat reduced for large fragments (the net increase is 6%). The structure of neutral adenine is shown as an inset. Each mass bin is  u wide and the ion yield unit is chosen such that a delay-averaged mass spectrum has a sum of 1000.
u wide and the ion yield unit is chosen such that a delay-averaged mass spectrum has a sum of 1000.
Download figure:
Standard image High-resolution image
Figure 2. Dissociation paths of interest. (a) The dissociation requiring the least energy is ejection of neutral HCN [61, 62]. (b) The resulting 108 u ion can further dissociate by successive HCN/HNC-losses (solid arrows in figure 1) and possibly reach a charged HNC+ (the steps beyond the 81 u ion [62] are illustrated without precise theoretical support). (c) A 2H-migration is likely to occur in the 108 u ion, after which two dissociations are shown: neutral 38 u with a 70 u ion [63] or neutral 43 u and a 65 u ion [62] (from which ionic 38 u would only require a single bond-breakage). (d) By splitting the 108 u in half [61] another 54 u isomer than in (b) can be produced. (e) Dissociations involving loss of the NH2-group (here assumed as first step) and any number of HCN-groups (dashed arrows in figure 1), leading to the 38 u ion [63]. Throughout the figure, shaded (dashed) ellipses indicate alternative origins of 38 u (ions) that require breaking one or two bonds.
Download figure:
Standard image High-resolution imageDashed arrows in figure 1 indicate a sequence where also the neutral—NH2 group is lost, explained in figure 2(e). This sequence ends with the 38 u C2N+ fragment whose relative yield varies considerably in the literature [6, 54, 64–66], most likely related to its higher appearance energy of 20–23 eV [54]. Simulations of 70 eV electron-impact-ionization [61] failed to reproduce both 38 u and 27 u ions, while they were stronger than the 39 u in an experiment using 14 keV projectiles [66]. While 38 u was seen for 3 keV Cl+ projectiles, it was not for 6 keV F2+ [67] (and 27 u is not seen in any of their mass spectra next to the strong 28 u). In figures 2(b) and (e), the 27 and 38 u ions are assigned as the end products of long dissociation sequences, where small perturbations along the way might lead to other outcomes instead. In summary, the production of C2N+ can be significant under the right conditions (see e.g. [54, figure 7]) but difficult to reproduce in simulations, making it interesting to try to better understand the relevant kinetics by examining the ionization of adenine also with higher photon energies (above 22 eV [63]).
As is evident from figure 1, XUV-photoionization leads to a high level of fragmentation (81% of the total yield) regardless of when (and if) probed by the NIR-pulse, indicating the moderate photostability of adenine in the 17–35 eV energy range [54]. The dashed blue curve with the NIR pulse before the XUV has the same fragment distribution as the XUV gives on its own [6, supplementary information] but when the NIR pulse is sent after the XUV (solid red curve) it deposits further energy to the molecules ionized or highly excited by the XUV and helps to fragment them into smaller pieces. This is seen more clearly in the difference mass spectrum, as strong enhancements of many small ionic fragments and reduced yields of the larger fragments. The net change is positive, indicating that the NIR not only dissociates but also photoionizes. On its own, the NIR-pulse can produce parent cations through multi-photon ionization, but essentially without fragmentation (1% of the total yield was 108 u).
To extract physical information from a pump–probe experiment, we will use a curve model based on a differential equation system where the probed state is being populated via transitions with a total rate of  and simultaneously decaying with a total rate of
and simultaneously decaying with a total rate of  to non-observable states. Under the assumption that
to non-observable states. Under the assumption that  , the solution has two exponential terms and can be analytically convoluted with a Gaussian to represent the finite pulse durations [6, supplementary information]. In the limiting case of
, the solution has two exponential terms and can be analytically convoluted with a Gaussian to represent the finite pulse durations [6, supplementary information]. In the limiting case of  , the model simplifies to a step function that decays with lifetime τ2, and represents probing from a state immediately populated by the short XUV-pulse. In the opposite case of probing a slowly populated final-state, which does not decay within the scan range (
, the model simplifies to a step function that decays with lifetime τ2, and represents probing from a state immediately populated by the short XUV-pulse. In the opposite case of probing a slowly populated final-state, which does not decay within the scan range ( ), the model can instead reduce to
), the model can instead reduce to  . The step height h may be either positive or negative. Further details can be found in appendix
. The step height h may be either positive or negative. Further details can be found in appendix
3.2. Correlated electron dynamics in the adenine cation
In addition to the enhanced dissociation described in the previous section, we find that a delayed NIR-pulse also leads to the production of adenine2+, the parent dication where no bond is broken but an extra electron is removed. Energetically, this outcome should be allowed already by the 17–35 eV XUV-pulse on its own, since, a dication appearance energy of  eV is known from electron impact ionization [54]. However, we are not aware of any reports of adenine dications produced by photons, prior to our own [6].
eV is known from electron impact ionization [54]. However, we are not aware of any reports of adenine dications produced by photons, prior to our own [6].
