

Abstract
Here we present the optoelectronic properties of pure inorganic lead-free halide perovskites in the form of Cs2AgBiX6 (X = Br, Cl, F, I) using the density functional theory calculations on cubic phase (Fm  m) and tetragonal phase (I4/m). First, all the structures of the two phases were optimized at the PBE level. Structural, electronic, optical properties, phonon, and thermal properties of Cs2AgBiX6 in cubic (Fm
m) and tetragonal phase (I4/m). First, all the structures of the two phases were optimized at the PBE level. Structural, electronic, optical properties, phonon, and thermal properties of Cs2AgBiX6 in cubic (Fm  m) and tetragonal phases (I4/m) were obtained using the VASP code. Tetragonal phases of all compounds of the form Cs2AgBiX6, except Cs2AgBiBr6, are reported here for the very first time. Among all the Cs2AgBiX6 (X = F, Cl, Br, I) structures, the cubic phase of Cs2AgBiBr6 was seen to have the highest absorption coefficient along with prominent electronic features that are favorable for optoelectronic applications. Thus, the cubic phase of Cs2AgBiBr6 was selected as the host lattice and bromine atoms were partly replaced with chlorine and iodine atoms. Electronic and optical properties of these mixed halide compounds of Cs2AgBiBr6−xFx, Cs2AgBiBr6−xClx, and Cs2AgBiBr6−xIx where x = 1, 2, 3, 4, 5 are investigated with hybrid functional HSE06 level. The electronic structure revealed that these mixed compounds exhibited indirect band gap nature regardless of the halide substitution (different x concentration) and the band gap of Cs2AgBiBr6 could be varied with the substitutions of fluorine, chlorine, and iodine atoms. Our in-depth analysis shows that Cs2AgBiBr6 and their mixed halides have the potential to become active double perovskite materials for photovoltaic applications and as photocatalysts for water splitting.
m) and tetragonal phases (I4/m) were obtained using the VASP code. Tetragonal phases of all compounds of the form Cs2AgBiX6, except Cs2AgBiBr6, are reported here for the very first time. Among all the Cs2AgBiX6 (X = F, Cl, Br, I) structures, the cubic phase of Cs2AgBiBr6 was seen to have the highest absorption coefficient along with prominent electronic features that are favorable for optoelectronic applications. Thus, the cubic phase of Cs2AgBiBr6 was selected as the host lattice and bromine atoms were partly replaced with chlorine and iodine atoms. Electronic and optical properties of these mixed halide compounds of Cs2AgBiBr6−xFx, Cs2AgBiBr6−xClx, and Cs2AgBiBr6−xIx where x = 1, 2, 3, 4, 5 are investigated with hybrid functional HSE06 level. The electronic structure revealed that these mixed compounds exhibited indirect band gap nature regardless of the halide substitution (different x concentration) and the band gap of Cs2AgBiBr6 could be varied with the substitutions of fluorine, chlorine, and iodine atoms. Our in-depth analysis shows that Cs2AgBiBr6 and their mixed halides have the potential to become active double perovskite materials for photovoltaic applications and as photocatalysts for water splitting.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
The silicon solar cells-based research community gave a new phase to photovoltaic devices in the 1950s [1]. Search for new concepts in the development of photovoltaics (PV) has resulted in the rise of organic–inorganic hybrid halide perovskite as alternative PV materials. Perovskites have become promising candidates for cheap and flexible PV technology with a much-improved power conversion efficiency (PCE) from 3% to 28% within a few years [2]. Lead-based halide perovskites exhibit great potential for the new generation of solar cells due to their attractive features such as narrow direct band gap, long carrier diffusion length, high absorption coefficient, the small effective mass of electrons and holes, small exciton binding energy and high carrier mobility [3]. However, lead toxicity and instability in ambient conditions are the main barriers faced by lead-based perovskites when it comes to practical applications. Owing to these difficulties, researchers have been seeking alternative environmentally friendly photovoltaic materials with similar optoelectronic properties as Methyl Ammonium Lead Iodide (CH3NH3PbI3) perovskite. Researchers have been focusing on perovskites of the form ABX3 where A = CH3NH3, CH(NH2)2, Cs, Rb; B = Pb, Sn, Ge; X = F, Cl, Br, I. New findings suggest that lead-free halide perovskites, such as Rb2InGaX6 (X = Cl, Br, I)) and Na2YCuX6 (X = Br, Cl) hold as potential candidates for applications in optoelectronic and thermoelectronic devices [4, 5]. Additionally, the perovskites with the configuration in the form of A2B'B''
X6 have gained much attention with different combinations of organic and inorganic cations. For instance, A site cation could be substituted with a monovalent inorganic or organic cation, and the different oxidation states of metals can be used for B'/B'' and X could be varied with different halides [6]. Incorporation with the inorganic metal cations which could match the A2B'B''X6 structure has become much more attractive in recent times. Successful replacement of Pb with Bi and Ag has been reported [6–8]. One of the compounds namely, Cs2AgBiX6 (X = Cl, Br, I) has been studied experimentally and theoretically in different application fields [9, 10]. The electronic properties, specifically the band structures of the Cs2AgBiX6 and lead-halide systems contain many similarities except for the type of band gap. The double perovskites, Cs2AgBiX6 possess an indirect band gap as opposed to the direct band gap seen for the lead halide perovskites such as CsPbBr3 and CsPbCl3 [7]. Salvney et al reported that the first synthesized cubic phase of Fm  m Cs2AgBiBr6 consisted of the indirect band gap and they discussed the photoluminescence (PL) behavior by mentioning its PL decay time of 660 ns.[6] McClure et al synthesized Cs2AgBiBr6 and Cs2AgBiCl6 and the optoelectronic properties were measured both experimentally and theoretically. They showed those materials possessed indirect band gaps. Experimentally the band gap values for the Cs2AgBiBr6 and Cs2AgBiCl6 were found as 2.19 eV and 2.77 eV respectively and these band gap values match with the theoretically derived values of 2.06 eV and 2.62 eV [7]. Volonakis et al discussed the electronic properties of the Pb-free double perovskites using density functional theory (DFT). They found that the Cs2
B'
B''
X6 (B' = Sb, Bi B' = Cu, Ag Au X = Cl, Br, I) exhibit small carrier effective masses between 0.1 and 0.4 me. Their band gap values were found to be less than 2.7 eV, which could cover the visible and near-infrared optical spectrum [8]. Marina et al calculated the quasi-particle band gaps by obtaining the indirect band gap values of 1.8 eV for Cs2AgBiBr6 and 2.4 eV for Cs2AgBiCl6, which is in agreement with another experimental study [9].
m Cs2AgBiBr6 consisted of the indirect band gap and they discussed the photoluminescence (PL) behavior by mentioning its PL decay time of 660 ns.[6] McClure et al synthesized Cs2AgBiBr6 and Cs2AgBiCl6 and the optoelectronic properties were measured both experimentally and theoretically. They showed those materials possessed indirect band gaps. Experimentally the band gap values for the Cs2AgBiBr6 and Cs2AgBiCl6 were found as 2.19 eV and 2.77 eV respectively and these band gap values match with the theoretically derived values of 2.06 eV and 2.62 eV [7]. Volonakis et al discussed the electronic properties of the Pb-free double perovskites using density functional theory (DFT). They found that the Cs2
B'
B''
X6 (B' = Sb, Bi B' = Cu, Ag Au X = Cl, Br, I) exhibit small carrier effective masses between 0.1 and 0.4 me. Their band gap values were found to be less than 2.7 eV, which could cover the visible and near-infrared optical spectrum [8]. Marina et al calculated the quasi-particle band gaps by obtaining the indirect band gap values of 1.8 eV for Cs2AgBiBr6 and 2.4 eV for Cs2AgBiCl6, which is in agreement with another experimental study [9].
The above-mentioned studies are mainly based on the cubic phase of Cs2AgBiX6 (X = Br, Cl) and only a limited number of reports on those materials with a tetragonal phase. The tetragonal phase formation in Cs2AgBiX6 that we focus on, could be explained by its crystal structure. This inorganic double perovskite consists of a three-dimensional framework of corner-connected octahedral referring to a rock salt order. McClure et al suggested the octahedral rotations could happen at room temperature due to the low bond valence sum for the Cs+ ion and large values of the displacement parameters. This octahedral tilting could affect the phase transition below room temperature [7]. Laura et al showed the temperature could affect the phase transition on Cs2AgBiBr6. At 144 K, the low temperature of the tetragonal structure(I4/m) could appear from the cubic structure of space group Fm  m [11].
m [11].
Applying pressure could be a green method to explore the structure–property relationships and phase transitions [12]. Zhang et al showed that the pressure could influence the phase transition of Cs2AgBiCl6. At 5.6 GPa the phase transition occurred from cubic Fm  m structure to a tetragonal one with space group I4/m [13]. Cs2AgBiBr6 crystallizes in space group Fm
m structure to a tetragonal one with space group I4/m [13]. Cs2AgBiBr6 crystallizes in space group Fm  m and it transforms to a tetragonal phase I4/m at 4.5 Gpa [14].
m and it transforms to a tetragonal phase I4/m at 4.5 Gpa [14].
The theoretical investigations based on structural, mechanical, electronic, and optical properties were calculated for the cubic and tetragonal phase of Cs2AgBiBr6 using a full-potential linearized augmented plane wave approach implemented in the WIEN2k code [15]. These findings encouraged us to seek the optoelectronic properties of the tetragonal phase of Cesium-based lead-free halide double perovskites. There are no other theoretical or experimental findings on tetragonal phases of Cs2AgBiX6 (X = F, Cl, Br, I) up to our knowledge. This study would be the first detailed investigation of optical and electronic property calculations considering both phases of Cs2AgBiX6 based on first principles. For the successful utilization of photovoltaic application, we predict the set of materials' properties on Cs2AgBiX6 (X = F, Cl, Br, I) for the cubic phase of (Fm  m) and for the tetragonal phase (I4/m). The optoelectronic properties, mechanical properties, and phonon calculations are carried out based on density functional theory calculations for the Cs2AgBiX6. And also in this study, we demonstrate a way to tune the bandgap of halide perovskites for solar cell applications. The best-performing solar materials should possess a band gap value between the range of 1.10–1.55 eV as shown by the Shockley-Quiesser limit [16]. Mixing the halides in double perovskites will enable tuning of the band gap. The theoretical and experimental investigations have earlier shown that the I– and Cl– ions substitutions in the cubic phased Cs2AgBiBr6 structures resulted in tuning the band gap [17–19]. However, there is a lack of detailed study of the structural, optical, and electronic calculations for these mixing halides. Thus, we report in detail the first principle studies on lead-free mixed halide double perovskites by substituting the halogen atoms with the bromine atoms in Cs2AgBiBr6 with the fluorine, chlorine, and iodine atoms. The fluorine atoms substitutions for the Cs2AgBiBr6 compound are the first-time computed results. We chose the cubic phase of Cs2AgBiBr6 for the substitution because it shows the best photovoltaic properties among these halide components. The optoelectronic characteristics are carried out for the Cs2AgBiBr6−xFx, Cs2AgBiBr6−x
Clx, and Cs2AgBiBr6−x
Ix
where x = 1, 2, 3, 4, 5. We perform a detailed study of electronic calculations including electronic band structures, the density of states, and effective mass calculations using the hybrid functional method as well the optical absorption calculations like absorption coefficient, refractive index, reflectivity, energy loss, and extinction coefficient. When it comes to the photocatalytic application, we analyze the band edge positions for these compounds relevant to the redox potentials of the water splitting.
m) and for the tetragonal phase (I4/m). The optoelectronic properties, mechanical properties, and phonon calculations are carried out based on density functional theory calculations for the Cs2AgBiX6. And also in this study, we demonstrate a way to tune the bandgap of halide perovskites for solar cell applications. The best-performing solar materials should possess a band gap value between the range of 1.10–1.55 eV as shown by the Shockley-Quiesser limit [16]. Mixing the halides in double perovskites will enable tuning of the band gap. The theoretical and experimental investigations have earlier shown that the I– and Cl– ions substitutions in the cubic phased Cs2AgBiBr6 structures resulted in tuning the band gap [17–19]. However, there is a lack of detailed study of the structural, optical, and electronic calculations for these mixing halides. Thus, we report in detail the first principle studies on lead-free mixed halide double perovskites by substituting the halogen atoms with the bromine atoms in Cs2AgBiBr6 with the fluorine, chlorine, and iodine atoms. The fluorine atoms substitutions for the Cs2AgBiBr6 compound are the first-time computed results. We chose the cubic phase of Cs2AgBiBr6 for the substitution because it shows the best photovoltaic properties among these halide components. The optoelectronic characteristics are carried out for the Cs2AgBiBr6−xFx, Cs2AgBiBr6−x
Clx, and Cs2AgBiBr6−x
Ix
where x = 1, 2, 3, 4, 5. We perform a detailed study of electronic calculations including electronic band structures, the density of states, and effective mass calculations using the hybrid functional method as well the optical absorption calculations like absorption coefficient, refractive index, reflectivity, energy loss, and extinction coefficient. When it comes to the photocatalytic application, we analyze the band edge positions for these compounds relevant to the redox potentials of the water splitting.