The difference-spectrum in figure 1 shows that the dication with a mass/charge ratio of  u/e has the fourth strongest dependence on NIR-delay, and is much stronger than any other ion between 65 and 70 u/e. The existence of a 67 u ion from the XUV-photon alone, however, makes it difficult to determine the exact baseline level of adenine dications. An estimate, after adjusting for the partly overlapping peak shapes, is that the NIR-enhancement step height parameter, h, is
u/e has the fourth strongest dependence on NIR-delay, and is much stronger than any other ion between 65 and 70 u/e. The existence of a 67 u ion from the XUV-photon alone, however, makes it difficult to determine the exact baseline level of adenine dications. An estimate, after adjusting for the partly overlapping peak shapes, is that the NIR-enhancement step height parameter, h, is  % of the baseline, which is more than twice as large as the relative enhancement for any fragment ion.
% of the baseline, which is more than twice as large as the relative enhancement for any fragment ion.
Even more interesting is that the dication enhancement starts, as seen in figure 3, with a small but certain delay compared to the changes in most of the cationic fragment yields, when fitting the pump–probe scans data as described in the Methods section. The zeroing of the delay-axis was described in the supplement of our previous publication [6]—briefly it is given by the simultaneous increase of many cationic fragments as well as atomic krypton (the Kr2+ yield). The onset-time for the dication is  fs in the previously reported experiment (one fit to the average of seven successive delay-scans) [6]. Here we present the results obtained by including additional data from three other experiments performed on different days under similar conditions. With this procedure, we can now extract a new onset time of
fs in the previously reported experiment (one fit to the average of seven successive delay-scans) [6]. Here we present the results obtained by including additional data from three other experiments performed on different days under similar conditions. With this procedure, we can now extract a new onset time of  fs (median and sample standard deviation of fits to four experiments). τ1 does not appear to depend on the NIR-intensity, although the enhancement most likely requires the absorption of two NIR photons (based on NIR-intensity scaling and the fitted Gaussian pulse widths) [6].
fs (median and sample standard deviation of fits to four experiments). τ1 does not appear to depend on the NIR-intensity, although the enhancement most likely requires the absorption of two NIR photons (based on NIR-intensity scaling and the fitted Gaussian pulse widths) [6].

Figure 3. Delayed production of stable adenine dications. Zoom-in on the central part of a XUV-pump–NIR-probe scan [6], with experimental data points as filled circles and the fitted curve showing a significantly delayed onset (see methods section for the definition of τ1) in the enhancement of the dication yield. The grey shaded areas show ±one standard error of the mean of seven successive scans in alternating directions. Open circles show the main part of the theoretical result, calculated using the non-equilibrium Green's function method. The theoretical signal remains zero when the pulses fully overlap and only starts to increase at positive delays, because shake-up to the selected LUMO+6 state takes time. The result via other states does not show such a strong and late increase [6, supplementary figure S18].
Download figure:
Standard image High-resolution imageTo investigate what mechanism can be responsible for the 2.4 fs delay between XUV-photoionization and the NIR-enhanced production of dications, we explored nuclear as well as electronic dynamics in different theoretical frameworks (see appendix

Figure 4. Bond elongation in the adenine cation. Time-dependent density functional theory Ehrenfest dynamics for the case of suddenly removing an electron from HOMO−3, illustrates the instability of the adenine cation even for near-valence excitation. In the first 4 fs, however, only 1% elongation is reached. Bond numbers shown over each curve are defined to the right, where also the shape of HOMO−3 is shown. The curves for bonds with smaller changes are omitted for clarity.
Download figure:
Standard image High-resolution imageConsidering instead the electronic dynamics, we identified that a multi-electronic process can fill the initial XUV-created inner-valence hole, while exciting at least one other electron to a normally unoccupied bound state. We refer to this as a shake-up state (although not requiring a shift in the kinetic energy of the primary photoelectron), but one can also refer to it as a frustrated Auger–Meitner-state (although no secondary electron has been emitted yet) [68, 69]. The cross-section for further photoionization by the NIR-pulse would then vary depending on the shake-up state, favoring those with one highly excited electron whose ejection is allowed in a single-active-electron picture.
We examined this proposed mechanism using two complementary theoretical frameworks while keeping the nuclei fixed. A rate-based approach, consistent with the experimental curve model, used Fermi's golden rule and first-order perturbation theory to obtain exponential time-constants for each final-state of the highly excited electron. More sophisticated many-body time-dependent simulations were made using a method [6, 70, 71] based on the non-equilibrium Green's function that can handle both the shake-up electron dynamics triggered by the XUV photoionization and the subsequent absorption of a delayed NIR pulse. Both these methods singled out LUMO+6 as a unique shake-up state due to rising relatively slowly but surely, while being in the energy range favored by two-photon NIR-photoionization. The rate-equation approach gives a time-constant of about 3 fs for shake-up to LUMO+6 [6, figure 2]. The many-body-simulated XUV + NIR signal in figure 3 shows that the LUMO+6 contribution remains at zero when the pulses fully overlap (in contrast to an instantaneous transition which would have reached half of its peak-value) and then starts to grow nearly linearly with delay. After 4 fs it surpasses other states whose contributions stayed at a plateau reached within the first femtosecond [6, figure S18]. Even without discussing individual states of a particular basis set, the Green's function method allows visualization of the overall net change in electron density caused by intrinsic electronic relaxation: figure 5(b) shows snapshots with the change in electron density as function of time, with respect to just after XUV-ionization, visualized as red (increased) or blue (decreased). The electron density more than 3 Å from the plane of the molecule is integrated and shown as a curve in figure 5(c). It clearly grows with time. We label the vertical axis simply as 'inflation' to summarize that what this observable represents is a charge migration from the plane of the molecule to more than 3 Å outside. Although multiple states contribute, the importance of LUMO+6 is hinted by the fact that its orbital in panel (d) has large π-like lobes on either side of the molecular plane, like the net charge migration and intuitively a large electric-dipole transition matrix element for photoionization by the NIR-pulse.

Figure 5. Charge migration in the adenine cation. (a) Illustration of the planes at 3 Å distance on either side of the molecular plane, which are used as integration boundaries for (c) and shown with dashed lines in (b) and (d). (b) Snapshots of the net change in charge density, with respect to just after the XUV-photoionization. (c) Integrated electron density more than 3 Å away from the molecular plane. The vertical axis for this dimensionless number is for brevity labelled 'inflation', referring to the trend of charge migration from the central plane of the molecule to a larger volume. (d) Shape of the LUMO+6 orbital, responsible for a slowly-increasing but strong contribution to the charge migration.
Download figure:
Standard image High-resolution imageTo summarize, we deduced that a specific electronic rearrangement in inner-valence-ionized adenine opens up the possibility for the NIR-pulse to create stable dications of the intact molecule. Compared with any rapidly dissociating dications [72] that might be created by the XUV-pulse directly, the route involving shake-up and NIR-photoionization must be creating dications with less excitation energy, perhaps helped by ejecting the second photoelectron. While the NIR-pulse can immediately contribute to further dissociation of adenine cations, the stabilization-enabling process takes a relatively long time (τ1 of a few femtoseconds) compared with other inner-valence electronic transitions, which makes this channel stand out in the experiment. The NIR-pulse then has to be sent before other processes, like the motion of the nuclei, close the window of opportunity—the dication signal decays with  fs. However, if this sequence is completed, the resulting dication remains stable for at least the 3 µs it takes to traverse the mass spectrometer.
fs. However, if this sequence is completed, the resulting dication remains stable for at least the 3 µs it takes to traverse the mass spectrometer.
This combined theoretical and experimental work constitutes, to our knowledge, the first demonstration of correlation-driven charge migration. Conceptually, it is also more of a one-way charge migration than the back-and-forth oscillation so far observed in isolated amino acids [7, 39–41] and smaller systems [8, 9]. The mere ability to resolve a transition time of a few femtoseconds as non-instantaneous is testament to the usefulness of isolated attosecond pulses to investigate even molecules with as many as 15 atoms.
3.3. Time constants for dissociation channels
While some fragment ion signals were already used as a time reference for accurate measurement of the intact dication's delay in [6], we here report a careful analysis of the time-constants extracted from the dissociative photoreactions, including data from many more ions. The delay-dependent yields of several fragments are shown in figure 6 and it is clear that they vary in different ways as a function of the XUV–NIR delay.

Figure 6. Delay-dependence of selected ions. The data points show experimental ion yields as function of XUV–NIR delay, with baselines shifted vertically for better display. The fitted model functions (see text) are shown with solid curves. The grey shaded areas show ±one standard error of the mean of seven successive scans in alternating directions.