2. Computational details
The projector-augmented wave (PAW)[20] method describes the interaction between the core and the valence electrons (treating 5s2 5p6 6s1 of Cs, 4d10 5s1 of Ag, 5d10 6s2 6p3 of Bi, 2s2 2p5 of F,3s2 3p5 of Cl, and 4s2 4p5 of Br and 5s2 5p5 of I as valence electrons) was implemented in the VASP code. These calculations are based on the density functional theory with the Perdew–Burke–Ernzerh of (PBE) exchange–correlation functional and hybrid functional (HSE06). The HSE06 functional, which matches the experimental studies was used for computing the electronic structure and the associated optical properties. This computation was first done at the PBE-GGA level to understand the behavior and nature of the materials, after that HSE06 was employed for a more accurate calculation. The optimized results were obtained with a kinetic cut-off energy of 600 eV, and a 6 × 6 × 6 Г-centered Monkhorst–Pack grid for integration over the first Brillouin zone (BZ). A three-dimensional visualization program VESTA is used to visualize the volumetric data, calculated for the equilibrium structures [21]. The elastic constant was computed by the finite strain method using the post-pre-processing tool VASPKIT [22]. Our phonon calculations are performed through the supercell approach [23]. Force constants of supercells are prepared and the PHONOPY code is used to calculate the phonon frequencies [24]. More details were elaborated regarding the optoelectronic property calculations in the supplementary document.
3. Results and discussion
3.1. Structural properties
The optimization could be the very first important step to obtaining a relaxed structure with zero stresses. The relaxed structure could be used as the initial structure for the properties we are finding afterward. During the optimization, the obtained energy and volumes of the structure are fit to the Birch-Murnaghan equation of state as shown in equation (1) to obtain equilibrium cell parameters.
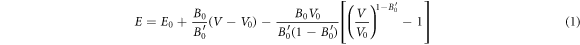
where V is the primitive-cell volume, B is the bulk modulus, which provides the behavior of the crystal volume under hydrostatic pressure, [25] and B' is its first pressure derivative. The zero indexes are the values at zero pressure [26]. Figure 1 represents the variation of total energy with volume for Cs2AgBiX6 in cubic and tetragonal phases. We found there is a tendency for the tetragonal phase to become more stable when the halogen component is changed from F towards I.
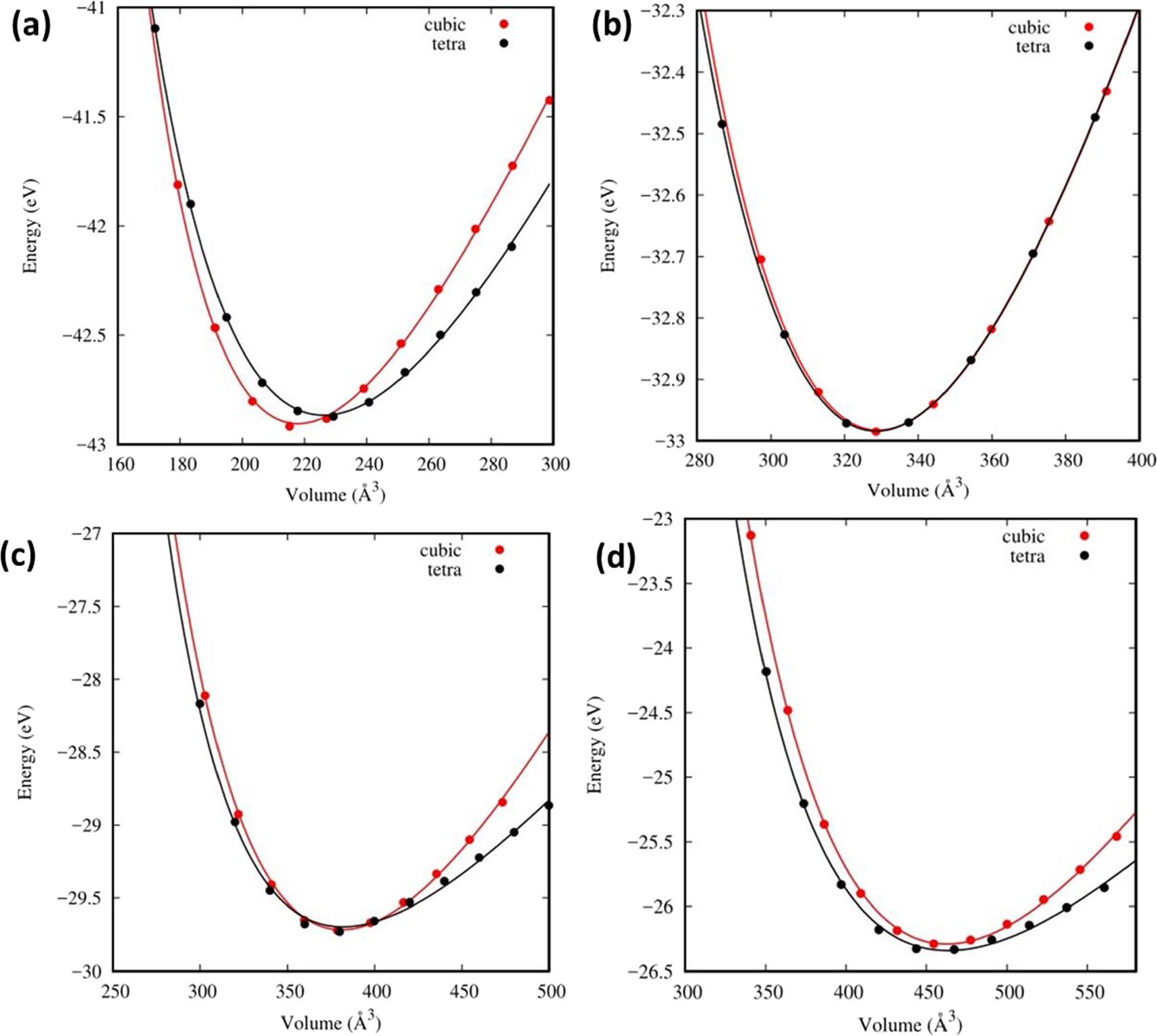
Figure 1. Total energy variation with volume of both phases of Cs2AgBiX6 for (a) Cs2AgBiF6 (b)Cs2AgBiCl6 (c)Cs2AgBiBr6 (d)Cs2AgBiI6.
Download figure:
Standard image High-resolution imageLattice parameters of the optimized Cs2AgBiX6 structures are calculated using GGA approximation and obtained values are tabulated in table 1. For the sake of completeness, these calculated lattice parameters are tabulated with other earlier reported theoretical and experimental studies. The comparison shows that our results for other known compounds are in good agreement with the reported literature values for cubic-phased Cs2AgBiX6 structures. No previous reports are available on lattice parameters for Cs2AgBiF6 structures. According to past theoretical studies, the deviation of the value of lattice constant of the cubic phase in bromide and chloride compounds is in the range of 0.1%–0.5%, [15, 27–29] and also experimentally it is between 1%–2% [6, 7] variations can be found. Thus the deviation is less than 5% in both previous experimental and theoretical works, we could rely on the lattice parameters of our relaxed structure and this implies the accuracy of our geometry optimization and the computational simulation method along with the input parameters which are mentioned under the computational methodology. The lattice constant and the unit cell elongation in the c direction for the tetragonal structure of the bromide compound differ from 1.6% to 5% from the experimental findings which were done under the low temperature, 122 K condition of phase transition from cubic to tetragonal [2]. The computed lattice constant for the iodine compound in the cubic phase deviates from 0.5% - 1.4% from other theoretical findings [30, 31]. When compared to the other double perovskites, cubic phases of the Cs2NaMCl6 (M = In, Tl, Sb, Bi) exhibited the lattice constants which vary from 0.8%–3.67% which gives some closer values to the chloride-based, Cs2AgBiCl6 compound [32]. Another theoretical study was done for the cubic phases of Cs2NaBX6 (B = Sb, Bi; X = Cl, Br, I), where the Cs2NaBiCl6, and Cs2NaSbCl6 lattice constants are different from 0.85% and 2.54% when compared to Cs2AgBiCl6. The other compounds' lattice parameters of Cs2NaBiBr6 and Cs2NaSbBr6 vary from 1% to 2% from Cs2AgBiBr6 respectively and finally, the lattice constant of the iodide compound of Cs2AgBiI6 differs from 1% to the Cs2NaSbI6 [33]. By examining those previous theoretical studies, we can say that changing the B site cation and the X site anion will not affect very much with our computed lattice parameters.
Table 1. Calculated structural parameters for Cs2AgBiX6 (where X = F, Cl, Br, and I) along with other experimental (exp.) and theoretical (Theo.) studies.
| Compound | Cubic (Fm 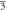 m) m) | Primary | Tetragonal (I 4/m) | Primary |
|---|---|---|---|---|
| Lattice parameters (Å) | cell volume | lattice parameters | cell volume | |
| Cs2AgBiF6 | 9.5038 Å | 214.60 Å3 | a = 6.5267 Å | 229.29 Å3 |
| c = 10.8009 Å | ||||
| Cs2AgBiCl6 | 10.9333 Å | 326.74 Å3 | a = 7.7052 Å | 333.68 Å3 |
| 10.965 [27] (Theo.) | c = 11.2410 Å | |||
| 10.777 [27] (exp.) | ||||
| 10.944 [28] (Theo.) | ||||
| 10.99 [29] (Theo.) | ||||
| Cs2AgBiBr6 | 11.4836 Å | 378.59 Å3 | a = 8.0127 Å | 384.96 Å3 |
| 11.462 [15] (Theo.), 11.25 [6] (exp.),11.53 [29] (Theo.), 11.271 [7] (exp.), 11.496 [27] (Theo.), | c = 11.9938 Å | |||
| a = 7.879, c = 11.3236 [2] (exp.) | ||||
| a = 7.844, c = 11.425 [15] (Theo.) | ||||
| Cs2AgBiI6 | 12.2054 Å | 454.56 Å3 | a = 8.4932 Å | 467.08 Å3 |
| 12.27 [30] (Theo.) | c = 12.9512 Å | |||
| 12.03 [31] (Theo.) | ||||
| 12.28 [29] (Theo.) |
The structural parameters of the tetragonal phases of Cs2AgBiF6, Cs2AgBiCl6, and Cs2AgBiI6 are also reported for the first time here. As table 1 shows, both the lattice parameters and the cell volume increase when the halide compound is varied from fluoride to iodide atoms. This can be attributed to the increase in the size of the p orbital of the halogen atom and the decrease in electronegativity when going from F to I. However, the primary cell volume appears larger in the tetragonal phase than in the cubic phase for all these halide compounds. The DFT calculations were done at standard parameters. The specific reasons for this structural difference can vary depending on the materials and conditions, such as external temperature and pressure, but generally, it involves changes in the arrangement of atoms or molecules within the crystal lattice. The elongation in the c-direction in tetragonal phases contributes to a larger overall volume compared to the more symmetrical cubic phases.