Download figure:
Standard image High-resolution imageStarting with the τ1-parameter that tells whether the probe's effect starts like a step function or gradually via a transition (defined on page 6), table 1 shows a clearly nonzero τ1 for the 70 u cation C2H4N , in addition to the previously described 2.4 fs electronic dynamics leading to the dication. The 70 u ion yield is hardly influenced by the NIR-probe at zero delay in figure 6 but is then gradually depleted, with the time constant
, in addition to the previously described 2.4 fs electronic dynamics leading to the dication. The 70 u ion yield is hardly influenced by the NIR-probe at zero delay in figure 6 but is then gradually depleted, with the time constant  fs. The uncertainty mainly represents the possibility of a continued slope outside the scan range and an even longer τ1 (the four experiments summarized in table 1 gave τ1 from 8.7 to 19.2 fs), while a shorter τ1 would fail to fit the data. The 70 u fragment is one of the few that require a concerted two-hydrogen migration [63], which turns the 108 u precursor ion into its lowest-energy isomer [62] and enables the dissociation path shown in figure 2(c). We can therefore speculate that the XUV-ionized molecule evolves in a way that eventually allows the NIR-absorption to suppress fragmentation into 70 u by preventing the initial hydrogen migration.
fs. The uncertainty mainly represents the possibility of a continued slope outside the scan range and an even longer τ1 (the four experiments summarized in table 1 gave τ1 from 8.7 to 19.2 fs), while a shorter τ1 would fail to fit the data. The 70 u fragment is one of the few that require a concerted two-hydrogen migration [63], which turns the 108 u precursor ion into its lowest-energy isomer [62] and enables the dissociation path shown in figure 2(c). We can therefore speculate that the XUV-ionized molecule evolves in a way that eventually allows the NIR-absorption to suppress fragmentation into 70 u by preventing the initial hydrogen migration.
Table 1. Summary of delay-dependence fitted for selected ions. The step height parameter expresses the enhancement (+) or suppression (−) with respect to the baseline signal at negative NIR-delay (approximately the XUV-only signal, but for 108 u and the parent cation the baseline also has a NIR-only contribution), before convolution with a Gaussian and not limited by the scan range. The table shows median values of fits to four experiments from different days with similar but not identical conditions, each containing 5, 6, 7 or 10 delay-scans. Uncertainties indicate the sample standard deviation between the four experiments and table cells with values smaller than their uncertainty are italicized. No time constants are shown for ions without clear delay-dependence (uncertain sign of the step height). For very long decay times,  is shown rather than arbitrary values (
is shown rather than arbitrary values ( 443 fs) influenced by the chosen fitting bounds.
443 fs) influenced by the chosen fitting bounds.
| Mass (u) | Sum formula | τ1 (fs) | τ2 (fs) | Step height (arb. u.) | Relative step height (%) |
|---|---|---|---|---|---|
| 27 | CHN+ |

|
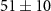
|
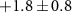
|
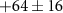
|
| 38 | C2N+ | 0.6 ± 0.7 |
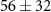
|
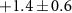
|
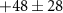
|
| 43 | CH3N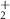
| 0.9 ± 19 |

|
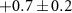
|
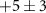
|
| 53 | C2HN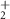
| 0.7 ± 0.8 |
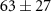
|
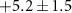
|
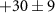
|
| 54 | C2H2N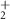
| — | — | −1.7 ± 2.4 | −6 ± 9 |
| 65 | C3HN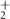
| 0.4 ± 0.7 |

|
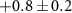
|
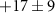
|
| 66 | C3H2N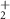
|
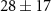
|
|
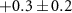
|
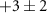
|
| 70 | C2H4N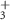
|
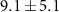
| 117 ± 326 |
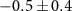
|
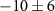
|
| 81 | C3H3N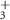
| 3.0 ± 7.4 |
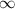
|

|
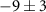
|
| 92 | C4H2N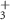
|
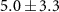
|
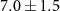
|
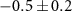
|

|
| 108 | C4H4N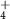
| 0.4 ± 4.0 |
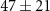
|
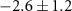
|
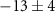
|
| 135 | C5H5N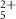
|

|
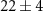
|
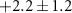
| (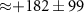 ) ) |
| 135 | C5H5N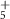
| — | — | −2.4 ± 3.4 | −3 ± 4 |
The lifetime τ2 tells how quickly the effect of the NIR-probe decays or recovers, and values between 40 and 70 fs are found for many of the fragment ions in table 1. Shorter lifetimes are found for the dication ( fs) and for the weak dip in the 92 u yield (
fs) and for the weak dip in the 92 u yield ( fs).
fs).