Among the halide materials in table 1, Our attention is directed toward the Cs2AgBiBr6 material due to its demonstrated favorable absorption properties (refer to the optical studies section for further information). The bromine atoms were replaced gradually with other halide atoms (F, Cl, I) in different concentration levels. The calculated structural parameters for Cs2AgBiBr6−x Xx where X = (F, Cl, I) are presented in (table 2). The symmetry of the mixed halides' crystal changes from higher symmetry (cubic phase) to lower symmetry (tetragonal phase) with the increasing amount of the substituted halide atoms of chlorine and iodine (x = 1, 2, 3, 4, 5). This can be attributed to the octahedral (BiBr6) tilting in Cs2AgBiBr6. As the ionic radius of the Cl− ion is smaller than the Br− ion and the electron negativity is higher than for the Br− ion, these factors lead to a decrease in the cell volume of Cs2AgBiBr6−xClx as x is increased from 1 to 5.
Table 2. The equilibrium lattice parameters for Cs2AgBiBr6−xFx, Cs2AgBiBr6−xClx, and Cs2AgBiBr6−xIx (x = 0, 1, 2, 3, 4, 5, 6).
| Cs2AgBiBr6−xFx | Cs2AgBiBr6−xClx | Cs2AgBiBr6−xIx | ||||
|---|---|---|---|---|---|---|
| Lattice | Primary | Lattice | Primary | Lattice | Primary | |
| Substitutions | Constants(Å) | Cell volume | Constants(Å) | Cell volume | Constants(Å) | Cell volume |
| Space group | (Å)3 | (Å)3 | Space group | (Å)3 | ||
| (PBE) | Space group | (PBE) | (PBE) | |||
| x = 0 | a = b = c = 11.4836 Å | 378.59 | a = b = c = 11.4836 Å, 11.4601 Å [17] | 378.59 | a = b = c = 11.4836 Å | 378.59 |
| Cubic | Cubic | Cubic | ||||
(Fm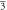 m) m) | (Fm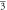 m) m) | (Fm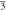 m) m) | ||||
| x = 1 | a = 8.1390 Å | 352.02 | a = 8.1255 Å, 8.0986 Å [17] | 370.91 | a = 8.1835 Å | 395.08 |
| c = 10.6280 Å | c = 11.2358 Å, 11.2104 Å [17]. | c = 11.7988 Å | ||||
| Tetragonal | Tetragonal | Tetragonal | ||||
| (I 4 m m) | (I4/mm) | (I4mm) | ||||
| x = 2 | a = 11.3738 Ǻ | 357.63 | a = 11.4889 Å, 11.4095 Å [17] | 363.24 | a = 8.4099 Å | 409.01 |
| b = 7.9349Ǻ | b = 7.9556 Å, 7.9382 Å [17]. | b = 11.6165 Å | ||||
| c = 7.9250Ǻ | c = 7.9477 Å, 7.9334 Å [17]. | c = 8.3731 Å | ||||
| Orthorhombic | Orthorhombic | Orthorhombic (Imm2) | ||||
| (Imm2) | (Imm2) | |||||
| x = 3 | a = 11.3230 Ǻ | 317.02 | a = 7.9516 Å | 356.07 | a = 11.6328 Å | 421.78 |
| b = 9.6876Ǻ | alpha = 60.07° | b = 12.1612Å | ||||
| c = 11.5606Ǻ | Trigonal | c = 11.9259 Å | ||||
| orthorhombic | (R3m) | Orthorhombic | ||||
| (Fmm 2) | (Fmm2) | |||||
| x = 4 | a = 9.9213Ǻ | 276.74 | a = 10.9787 Å, 10.9418 Å [17]. | 345.16 | a = 12.1987 Å | 434.28 |
| b = 7.5348Ǻ | b = 7.9333 Å, 7.9274 Å [17] | b = 8.4524 Å | ||||
| c = 7.4032Ǻ | c = 7.9250 Å, 7.9053 Å [17] | c = 8.4230 Å | ||||
| orthorhombic | Orthorhombic | Orthorhombic (Imm2) | ||||
| (Imm 2) | (I mm2) | |||||
| x = 5 | a = 7.1208 Å | 254.23 | a = 7.7566 Å, 7.7527 [17]. | 336.42 | a = 8.6429 Å | 445.02 |
| c = 10.0277 Å | c = 11.1835 Å, 11.1565 Å [17]. | c = 11.9150 Å | ||||
| Tetragonal | Tetragonal (Imm2) | Tetragonal | ||||
| (I 4 m m) | (I4mm) | |||||
| x = 6 | a = b = c = 9.5038 Å | 214.60 | a = b = c = 10.9333 Å, 10.9354 Å [17]. | 326.74 | a = b = c = 12.2054 Å | 454.56 |
| Cubic | Cubic | Cubic | ||||
(Fm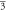 m) m) | (Fm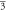 m) m) | (Fm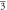 m) m) | ||||
The structural parameters and cell volumes of Cs2AgBiBr6−xClx are almost the same as reported in previous theoretical and experimental studies on mixed halide compounds [17, 19, 27]. Jing Su et al performed a theoretical study from the VASP code based on the Cl atoms mixed Cs2AgBiBr6, the deviation (%) of their lattice parameters' values from our computed values is negligible i.e. (x = 1; a = 0.33%, c = 0.22%; x = 2; a = 0.69%, b = 0.22%, c = 0.18% and x = 4; a = 0.33%, b = 0.07%, c = 0.25%; x = 5; a = 0.24%) [17].
In table 2, the opposite structural behavior of the cell volume of Cs2AgBiBr6−xClx is seen in Cs2AgBiBr6−xIx. As expected, the cell volume increases when more iodine atoms are substituted instead of bromine atoms in the parent compound, Cs2AgBiBr6 because the ionic radius of the I− ion is larger than the Br− ion, and the I− ion exhibits lower electron negativity than the Br− ion. In both cases, the cubic phase can be seen in the pure Cs2AgBiBr6 but it tends to have a lower symmetry with the added iodine atoms and ended up with the tetragonal structure when x = 5. A similar facial transition effect is seen with chlorine substitutions.
The mixed halides with the fluorine atoms gave the lowest cell volume for all the number of substitutions compared to the chlorine and iodine atoms substitutions because the fluorine atom has the highest electron negativity and the smallest anion radius.
3.2. Elastic properties
The elastic constant could describe the material's behavior when stress is applied to it. We have computed the single crystal elastic constant using the finite strain technique shown in table 3 for the cubic phase of Cs2AgBiX6 (X = F, Cl, Br, I) within the GGA approach. Within the elastic region, stress and strain could be expressed in Voigt notation which satisfies Hooke's law, [22]
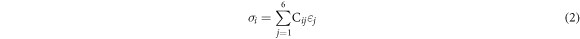
where  and
and  are respectively the stress and strain. Both stress and strain have three tensile and three shear components. i.e. 1 ≤ i,j ≤ 6.
are respectively the stress and strain. Both stress and strain have three tensile and three shear components. i.e. 1 ≤ i,j ≤ 6.  is the second-order elastic stiffness tensor in the 6 × 6 matrix and it could be derived from the first-order derivative of the stress–strain curves as represented in equation (2). The
is the second-order elastic stiffness tensor in the 6 × 6 matrix and it could be derived from the first-order derivative of the stress–strain curves as represented in equation (2). The  values depend on the symmetry of the crystal structure. The cubic crystal which has the higher symmetry consists of three independent elastic constants but for lower symmetry like the triclinic structures associated with 21 single elastic constants, when the symmetry reduces, the number of independent elastic constants increases. We calculate elastic constants for the cubic phase (C11, C12, and C44) using the finite strain method which satisfies the mechanical stability criteria as shown below.
values depend on the symmetry of the crystal structure. The cubic crystal which has the higher symmetry consists of three independent elastic constants but for lower symmetry like the triclinic structures associated with 21 single elastic constants, when the symmetry reduces, the number of independent elastic constants increases. We calculate elastic constants for the cubic phase (C11, C12, and C44) using the finite strain method which satisfies the mechanical stability criteria as shown below.
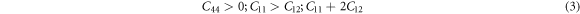
Using the calculated single-crystal elastic constants  s could predict the elastic moduli of the macroscopic mechanical properties such as bulk modulus, Young's modulus, Poisson ratio, etc. through the Voigt-Reuss-Hill approximations using the following relationships [34, 35].
s could predict the elastic moduli of the macroscopic mechanical properties such as bulk modulus, Young's modulus, Poisson ratio, etc. through the Voigt-Reuss-Hill approximations using the following relationships [34, 35].
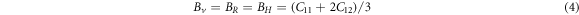
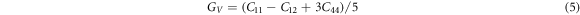
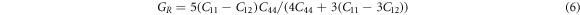
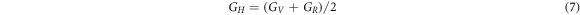

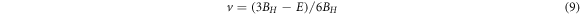
The subscripts R, V, and H represent the Reuss, Voigt, and Hill approximations, respectively.
Table 3. The calculated single-crystal elastic constants Cij (in GPa), bulk modulus B (in GPa), shear modulus G (in GPa), Poisson's ratio (v), Young's modulus E (in GPa), compressibility(GPa−1), Anisotropy(A) for Cs2AgBiX6 (X =, Cl, Br, I) phases. Subscript V indicates the Voigt bound, R indicates the Reuss bound, and H indicates the Hill average.
| Properties | Cs2AgBiF6 | Cs2AgBiCl6 | Cs2AgBiBr6 | Cs2AgBiI6 |
|---|---|---|---|---|
| Cij | C11 = 47.614 | C11 = 32.474,39.378 [36],50.03 [35] | C11 = 32.674,34.09 [38] 24.55 [36],38.74 [37], 44.05 [35] | C11 = 29.568 |
| C12 = 23.632 | C12 = 15.645,13.76 [36],19.96 [35] | C12 = 14.470,13.56 [36],7.58 [37], 16.36 [35], 7.18 [38] | C12 = 12.742 | |
| C44 = 11.060 | C44 = 7.103,8.71 [36], 7.76 [35] | C44 = 6.880,6.75 [38] 7.53 [36], | C44 = 6.221 | |
| 7.46 [37], 6.56 [35] | ||||
| BH | 31.626 | 21.175, 29.98 [35] | 20.538, 25.59 [35] | 18.35 |
| Gv | 11.43 | 6.59 | 7.77 | 7.10 |
| GR | 11.415 | 6.425 | 7.625 | 6.945 |
| GH | 11.424 | 6.508, 10.14 [35] | 7.697, 8.89 [35] | 7.021 |
| EV | 30.61 | 17.91 | 20.70 | 18.86 |
| ER | 30.567 | 17.504 | 20.355 | 18.50 |
| EH | 30.588 | 17.709 | 20.526 | 18.681 |
| vv | 0.34 | 0.36 | 0.33 | 0.33 |
| vR | 0.339 | 0.362 | 0.335 | 0.332 |
| vH | 0.339 | 0.361 | 0.333 | 0.330 |
| (B/G) | 2.77 | 3.25, 2.96 [35] | 2.67,2.59 [36], 2.88 [35], 6.8 [38] | 2.61 |
| A | 0.922 | 0.844 | 0.756 | 0.739 |
| Debye Temperature(K) | 175.1 | 133,164 [35] | 127.9,217 [36],136 [35], 137.64 [38] | 111.9 |
The obtained Cij values and all the other macroscopic elastic moduli increase from iodide compound to fluoride compound. For Cs2AgBiX6 (X = Cl, Br, I), our calculated elastic constant values show good agreement with the past theoretical results [36–38]. This is the first time elastic constant calculations have been carried out for Cs2AgBiF6. Anisotropy is defined by A = 2C44/(C11–C12), the material is isotropic if A = 1, otherwise, it will be anisotropic, which is the case for all cubic Cs2AgBiX6 structures as illustrated in table 3. The anisotropic nature could be seen along the principle directions in our compounds as C11 ≠ C12 ≠ C44 [38]. The derived bulk and shear moduli could explain the hardness of the material under different strains. Pugh's criterion (B\G) could be used to determine a material's ductile or brittle nature. When B/G > 1.75 material shows the character of ductility when it is less than 1.75, the material is brittle. Our halide compounds show the nature of ductility. The stiffness of the material described by Young's modulus, and the compressibility of the compound increase from the iodide compound to the fluoride compound. The bonding nature of the material could be described from the Poisson ratio (ν). If ν is less than 0.1, the material shows covalent characteristics, and the tabulated ν values are greater than 0.25, thus the materials are ionic as ν > 0.25 means ionic nature [15].
3.3. Electronic properties
The band gap of the material determines the material's suitability for photovoltaic applications. We employ GGA and the most accurate HSE06 methods to calculate electronic band structures for materials Cs2AgBiX6 and the results for the band gaps are shown in table 4. We found that the GGA band gap value underestimates the experimentally verified band gap values reported in the literature. However, HSE06 band gap values are in good agreement with the past theoretical and experimental findings for the cubic phases of Cs2AgBiX6 (X = Cl, Br, I). However, there we can see the overestimation of the experimental band gap value because we used the standard 25% Hatree Fock (HF) exchange with the screening parameter of 0.2. These standard values were used for the rest of the electronic property calculation in both pure halides and mixed halides. Deepika et al obtained the band gap value of 3.15 eV for the Cs2AgBiCl6, there they implemented the spin–orbit coupling (SOC) along with the HSE06 and obtained the band gap value of 2.60 eV which is closer to the experimental value of 2.77 eV [39]. Also, they observed when increasing the HF exchange term (α) the band gap values are decreasing. McClure et al used the α as 26% and different cutoff energies which vary from our computational method with the incorporation of SOC and they obtained the band gap values of 2.62 eV and 2.06 eV which are close to the experimentally derived values of 2.77 eV and 2.19 eV for Cs2AgBiCl6 and Cs2AgBiBr6 respectively [7]. As we observed from the past theoretical studies, they employed the SOC along with the exchange–correlation term due to the heavy metal, Bi included in these perovskites [7, 39, 40] which can influence the band degeneracy or the band splitting of Bi-p orbital [17]. Hence this could lead to a decrease in the band gap value than the HSE06 without SOC. Due to the high computational cost and time consumption, we implemented our DFT calculations only with the HSE06 excluding the SOC, hence this can overestimate our bandgap values compared to the experimental findings. The same phenomena influenced the Cs2AgBiI6 where we obtain its band gap value of 1.78 eV which is overestimated from the past theoretical studies which are reported as 1.08 eV and 1.32 eV [7, 40]. However, SOC will not be affected greatly for the calculations based on the VASP code as Feng et al discussed that SOC can be addressed by the PAW method which is the accurate pseudopotential approximation [41]. As emphasized earlier, this is the first time the hybrid calculations for the cubic phase of Cs2AgBiF6 and GGA and HSE06 calculations on tetragonal structures of Cs2AgBiX6 (X = F, Cl, I) are reported. Figure S1 and figure 2 illustrate the electronic band diagrams of the Cubic and Tetragonal phases of Cs2AgBiX6 (X = F, Cl, Br, I) respectively. The band structures E(k) were computed on a discrete k mesh along the high-symmetry directions in the BZ.
Table 4. Calculated band gap values (Eg; in eV) of the halide compounds using GGA and HSE06 method.
| Compound | Eg (GGA/PBE) | Eg (HSE06) | ||
|---|---|---|---|---|
| Cubic | Tetragonal | Cubic | Tetragonal | |
| Cs2AgBiF6 | 2.79 | 2.60 | 4.53 | 4.28 |
| 2.36 [44] | ||||
| Cs2AgBiCl6 | 1.92 | 1.97 | 3.17 | 3.25 |
| 1.92 [44], 1.91 [45] | 3.15 [39], 2.62 [7], 2.35 [40] | |||
| Cs2AgBiBr6 | 1.42 | 1.56 | 2.51 | 2.70, 2.3 [48] |
| 1.41 [44], 1.42 [45],1.33 [46] | 2.06 [15] | 2.06 [7], 1.79 [40], 1.98 [47] | ||
| Cs2AgBiI6 | 0.91 | 1.23 | 1.78, 1.08 [40], 1.32 [28] | 2.16 |
| 0.76 [44], 0.89 [45] | ||||
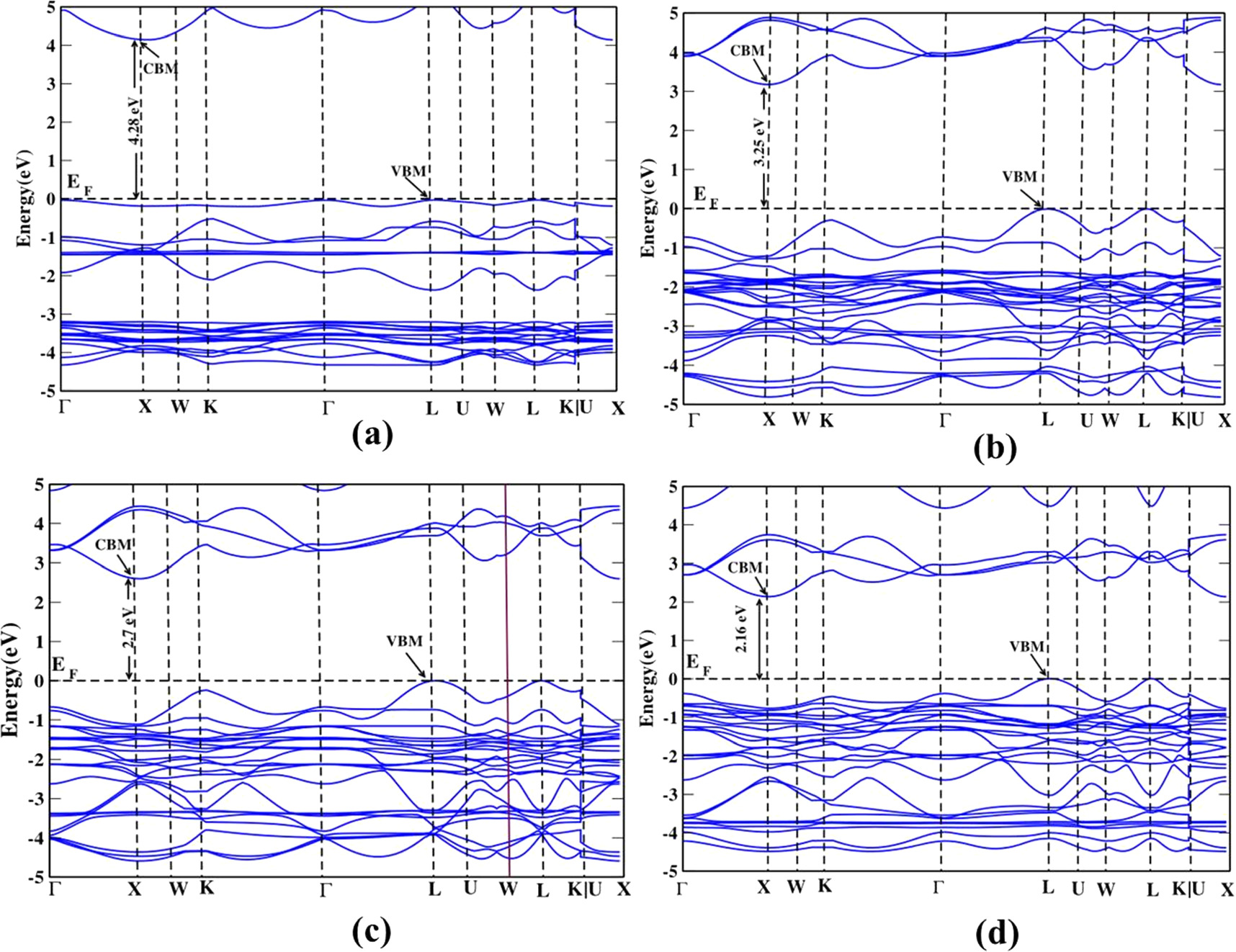
Figure 2. Calculated electronic structure of tetragonal phase in HSE06 method for (a) Cs2AgBiF6 (b) Cs2AgBiCl6 (c) Cs2AgBiBr6 (d) Cs2AgBiI6. The Fermi level is set at zero energy and marked as EF.
Download figure:
Standard image High-resolution imageIt is important to note that the band structures of these double perovskites in both cubic and tetragonal phases show an indirect band gap nature. The computed band gap values are listed in table 4 along with the references. The conduction band minimum (CBM) lies at the L-point whereas the valence band maxima (VBM) lies at the X-point in cubic phase materials (see figure S1). This is in agreement with the work done by Xinbo et al regarding the location of the band edge positions [42].
For the tetragonal phase (figure 2) CBM lies at X and VBM lies at L high symmetric points in the BZ. The band gap decreases from fluoride compound to iodide compound as we change the halogen atom. This could be related to the lattice parameters represented in table 1. The lattice parameters of the Cs2AgBiX6 in both cubic and tetragonal phases increase and also the bond length increases when the halogen atom of the material changes from F to I which could influence the reduction of the bandgap values accordingly. Typically, smaller bond lengths mean the electrons are more tightly bound to the atom, and hence it requires more energy to excite the electron from the valence band to the conduction band. Thus fluoride-based compound needs to absorb higher photon energy compared to other compounds as discussed by Rajeev et al [29] Volonakis et al also showed that indirect band gap values increase when moving up the halogen column in the periodic table [8]. The band gap values in the cubic phase are smaller than the tetragonal phases of these relevant double perovskites as seen in table 4. This could be explained by symmetry characterization. The symmetry is reduced from cubic to tetragonal with the influence of structural distortion [43]. This could lead to small band gap values in the cubic phase compared to the tetragonal phase.
We calculated band gap values employing both GGA/HSE methods for a variety of mixed halides of the form Cs2AgBiBr6−x Xx (X = F, Cl, I) in table 5. The mixed halide compounds show an indirect band gap nature when the bromine atoms are substituted by chlorine, iodine, and fluorine atoms. The band gaps of Cs2AgBiBr6−xClx mixed halides varied between the values of pure halide 3.17 eV (Cs2AgBiCl6) and 2.51 eV (Cs2AgBiBr6) as shown in table 5. The lowest bandgap value was found at 2.35 eV when the one bromine atom was substituted by the chlorine atom. After that band gap increases to 3.01 eV for the substitutions of five bromide atoms by the chloride atoms. Our HSE06 results for Cs2AgBiBr6−xClx are in agreement with the experimental findings [49]. Matthew et al observed the band gap linearly increases in Cs2AgBiBr6−x Clx in between the bandgap values of Cs2AgBiBr6 (2.19 eV) and Cs2AgBiCl6 (2.77 eV) experimentally. However, they observed, the deviation of the law of Vegard when increasing the chlorine concentration to the bromide compound where the chlorine content exceeds 85% [49].