The fragment ions most strongly affected by the NIR-probe are 53 u, 108 u, 27 u and 38 u (all shown in [6]). We will here focus on 27 and 38 u, which have the largest relative step heights: 64% and 48% of their negative-delay (or XUV-only) baselines, and whose relative yield has varied between types of experiments in the literature (see section 3.1). In the following section we will also examine possible oscillations in their fragment yields.
Neutral 27 u HCN or HNC fragments appear in many dissociation paths of adenine (e.g. 40% of those in [64]), with several examples shown in figure 2 based on assignments for ionization by a single photon or projectile. It is, however, not clear which dissociation path is the most likely to yield charged 27 u fragments for one-photon ionization and whether the delayed NIR probe enhances the same or another path. Since the 38 u ion has been investigated less in the previous literature, we drew ellipses in figure 2 to mark where C2N could be produced in other ways than via the known—NH2-loss sequence in figure 2(e) [63]. We then included the suggested precursors or competing products in table 1 to look for similarities in delay-dependence. Seeing that the 54 u yield is not influenced by the probe-pulse allows us to exclude figures 2(b) and (d), and that the time constants for 70 u and 43 u are very different from those for 38 u speaks against figure 2(c). We thus conclude that 65 u is the most plausible precursor for 38 u, and it is remarkable that the complementary part of 65 u would be 27 u, i.e. 65 u could be a precursor for both of the fragments with the largest relative NIR-enhancement. Different isomers of 65 u appear in the (c) and (e) dissociation paths, in both cases with the breakage of a CN-bond as a final step.
3.4. Oscillating fragment yields
To examine whether there is any oscillation in the ion yields on top of the time dependence fitted in the previous section, a single-frequency oscillation was fitted to the residual data points, using only the part of the scan at positive NIR delay to select oscillations excited by the XUV-ionization. Such oscillations are found for the 27 u HCN+/HNC+ and 38 u C2N+ fragment yields, as shown by dashed lines in figure 7(a). The median and standard deviations for the periods in four experiments under similar conditions are  fs and
fs and  fs, respectively. Oscillation periods for the other ions were not consistent between experiments, with standard deviations of 6–7 fs for two of them and exceeding 11 fs for eight of them. The 27 u and 38 u oscillations have similar periods and are initiated with approximately the same phase, having the first cosine peak at NIR-delays of
fs, respectively. Oscillation periods for the other ions were not consistent between experiments, with standard deviations of 6–7 fs for two of them and exceeding 11 fs for eight of them. The 27 u and 38 u oscillations have similar periods and are initiated with approximately the same phase, having the first cosine peak at NIR-delays of  and
and  fs. This suggests that the modulating NIR-transition occurs as a common precursor rather than as a delay-dependent competition between the two fragmentation outcomes.
fs. This suggests that the modulating NIR-transition occurs as a common precursor rather than as a delay-dependent competition between the two fragmentation outcomes.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 7. Oscillating ion yields. (a) Points show ion yields as function of XUV–NIR delay and solid curves show the fitted model functions of section 3.3. Circles show the residuals after subtracting the fitted curve. Thick dashed lines show cosines fitted to the residuals at positive delays (thin dashes show the extrapolation to negative delays, where no oscillation is expected). The grey shaded areas show ±one standard error of the mean of seven successive scans in alternating directions. (b) Spectrograms using the same residual data points as in (a), but within a sliding 25-point Welch window (35 fs FWHM) [73]. The signal-to-noise ratio is defined as amplitude2 of the fitted single cosine divided by the average of all datapoints' squared uncertainty. The dotted lines in (b) mark the frequencies of the oscillations in (a).
Download figure:
Standard image High-resolution image{kind=link}
To investigate whether the oscillation frequency or amplitude vary during the scan, figure 7(b) shows spectrograms computed using a sliding Welch [73] window function. Its spectral properties lie between those of the Hann and the rectangular windows [74]. We find that the oscillations are stronger when the window is centered near the end of the scan (e.g., +25 fs) than when close to the start of the scan (e.g. −15 fs), which confirms that the oscillations occur in the XUV-ionized sample and are probed by the NIR-pulse (not excited by the NIR-pulse). The strongest signal/noise ratio is found at NIR-delays of 5–10 fs, near the peaks of the non-oscillating model curves in figure 7(a).