Table 5. Calculated band gap values (Eg; in eV) of the materials using GGA and HSE06 method for Cs2AgBiBr6−xFx, Cs2AgBiBr6−xClx, and Cs2AgBiBr6−xIx (x = 0, 1, 2, 3, 4, 5, 6).
| Substitutions | Cs2AgBiBr6−xFx | Cs2AgBiBr6−xClx | Cs2AgBiBr6−xIx | |||
|---|---|---|---|---|---|---|
| Eg (GGA/PBE) | Eg (HSE06) | Eg (GGA/PBE) | Eg (HSE06) | Eg (GGA/PBE) | Eg (HSE06) | |
| x = 0 | 1.42 | 2.51 | 1.42 | 2.51 | 1.42 | 2.51 |
| x = 1 | 1.63 | 2.55 | 1.46 | 2.35, 2.27 [49] | 1.32, 1.07 [50] | 2.03 |
| x = 2 | 1.89 | 2.86 | 1.50 | 2.44, 2.34 [49] | 1.15, 0.94 [50] | 2.21 |
| x = 3 | 2.02 | 3.1 | 1.62 | 2.85, 2.41 [49] | 1.00, 0.93 [50] | 2.02 |
| x = 4 | 2.26 | 3.5 | 1.68 | 2.68, 2.51 [49] | 0.96, 0.86 [50] | 1.68 |
| x = 5 | 2.32 | 3.65 | 1.80 | 3.01, 2.60 [49] | 0.90, 0.81 [50] | 1.87 |
| x = 6 | 2.79 | 4.53 | 1.92 | 3.17 | 0.91 | 1.78 |
Based on the accurate HSE06 calculations, it is noted that the iodine substitutions result in low band gap values with the lowest band gap of 1.68 eV when the four bromine atoms were substituted by the iodine atoms. The bromide and iodide ions show a good combination of halide compounds which result in band gap values of 2.03 eV, 2.21 eV, and 2.02 eV for the compounds of Cs2AgBiBr5I, Cs2AgBiBr4I2, and Cs2AgBiBr3I3 respectively which correspond to the visible light region in the solar spectrum. Experimental and theoretical works were carried out by Hua Wu et al on mixed halides of iodine mixed Cs2AgBiBr6. Experimentally, the halide exchange on Cs2AgBiBr6 was done using the post-treatment with methylammonium iodide (MAI) salt [50]. When the MAI concentration is 15 mg mL−1 which is correspondence to the substitution of the two bromine atoms in Cs2AgBiBr6 with the inclusion of iodine atoms (x = 2) resulted in a band gap value of 2.20 eV which is in good agreement with our work (2.21 eV) under HSE06 approximation [50]. However, their DFT calculations at the GGA level show much deviation between 7% - 18%. This much deviation can occur due to the different material simulational packages along with their own set of input parameters and simulation tools when determining the corresponding band gap values. The Cs2AgBiF6 has the highest band gap among the halide compounds. When it comes to the mixed halides with the replacement of bromine atoms by the fluorine atoms, the band gap of the Cs2AgBiF6 could be able to reduce with the incorporation of the bromine atoms. The lowest band gaps of 2.55 eV and 2.86 eV were obtained by substituting one and two bromine atoms with fluorine atoms respectively (table 5). The same type of band diagram could be seen for all the mixed halide compositions of Cs2AgBiBr6−xFx , Cs2AgBiBr6−xClx, and Cs2AgBiBr6−xIx and are shown to possess indirect band gaps. The VBM and CBM are located along the high symmetry points of X and L in the first BZ. Here we present the band diagrams of the Cs2AgBiBr5Cl, Cs2AgBiBr5I, and Cs2AgBiBr5F as shown in figure 3. The rest of the mixed halides' band structures could be seen in figure S2-S4 in the supplementary document. Almost the same electronic features can be seen in the recent theoretical work based on HSE06 for the Cs2AgBiBr6−x Clx (x = 1, 2, 3, 4, 5) [17]. Jing Su et al showed the bandstructure calculations of mixed halides on Cs2AgBiBr6−xClx (x = 0, 1, 2, 3, 4, 5, 6) have the indirect band gap nature and the calculated band gap values are in the range of 2.1–2.7 eV using the hybrid functional method, HSE06 [17].

Figure 3. Calculated electronic structure of mixed halides in HSE06 method for (a) Cs2AgBiBr5F (b) Cs2AgBiBr5Cl (c) Cs2AgBiBr5I. The Fermi level is set at zero energy and marked as EF.
Download figure:
Standard image High-resolution image3.4. Density of States (DOS) calculations
To have a better understanding of the orbitals of the elements that are responsible for the band edge positions we study their Total Density of States (TDOS) and partial density of states (PDOS) using the HSE06 approximation in both phases. The calculated PDOS depicted in figure S5 and figure 4 near the Fermi level. VBM consists of hybridization of the halide X-p states (Cl-3p, Br-4p, I-5p, and F-2p) and Ag-4 d orbitals with a little admixture of Bi-6s states. The CBM is mainly composed of the antibonding of Bi-6 p orbitals and slightly of X- p. The contribution of the Cs atom for the electron transition is very low near the EF because there is no obvious DOS of Cs+ ions. However, Cs+ ion is helpful for the charge neutrality of the structure. The electron transitions could appear as indirect from X-p and Ag-4d antibonding states to the Bi-6p states, in agreement with the previous DFT results [51].
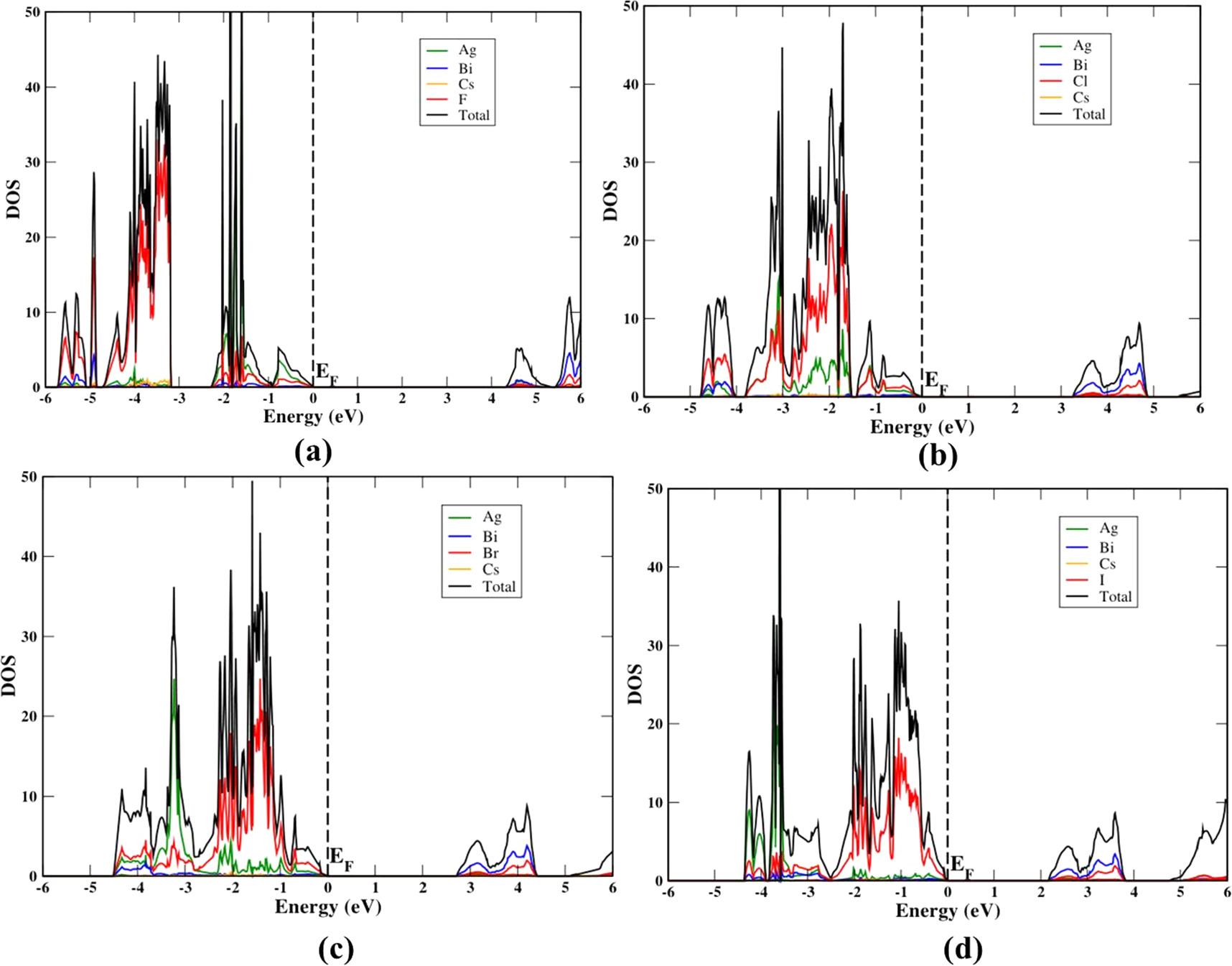
Figure 4. Calculated PDOS of the tetragonal phase in the HSE06 method for (a) Cs2AgBiF6 (b) Cs2AgBiCl6 (c) Cs2AgBiBr6 (d) Cs2AgBiI6. The Fermi level is set at zero energy and marked as EF.
Download figure:
Standard image High-resolution imageDOS calculations for mixed halide compounds were carried out employing HSE06. The PDOS of the mixed halide compounds of Cs2AgBiBr6−xClx, Cs2AgBiBr6−xIx, and Cs2AgBiBr6−xFx are computed near the Fermi level. The same density of states features could be noticed for the varied substitutions of F, Cl and I on the pristine Cs2AgBiBr6. We present the PDOS feature of mixed halide compound by considering Cs2AgBiBr5Cl, Cs2AgBiBr5I, and Cs2AgBiBr5F in figure 5 as examples to understand the detail states of the valence band and conduction band. The top of the valence band is mainly composed of Ag - 4d orbitals, Br-4p, and substituted (F / Cl / I) halogen (2p /3p / 5p) orbitals, while the conduction band is mainly contributed by Bi - 6p orbitals.
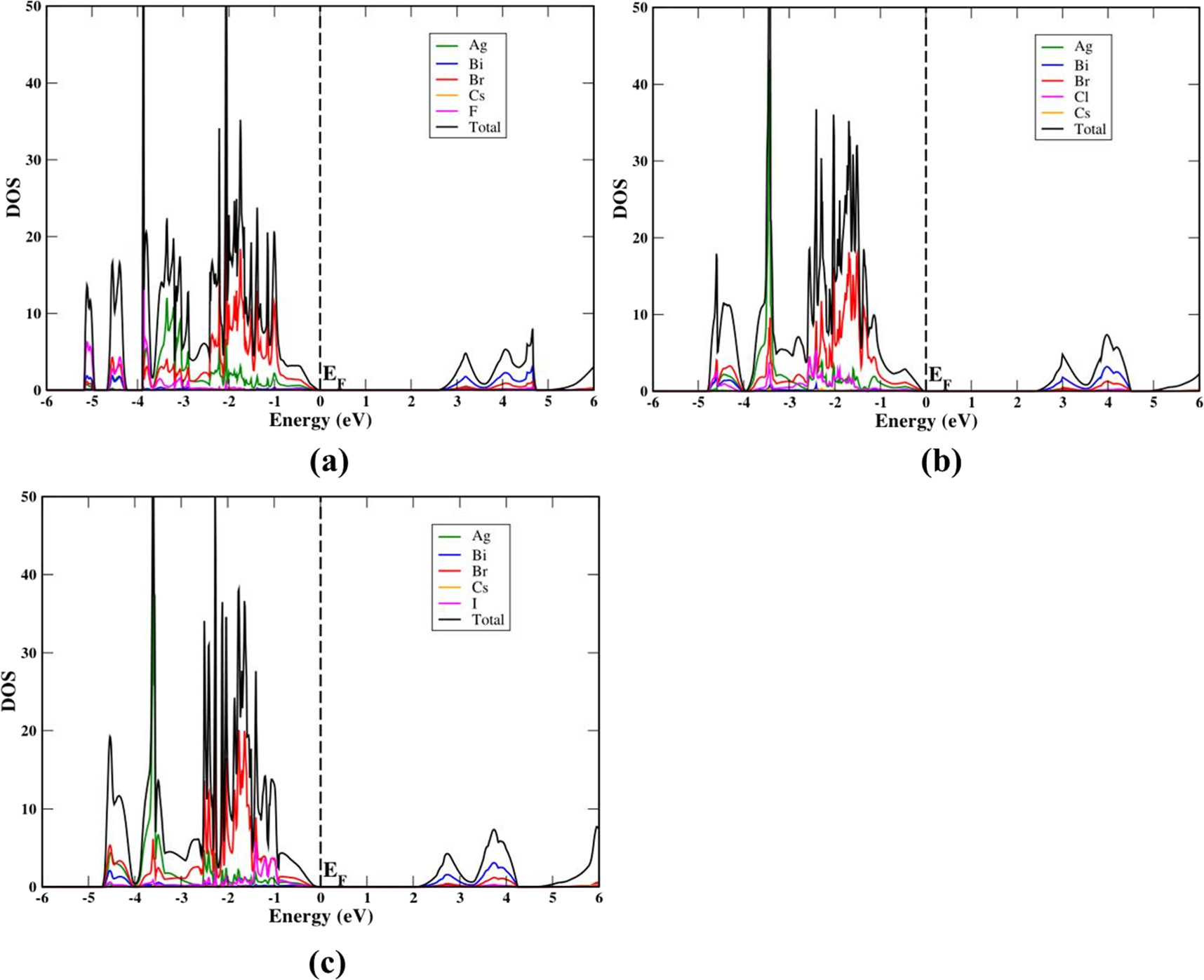
Figure 5. Calculated PDOS of mixed halides in HSE06 method for (a) Cs2AgBiBr5F and (b) Cs2AgBiBr5Cl (c) Cs2AgBiBr5I. The Fermi level is set at zero energy and marked as EF.