In the frequency range of the observed oscillation, neither coherent beating between electronic states nor coherently excited vibrational wavepackets can be excluded a priori. The onset of nuclear motion could change the spacing between electronic states and lead to decoherence [75, 76] but the oscillation amplitude remains roughly constant for at least as long as the τ2 lifetime of the non-oscillating step. While a time-dependent localization of charge on the 27 u-side in the dissociation figure 2(a) could probably modulate the 27 u ion yield, there does not appear to be any such immediate path toward charged 38 u fragments. Considering these arguments and the difficulty of simulating coherent electronic dynamics while letting the nuclei move, we will in the following focus on a vibrational assignment.
The observed oscillation frequency is consistent with the general band for CN-stretching vibrations, possibly in the 65 u precursor and certainly in the neutral 27 u and 38 u fragments (16 fs for HN–C [77] and HC–N [78], 17 fs for C–C–N asymmetric stretch [79, 80]). Although NIR-photoionization of electronically excited fragments can have a cross-section dependent on their bond lengths, such an assignment is difficult to reconcile with the immediate onset of the oscillations. In particular, the 38 u fragment shown in figure 2 cannot be formed without substantial nuclear motion [61, 63], which must be expected to take time.
To get an idea of which bonds start to elongate quickly, and better support a vibrational explanation, we revisit figure 4 and our TDDFT Ehrenfest dynamics calculations for the adenine cation [6, supplementary figure S10]. Two CN-bonds in the backbone show rapid elongation: bond #9 relating to figure 2(a) and bond #11 which concerns the—NH2-group which has already been highlighted in the dissociation path of figure 2(e). Figure 4 shows that the C–NH2 bond #11 elongates dramatically by 6.5% in 15 fs after the sudden removal of an electron from HOMO−3. The simulation as well as vibronic spectroscopy on adenine [81, 82] give period times of 30–33 fs for this bond, so that its second overtone would match the ion yield oscillations perfectly. A coherent comb beating at twice the fundamental frequency could be excited if only the even-numbered vibrational levels have nonzero Franck–Condon factors [83] (or classically by a transition occurring only at the equilibrium geometry twice per oscillation period). Within the experimental uncertainty, a directly matching fundamental 20 fs oscillation period has also been reported for a mode that involves both the C–NH2 stretch and NH2 scissoring [84–86]. (An even better matching peak at 17 fs, 1909 cm−1, in the calculations of Nowak et al [86] was weak and not assigned.)
4. Conclusions
Controlling photochemical reactions requires a thorough understanding of both electronic and vibrational dynamics. The greatest challenges for theoretical work can be found at ultrafast time scales, in energy-ranges where electronic correlation is important (inner-valence holes) and with large molecules. In this work, we time-resolved different types of relaxation processes in the nucleobase adenine (C5H5N5) after photoionization in the previously unexplored XUV energy range, also relevant for DNA-damage by secondary electrons in tissue.
Our XUV-pump—NIR-probe experiment provides onset- and decay-times of molecular processes which are revealed by how the yields of the intact adenine dication and several fragment ions are influenced by the NIR pulse at different time delays. Using this approach, we have been able to extract decay times for the 65, 66, 70 and 92 u channels (never reported before) and refine values for the recently reported stronger channels. Although the precise non-adiabatic mechanisms responsible for their decays have not been elucidated, assignments were given for the few-femtosecond delayed onsets discovered for the 70 u C2H4N fragment ion and the dication (C5H5N
fragment ion and the dication (C5H5N ). The former may be related to double hydrogen-migration that is required to form C2H4N
). The former may be related to double hydrogen-migration that is required to form C2H4N . The latter was assigned as a kind of charge migration with the help of first-principles theory and represents the first time that a multi-electronic process in a polyatomic molecule has been tracked in real time [6]. The time required for the charge migration allowed us to achieve a degree of control of the yield of intact dications via the XUV–NIR delay parameter and the ability to resolve the phenomenon as non-instantaneous demonstrates the potential of attosecond technology even for molecules with as many as 15 atoms.
. The latter was assigned as a kind of charge migration with the help of first-principles theory and represents the first time that a multi-electronic process in a polyatomic molecule has been tracked in real time [6]. The time required for the charge migration allowed us to achieve a degree of control of the yield of intact dications via the XUV–NIR delay parameter and the ability to resolve the phenomenon as non-instantaneous demonstrates the potential of attosecond technology even for molecules with as many as 15 atoms.