Download figure:
Standard image High-resolution image3.5. Effective mass calculations
The electron/hole motion and the materials' conductivity could be predicted by effective mass calculations [52]. The smaller (bigger) the effective mass (m*) is the faster (slower) the carriers could migrate. The low effective mass would be a sign of the low resistance and high conductivity of a material predicting it is a good material for practical applications. Here we use the finite difference method for the effective mass calculator (EMC) using PBE and HSE06 functional other than parabolic fitting to investigate the carrier mobilities. Parabolic fitting is not always applicable because it is quite complicated to fit the band to a quadratic polynomial. The computed band structures in figure S1 and figure 2 show the bands have different dispersion characteristics i.e. no proper parabolic bands at all. Therefore we computed our effective masses of electrons and holes using the Finite Difference Method (FDM) where the second and mixed derivates of the equation (11) are evaluated on a three-point stencil (h is step size) by equations (12) and (13) with the order of error estimation O(h2 ). This FDM method was used by other researchers and obtained valid effective mass values to compare with the experimental findings [52]. The effective mass(m*) of a charge carrier is defined as in equation (10).
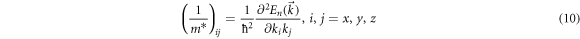
where indices i and j denote reciprocal components, k is the wave vector, and ħ is the reduced Planck constant.
x
,
y
, and
z
represent the reciprocal cartesian space (2π/A) and  is the dispersion relation for the n-th band. The explicit form of the symmetric tensor on the right-hand side of equation (10) is given below.
is the dispersion relation for the n-th band. The explicit form of the symmetric tensor on the right-hand side of equation (10) is given below.



the effective mass components are the inverse of the eigenvalues of the above equation (11) and the principal directions correspond to the eigenvectors.
Effective masses of electrons and holes of Cs2AgBiX6 in both the cubic and tetragonal phases are computed and presented in tables 6 and 7 with the three effective masse components (M1, M2, and M3). A similar method was utilized by Volonikas et al to calculate the effective masses of charge carriers using finite difference calculations for the cubic phases of the compounds Cs2AgBiX6 (X = Cl, Br, I) [53]. Our calculated results are in agreement with those results. The effective masses of the cubic phase mentioned in table 6 are derived with the step size of 0.01 (1/Bohr). In table 7, we present the effective masses for the tetragonal phase with the step size of 0.1(1/Bohr). The relevant k-points for the top of the valence band and bottom of the conduction band for both phases are found using the band structure plots of figure S1 and figure 2 where the electrons and holes transitions are taking place. The effective masses we obtain, for the cubic phase, are similar to the values obtained from parabolic fitting from the literature survey [30]. Except for the tetragonal phases of Cs2AgBiBr6 other halide compounds for tetragonal phases' effective masses are for the first time calculated values. The effective masses for pure and mixed halide compounds are calculated using GGA and HSE06 approximations and presented in table 6, table 7, table S1, table S2, and table S3. We found that most of the calculations in HSE06 show lesser values for effective masses compared to the values obtained with GGA.
Table 6. Computed effective masses for the cubic phase of Cs2AgBiX6 (X = F, Cl, Br, I) using a step size of 0.01 (1/Bohr), electrons effective masses were calculated at L-point (0.5, 0.5, 0.5) and for holes at X-point (0.5,0,0.5) in the units of the mass of electron at rest (me).
| Material GGA/HSE06 | Electrons | Holes | ||||
|---|---|---|---|---|---|---|
| M1 | M2 | M3 | M1 | M2 | M3 | |
| Cs2AgBiF6 | 1.158/2.097 | 0.589/0.993 | 0.589/0.610 | 0.459/0.284 | 1.524/1.045 | 1.524/1.482 |
| Cs2AgBiCl6 | 0.440/0.416 | 0.343/0.317 | 0.343/0.254 | 0.236/0.176 | 0.896/0.641 | 0.896/0.660 |
| Cs2AgBiBr6 | 0.347/0.343 | 0.261/0.248 | 0.261/0.240 | 0.209/0.155 | 0.833/0.440 | 0.833/0.487 |
| Cs2AgBiI6 | 0.273/0.292 | 0.186/0.195 | 0.186/0.187 | 0.188/0.123 | 0.785/0.559 | 0.785/0.574 |
Table 7. Computed effective masses for the tetragonal phase of Cs2AgBiX6 (X = F, Cl, Br, I) using a step size of 0.1(1/Bohr), electrons effective masses were calculated at X-point (0.5, 0, 0.5) and for holes at L -point (0.5,0.5,0.5) in the units of the mass of electron at rest (me).
| Material GGA/HSE06 | Electrons | Holes | ||||
|---|---|---|---|---|---|---|
| M1 | M2 | M3 | M1 | M2 | M3 | |
| Cs2AgBiF6 | 2.163/0.914 | 0.732/0.475 | 0.614/0.424 | 1.457/1.451 | 1.952/4.622 | 5.996/0.681 |
| Cs2AgBiCl6 | 0.551/0.507 | 0.419/0.362 | 0.379/0.335 | 0.343/0.248 | 0.584/0.451 | 1.915/1.305 |
| Cs2AgBiBr6 | 0.450/0.433 | 0.338/0.304 | 0.334/0.300 | 0.320/0.227 | 0.596/0.435 | 1.246/0.910 |
| Cs2AgBiI6 | 0.557/0.518 | 0.342/0.327 | 0.320/0.304 | 0.206/0.196 | 0.328/0.275 | 0.440/1.061 |
Overall, our effective masses calculated for both phases of Cs2AgBiX6 show that they have higher mobility of electrons than the hole mobility and the mobility increases as X (halogen atom) is changed from F to I. The recombination effect will be lower hence the corresponding diffusion length will be higher. The isotropic nature could be seen mostly in the cubic phase than in the tetragonal phase. Among these halide compounds in both phases, the cubic phase of Cs2AgBiI6 shows the low effective masses of both electrons and holes making it a promising photovoltaic material. The fluoride compound exhibits larger effective masses of charge carriers as shown in tables 6 and 7 compared to the other effective masses of halide compounds. This may be due to having flat valence bands in reciprocal space (see figure S1 and figure 2). The effective mass of holes is higher than the electrons' due to the band dispersion features in both phases. When considering the other kinds of double perovskites, Shuai et al studied the sodium-based double perovskites, Cs2NaBX6 (B = Sb, Bi, X = Cl, Br, I). They too observed the higher values of the holes' effective mass than the electrons' values and also the magnitude of those photocarriers' masses decreased when changing the halogen compound, X-site anion [33]. Wei et al calculated the effective masses using the GGA approximation for the lead-based perovskites which have direct band gaps, CH3NH3 PbI3 for the tetragonal phase. Their obtained values were inferior to our calculated values for the tetragonal phase of the Cs2AgBiI6. However, it shows quite closer values to the holes' effective masses as 0.28 and 0.30 me when compared to our values of 0.206 and 0.328 me. [54]. The effective masses of electrons and holes of the Cs2AgBiBr6 and Cs2AgBiCl6 show lower values than the values obtained by De yuan et al for the Cs2CuBiX6 (X = I, Br, Cl) [55]. The effective masses of the hole charge carriers of the tetragonal phase show similar values to the hole effective masses of tetragonal phases of CH3NH3 PbX3 (X = I, Br) [56]. The effective masses of mixed halide compounds are comparable, chlorine substitutions (table S1) show some higher values than the values in the host compound, Cs2AgBiBr6. However, the iodine substitutions (table S2) lead to lower values when compared to the parent compound. The fluorine substitutions (table S3) to the pure Cs2AgBiBr6 can able to reduce the effective mass of the pure Cs2AgBiF6 compound with the mixing of the bromine atoms. There the effective mass was reduced when more bromine atoms were present, comparatively low effective mass was found for the compound of Cs2AgBiBr5 F.
3.6. Optical properties
When the light irradiates on the surface of the material the phenomena of light reflection and absorption occur. Theoretically, this phenomenon is described by the complex dielectric function, ε(ω) = ε1(ω) + iε2(ω) for the optical properties of materials by Kramer–Kronig relationship. Here ε(ω) describes the interaction between photons and electrons. The real part of ε(ω) in the limit of zero energy (or infinite wavelength) is equal to the square of the refractive index. ε2(ω) is responsible for absorption performance, it reflects the electronic transitions from occupied valence bands to unoccupied conduction bands.
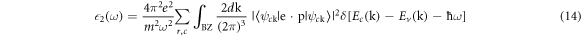
Equation (14) implies the sum of all conduction bands and valence bands where their energies are Ec and Ev respectively.
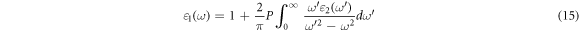
The ε1(ω) see equation (15) gives information on polarizability, From ε2(ω), the real part ε1(ω) can be derived by using the Kramers-Kronig relation. Using the dielectric constant we could compute the frequency-dependent linear optical properties such as refractive index n(ω), absorption coefficient α(ω), extinction coefficient K(ω), reflectivity R(ω), and electron energy-loss spectra, L(ω) [57, 58].
We investigate the optical properties including dielectric function and optical absorption for the considered double perovskites using HSE06 functional for both phases. The calculated optical features are in the range of energy 0 eV to 6 eV.
The absorption coefficient α(ω) describes the attenuation of the photon energy of the material and it plays a major role in identifying the suitability of the material for optoelectronic applications. The absorbance plots in figure 6 were calculated to the corresponding double perovskites in both the cubic phase and tetragonal phase and they did not show any major difference between those phases. The redshift can be seen in figure 6 when changing the halogen atom (X) from F to I. In figure 6, absorption spectra: there we can find the optical band gap. The absorption spectrum edge corresponds to the direct optical transitions from the top of the valence band to the bottom of the conduction band. We could find this direct optical transition of the Cs2AgBiX6 using the absorbance plot with the help of Origin software see table S4. The optical band gaps (table S4) are similar in both phases except for the little variations of magnitude. The absorption intensity is quite lower in low symmetry structure as shown in figure 6 other than that in general both phases show the same optical characteristics. The same feature was obtained for Cs2AgBiBr6 in cubic and tetragonal phases by Tahar et al [15].

Figure 6. Computed absorption of the lead-free halide double perovskites Cs2AgBiX6(X = F, Cl, Br, I) using HSE06.
Download figure:
Standard image High-resolution imageThe two phases of Cs2AgBiBr6 and Cs2AgBiI6 cover the visible and ultraviolet regions. Their optical band gap values (table S4) could be comparable to the reported values of CH3NH3PbBr3 and CH3NH3PbCl3 at 2.26 eV and 3.00 eV respectively [7]. These two halide compounds show the attenuation of solar irradiation high compared to the fluoride and chloride compositions. Cs2AgBiF6 and Cs2AgBiCl6 show the absorbed radiation falls to the ultraviolet region. The Cs2AgBiX6 in both phases achieves a high absorption coefficient of over 105 cm−1 in the regions of both ultraviolet and visible regions.