Furthermore, we propose that delay-dependent oscillations in the yields of the 27 u CHN+ and 38 u C2N+ fragment ions may be a consequence of C–NH2-stretching vibrations in the adenine cation. The XUV attosecond pulse excites a broadband coherent vibrational wave packet whose beating, as a function of delay before the NIR-probe is absorbed, may modulate the probability of entering the dissociation path in figure 2(e) where loss of NH2 is necessary. The similar oscillation amplitudes for HCN+ and C2N+ suggest that a similar number of steps are required, i.e., that the oscillating HCN+ is formed after hydrogen migration together with a neutral 65 or 38 u, rather than directly with 92 u. The multi-step dissociation paths that can lead to 27 or 38 u ions have not received much attention in previous studies (particularly with the C2N+ branching ratio varying substantially between different ionization methods and between experiment and theory), and our findings provide new insights toward the understanding of vibrational wave packet dynamics of this relatively large molecule.
In summary, by controlling when the NIR-pulse is sent with extreme time resolution we have been able to suppress and enhance different molecular dissociation channels, both in a conventional way by exploiting vibronic dynamics and in a novel way based on pure electronic dynamics. Further advances in the ability to control photochemistry at its natural timescale [10, 12] may come from the development of more advanced simulations for polyatomic systems going beyond the Born-Oppenheimer approximation and by combining them with experimental investigations, such as the present one, which covers the full range from electronic, vibrational to dissociative phenomena.
Acknowledgments
F Ca acknowledges support from the European Research Council under the ERC-2014-StG STARLIGHT (Grant Agreement No. 637756). F Ca and A R acknowledge support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—SFB-925—Project 170620586 and the Cluster of Excellence Advanced Imaging of Matter (AIM). F L and V W acknowledge the Fonds de recherche du Québec—Nature et technologies (FRQNT) and the National Science and Engineering Research Council (NSERC). V W acknowledges support from the Vanier Canada Graduate Scholarship (Vanier CGS) program. A T acknowledges support from the Helmholtz association under the Helmholtz Young Investigator Group VH-NG-1613. S L acknowledges support from the Alexander von Humboldt foundation. A R acknowledge financial support from the European Research Council (ERC-2015-AdG-694097). The Flatiron Institute is a division of the Simons Foundation. G S and E P acknowledge EC funding through the RISE Co-ExAN (Grant No. GA644076), the European Union project MaX Materials design at the eXascale H2020-EINFRA-2015-1, Grant Agreement No. 676598, Nanoscience Foundries and Fine Analysis-Europe H2020-INFRAIA-2014-2015, Grant Agreement No. 654360 and Tor Vergata University for financial support through the Mission Sustainability Project 2DUTOPI. E P, G S and M N acknowledge funding from MIUR PRIN, Grant No. 20173B72NB. J B G acknowledge support from the EPSRC (UK) Grant Number EP/M001644/1.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Appendix A.: Fitting and statistics
For each experiment, ion yields from multiple delay-scans are averaged and errorbars defined by the standard error of the mean. The curve model introduced on page 6 is expanded for a global fit of the delay-dependent signals for all the analysed ions, where the pulse duration is a shared parameter and each ion has individual parameters for baseline, step height, exponential onset-time τ1 and exponential decay-time τ2.
The Gaussian for each signal has a full-width at half-max  where W represents NIR pulse duration and ni
is the number of NIR-photons absorbed for the signal of ion i (usually 1 but chosen as 2 for 92 u, 108 u and the adenine dication). The zeroing of the delay-axis can be fitted as an additional common parameter but for the main experiment it is held fixed to agree exactly with our previous publication [6] (would otherwise vary on the 0.1 fs level depending on which ions are included in the global fit). Additionally, we have here considered three similar experiments to check the robustness and provide median-values and sample standard deviations of fitted parameters.
where W represents NIR pulse duration and ni
is the number of NIR-photons absorbed for the signal of ion i (usually 1 but chosen as 2 for 92 u, 108 u and the adenine dication). The zeroing of the delay-axis can be fitted as an additional common parameter but for the main experiment it is held fixed to agree exactly with our previous publication [6] (would otherwise vary on the 0.1 fs level depending on which ions are included in the global fit). Additionally, we have here considered three similar experiments to check the robustness and provide median-values and sample standard deviations of fitted parameters.
For oscillations fitted to residuals beyond the above curve model, significance can be judged via the signal-to-noise power ratio (computed as the fitted cosine's squared amplitude divided by the average of all datapoints' squared signal-errorbars). Its statistical distribution is known for the null hypothesis of no real signal, so for a p-value of 5% a signal-to-noise ratio of 6.6 is considered significant, including the correction [87] for examining multiple frequencies (37 here, in steps of 3 THz from 42 to 150 THz). Additionally, we compute the difference in corrected Akaike Information Criterion (dAICc) [88] with the sign chosen to be positive if the cosine model function is preferred over a single-parameter constant model. The residual oscillations in figure 7(a) for 27 u and 38 u have the respective signal-to-noise ratios 7.7 and 7.5 (corresponding to p-values of 0.017 and 0.020) and dAICc-values of 4.7 and 8.4.
Appendix B.: Theory
In this section we describe the basics of the theoretical approaches employed to simulate the photo-induced nuclear and electronic dynamics in adenine.
B.1. Nuclear dynamics
TDDFT, the extension to the time-domain of the ordinary density functional theory is a formally exact quantum mechanical ab-initio method able to describe the dynamics of many-body systems [89, 90]. The TDDFT standpoint is the effective one-particle potential called exchange-correlation potential. While TDDFT within standard exchange-correlation functionals flavours is not able to capture the level of electronic correlation required to describe the correct correlated electronic dynamics in ionized adenine, it can still be used as an affordable solution to simulate the qualitative dynamical nuclear behavior following XUV ionization. Specifically, we follow the nuclear motion after a sudden single (or double) ionization by means of TDDFT + Ehrenfest dynamics within a self-interaction-corrected PBE exchange correlation functional [91, 92]. The TDDFT + Ehrenfest dynamics method is a non-adiabatic molecular dynamics approach which couples the time-dependent propagation of the Kohn–Sham orbitals with classical equations of motion for the nuclei determined on the base of the electronic potential energy surface (PES). In practice, the combined electronic–nuclear dynamics is carried out by a time stepping procedure where first the electronic evolution is obtained from TDDFT to calculate the time-evolved density and the latter is then used to calculate the PES from which the nuclear forces are evaluated and used to displace the nuclei within classical equations of motion. While this approach allows to treat non-adiabatic effects, it is formally justified only as long as the nuclear propagation is well described by single PESs and no quantum mixing of nuclear trajectories is expected to take place, like in the case of conical intersections which we neglect in this work. We utilize the Octopus code [93, 94] to perform TDDFT + Ehrenfest dynamics on adenine following the single (or double) removal of an electron from a given Kohn–Sham state. As discussed in the text we are able to demonstrate that ionization of adenine without any stabilization mechanism initiated by electronic correlation inevitably leads to bond elongation (figure 4) and eventual molecular dissociation on a time scale ( fs) much longer than the one associated with the electronic charge migration.
fs) much longer than the one associated with the electronic charge migration.
B.2. Correlated electronic dynamics
As mentioned in the above section the available TDDFT exchange correlation potentials are not mature enough to capture the electronic correlation necessary to describe shake-up effects. For this reason we here employ a time-dependent density matrix propagation method based on non-equilibrium Green's function theory (NEGF) [95]. The NEGF formalism is based on a non-local non-hermitian self-energy operator comparable to the exchange-correlation potential in TDDFT, with the fundamental difference that one has a systematic perturbative approach to handle electron-electron and electron-photon interactions. The time-dependent equation for the density matrix reads:
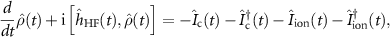
where  is the single particle Hartree–Fock Hamiltonian describing the system, and
is the single particle Hartree–Fock Hamiltonian describing the system, and  and
and  are the collision and ionization integrals respectively. The collision integral, which is expressed in terms of the correlation self-energy contains all the relevant electron-electron scattering processes. The ionization integral instead allows to include the laser induced ionization. Recent implementations of the density matrix propagation approach, such as the one in the CHEERS code [70], have made accurate simulations of realistic systems computationally accessible. Here we numerically solve the above equation for the charge dynamics of adenine induced by an experimental attosecond XUV pulse. Specifically, we construct the Hartree–Fock Hamiltonian starting from the Kohn–Sham wavefunctions and approximate the self-energy at the second-Born level; all details are provided in our recent report [6].
are the collision and ionization integrals respectively. The collision integral, which is expressed in terms of the correlation self-energy contains all the relevant electron-electron scattering processes. The ionization integral instead allows to include the laser induced ionization. Recent implementations of the density matrix propagation approach, such as the one in the CHEERS code [70], have made accurate simulations of realistic systems computationally accessible. Here we numerically solve the above equation for the charge dynamics of adenine induced by an experimental attosecond XUV pulse. Specifically, we construct the Hartree–Fock Hamiltonian starting from the Kohn–Sham wavefunctions and approximate the self-energy at the second-Born level; all details are provided in our recent report [6].