The experimental studies revealed that Cs2AgBiBr6, extends the absorption to the ultraviolet region as well the absorption begins around 400 nm wavelength which belongs to the ultraviolet region, [47] in agreement with our theoretical observations for bromide compound, Cs2AgBiBr6. Cs2AgBiBr6 of two phases shows high absorption among these halide components. The substitutions for the cubic phase of Cs2AgBiBr6 are done as it shows the highest absorption intensity among these halide compounds (see figure 7 where the absorption coefficient enlarged between the 4.0 eV—5 eV.)
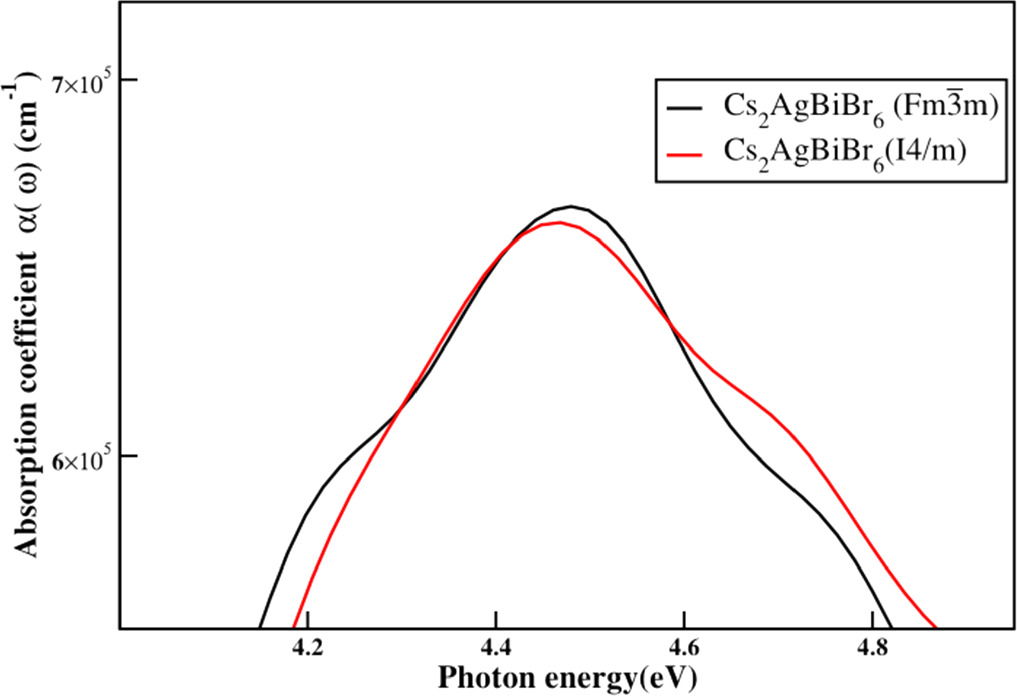
Figure 7. The absorption coefficient of Cs2AgBiBr6 for cubic and tetragonal phases.
Download figure:
Standard image High-resolution imageThe similar features of the absorption spectra for the substitutions of the cubic phase of Cs2AgBiBr6 with F, Cl, and I atoms can be seen in figure S6, figure S7, and figure S8 respectively. These substitutions consist of the indirect band gap nature. Figure S6 and figure S7 show that the blue shift of the absorption curve occurs when increasing the substitutions of the fluorine and chlorine atoms. Figure S8, the redshift occurs when increasing the iodine atoms in the bromide site. The absorption intensity of these mixed halides is situated in between the pure bromide and iodide compounds and it changes corresponding to the mixed halide ratios i.e. Br/F, Br/Cl, and Br/I.
The calculated real and imaginary parts of dielectric functions of Cs2AgBiX6 in both phases show similar features. Concerning the real part (ε1) of the dielectric function for both the cubic(tetragonal) phases which is shown in figure S9. The ε1 starts from 2.48 (2.38), 3.5 (3.41), 4.1 (4.05), and 5.37 (4.98) for Cs2AgBiX6 (X = F, Cl, Br, I). These values are the static dielectric function ε1 (ω→0). At these starting points, materials have the ability to interact with an electric field without absorbing the photon energy [57]. Afterward, the dielectric function increases with the photon energy and attains the maximum values. The maximum ε1(ω) values for Cs2AgBiF6 and Cs2AgBiI6 are at 2.47 eV (2.68 eV) and 5.13 eV (4.95 eV) were obtained for cubic(tetragonal) phases respectively. Cs2AgBiCl6 and Cs2AgBiBr6 are having two peaks of the ε1(ω). Cs2AgBiCl6, the first and second peaks occur at 3.88 eV (3.92 eV) and 4.48 eV (4.55 eV) for cubic (tetragonal) phases respectively. For the Cs2AgBiBr6 first peak occurs at 3.24 eV (3.31 eV) and the second peak at 3.82 eV (3.88 eV) corresponds to the cubic (tetragonal) phases. This implies there is a shift could occur from higher energy to lower energy from F to I. Tripathy et al observed that similar peak shifting occurs to lower energy in imaginary dielectric function by substituting larger halide ions in Cs2AgBiCl6, Cs2AgBiBr6, and Cs2AgBiI6 [30]. Typically these starting values are the dielectric constant where ε1(0). After reaching the imaginary function to these maxims it starts to decrease by increasing the photon energy. There are no negative ε1 values that occur in our computed dielectric function as shown in figure S9 which means it shows the prominent optoelectronic properties in both phases.
The Real part of the dielectric function (ε1) is plotted as shown in figure S10 for the mixed halide compounds. When increasing the substitutions of the fluorine and chlorine atoms to the pure Cs2AgBiBr6, the graphs figure S10 (a), and figure S10 (b) show the blue shift of the ε1 values. For the Cs2AgBiBr6−xIx in figure S10 (c), we could see the redshift of the real part of the dielectric function when increasing the iodine atoms. There are no negative values ε1 values in mixed halide components which indicates that the interior of these materials always exhibits prominent dielectric behavior. De-Yuan et al discussed the same observation for the Cs2CuBiX6 [55]. The computed static dielectric function for mixed halide compounds is given in table S5.
Using the Penn model, The dielectric constant value (ε1(0)) is inversely proportional to the band gap of the material (Eg ) as shown in the below equation (16) [59];
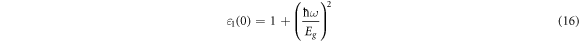
From equation (16), the higher band gap values correspond to the lower ε1(0) values in comparison with the band gap values shown in tables 4 and 5.
Imaginary part (ε2(ω)) describes the interband transitions i.e. electron transitions from occupied states to the unoccupied states, [58] where the electrons transitions could take place from occupied X - p states and Ag - 4d orbitals to the unoccupied Bi-6p states which could be verified with the density of states plots. ε2(ω), which is mentioned in equation (14) could be derived from the band structure and the dipole matrix elements [60]. This equation could be related to the absorption coefficient at a given frequency ω [60]. This could be the reason figure 6, figure S6, figure S7, and figure S8 appear with the same optical features as figure S11 and figure S12 i.e. The imaginary part of the dielectric function and absorption spectra show the same optical behavior in our pure and mixed halide compounds.
Overall, the results suggest that the material Cs2AgBiI6 and Cs2AgBiBr6 have a higher dielectric constant (figure S9) in low energy region (from 2 to 4 eV) following broad optical spectra which covers most of the region in the solar spectrum including visible and ultraviolet regions compared to other compounds. Therefore, the high value of the dielectric function and absorption intensity in the solar spectrum implicated these two compounds could be promising candidates for photovoltaic applications. For the mixed halides, the substitution of fluorine and chlorine atoms increases in Cs2AgBiBr6, figures S12 (a) and S12 (b) demonstrate a clear blue shift in the ε2 values. On the other hand, in the instances of Cs2AgBiBr6−xIx, illustrated in figure S12 (c) there is a discernible redshift observed in the imaginary part of the dielectric function as the number of iodine atoms increases.
The Refractive index is also an important physical parameter related to microscopic atomic interactions. This could measure the transparency of the incident photon [61]. The obtained static refractive index n(0) is related to the static real part of the dielectric function as shown in below equation (17) [62].
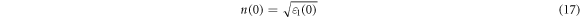
n(0) values are listed in table S6 for the Cs2AgBiX6 in both phases, the plot in figure S13 (a). They are in good agreement with past theoretical works [57, 58, 62]. The n(0) value increase when changing the halogen (X) compound from F to I. The computed n(0) values for the mixed halide compounds are listed in table S7. Fluorine and chlorine atoms substituted compounds show the n(0) values are decreasing with the increase of fluorine and chlorine atoms in figure S16 (a) and figure S14 (a). However n(0) values of figure S15 (a) increase when the iodine atoms are substituting to the pristine, Cs2AgBiBr6 compound.
The scattering and reflection of light from materials are related to the R(ω). The computed R(0) values are listed for pure and mixed halide compounds see table S6 and table S7. Reflectivity spectra for the lead-free double perovskite, Cs2AgBiX6 for both phases are shown in figure S13 (b). The reflectivity R(0) of the mixed halide compounds decreases from the 9% and 11.3% reflectivity to 5.17% and 9.25% when 'F' and 'Cl' atoms substitute in place of 'Br' [figure S16 (b) and figure S14 (b)]. However, reflectivity increases up to 15.42% when the iodine substitutions take place [figure S15 (b)].
The extinction coefficient (K(ω)) implies the ability to absorb the photon energy by the material. The higher values of extinction coefficient could be seen in the lower energy range compared to the higher energy range in pure halide compounds see figure S13 (c). For the mixed halide compounds when the fluorine and chlorine atom substitutions are increasing the spectra shift to a higher energy range [figure S14 (c) and figure S16 (c)], nevertheless, the iodine substitutions shift the spectra to a lower energy range [figure S15 (c)]. In these lower regions, the material could absorb more photon energy which is described by their absorption spectra, thus absorption coefficient and extinction coefficient are related by the following equation (18) [63].
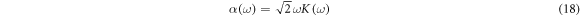
Higher values of extinction coefficient relate to higher absorption.
The energy loss function is an important factor in photovoltaic applications. It describes the energy loss of the moving charge carriers inside the material. Here we computed the energy loss function for pure and mixed halide components in figure S13 (d), figure S14 (d), figure S15 (d), and figure S16 (d). The peaks of energy loss functions are related to the corresponding abrupt reduction values of R(ω).
3.7. Transition dipole moment
The transition dipole moment (TDM) is typically expressed as the probability of electron transition between states in the valence band and the conduction band [22]. The TDM calculations provide information regarding the height of the intensity of absorption spectra instead of the transitions of electrons between two states which mainly depends on the selection rules. We performed the optoelectronic properties on the cubic and tetragonal phases of Cs2AgBiX6 perovskite and present the HSE06 calculated band structures and corresponding TDM diagrams as shown in figures 8 and 9 below. In this configuration, there are a total of 76 valence electrons. Following that, we proceed to calculate the square of the transition dipole moment originating from the highest valence band (indexed at 38) to the lowest conduction band (indexed at 39). This computation yields the square modulus of the electronic wave functions at the edges of these bands. As we can see the resultant absorption spectra show the different peaks are visible in the absorption region that arises as a result of transitions of electrons between VBs and CBs. More transitions occur along the high symmetry k-points; G to L, G to K, and G to X for both phases. The intensity of the height increases when it moves from F to I because the band gap decreases as more transitions could happen from the valence band to the conduction band. The transition dipole moment occurs from Ag - d (VBM) to Ag - s (CBM) states and also more electron transitions are strong and allowed at the Gamma point for all the compounds including the mixed halides. This results in strong intra-atomic transitions. Cs2AgBiI6 in both phases shows a higher intensity of the TDM compared to other double perovskite materials as it shows the lowest band gap values compared to other materials.

Figure 8. Transition dipole moment of cubic phases (a) Cs2AgBiF6 (b) Cs2AgBiCl6 (c) Cs2AgBiBr6 (d) Cs2AgBiI6. The top of the VB and bottom of the CB bands are highlighted by bold black color. The Fermi level is set as zero energy and marked as EF..
Download figure:
Standard image High-resolution image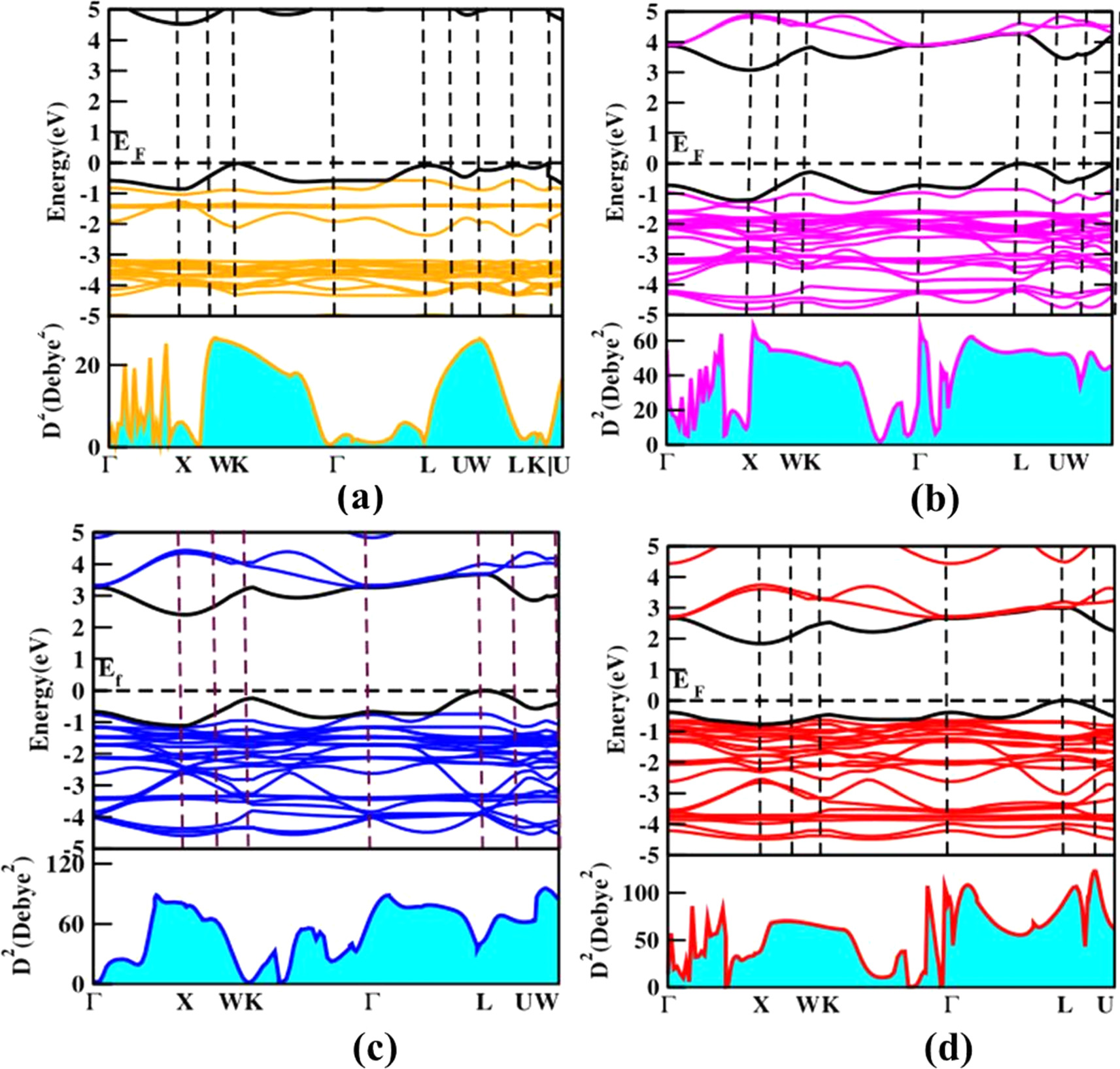
Figure 9. Transition dipole moment of tetragonal phases (a) Cs2AgBiF6 (b) Cs2AgBiCl6 (c) Cs2AgBiI6. The top of the VB and bottom of the CB bands are highlighted by bold black color. The Fermi level is set as zero energy and marked as EF.
Download figure:
Standard image High-resolution imageCompared to cubic phase compounds, the tetragonal phase compounds of Cs2AgBiF6, Cs2AgBiCl6, and Cs2AgBiBr6 show a higher value of transition probabilities than the cubic phase for these compounds. However, the cubic phase of Cs2AgBiI6 shows more electron transitions among all the halide compounds of both phases as it shows the lowest band gap value of 1.78 eV (table 4).
The Fluorine substitutions mixed halides (figure S17) represent the more electron transitions that take place on Cs2AgBiBr5F, Cs2AgBiBr4F2, and Cs2AgBiBr3F3. The pure Cs2AgBiF6 has low electrons' transitions for both phases but with the substitution of the bromine atoms with the fluorine atoms, we were able to increase the height of TDM resulting in more transitions taking place from VB to CB. When it comes to the chlorine atom substitutions, the lowest band gaps were obtained for the compounds of Cs2AgBiBr5Cl and Cs2AgBiBr4Cl2 (table 5) showing that more transitions occurred in our TDM figures (figure S18). These two compounds carry the same height of intensity of the absorption spectrum as well for the TDM. All the iodine-substituted compounds (figure S19) gave more transition probabilities compared to the fluorine and chlorine substitutions. Among them, Cs2AgBiBr2I4 and Cs2AgBiBrI5 show the high intensity of TDM because their computed band gaps were lower compared to the other iodine substitutions.
3.8. Phonon spectrum
The phonon dispersion relations are defined as the k dependency of the frequencies for all branches and selected directions in the crystal. The phonon dispersion relations are drawn along the crystal high-symmetry points of the Brillouin zone. Phonon calculations are done for all polytypes to understand the dynamical stability. The dynamical matrices are calculated from the force constants, and also the phonon dispersion curves, at the theoretical equilibrium volume were computed (see figures 10 and 11). We found that two of the tetragonal structures for Cs2AgBiBr6 and Cs2AgBiCl6 are dynamically stable. While all the cubic structures show negative frequencies and are dynamically unstable. Such instability causes the entire lattice to undergo a structural change. From figures 10 and 11, it is clear that a phonon band gap between the acoustic and optical modes is found in all compounds (cubic Cs2AgBiI6) except in the cubic and tetragonal phases. This separation affects the phonon scattering processes and the lattice thermal conductivity.

Figure 10. Calculated phonon dispersion curves of cubic phase (a) Cs2AgBiF6 (b) Cs2AgBiCl6 (c) Cs2AgBiBr6 (d) Cs2AgBiI6.
Download figure:
Standard image High-resolution image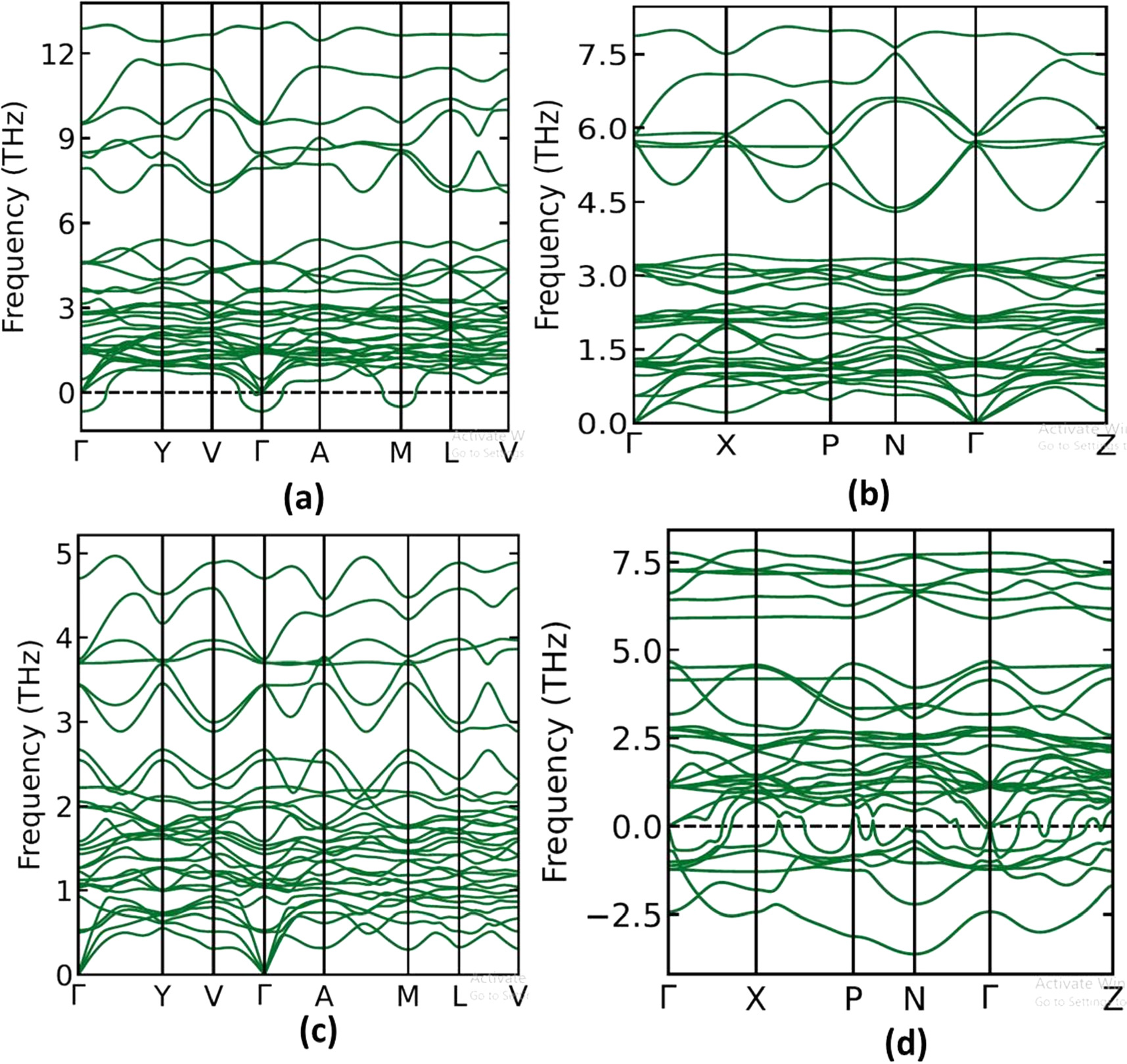
Figure 11. Calculated phonon dispersion curves of tetragonal phase (a) Cs2AgBiF6 (b) Cs2AgBiCl6 (c) Cs2AgBiBr6 (d) Cs2AgBiI6.
Download figure:
Standard image High-resolution image3.9. Thermodynamical properties
Thermal properties of the solids can be derived from lattice vibrations in terms of phonons. Here we examined the thermodynamical behavior of the Cs2AgBiX6 (X = F, Cl, Br, and I) in both phases. For these double perovskites, we computed the thermodynamic properties such as vibrational energy (kJ/mol), free energy (kJ/mol), heat capacity (J/K/mol), and entropy (J/K/mol). The temperature range was set for these compounds from 0 to 1000 K to compute the above-mentioned thermal properties. Figures 12 and 13 show that the vibrational energy increases up to 200 kJ mol−1 where the temperature meets 1000 K and the free energy decreases exponentially which satisfies Debye's law [64]. Entropy increases in both phases when the temperature is increasing which proves the third law of thermodynamics and also it reaches the absolute zero value at very low temperatures [64]. A recent theoretical study evaluated the thermodynamic properties such as free energy, enthalpy, and entropy on the cubic phase of Cs2AgBiX6 (X = Cl, Br, and I) using the phonon vibrational modes. They observed when raising the temperature, those thermodynamic properties approach zero at extremely low temperatures which corresponds to our findings [65]. At the constant volume, the heat capacity was computed as CV for both phases. All the compounds exhibit high CV which is around 200 J K−1 mol−1 which does not change with the rising of the temperature.

Figure 12. Computed thermodynamic parameters Free energy, Entropy, Heat capacity (CV), and Vibrational energy of cubic phases (a) Cs2AgBiF6 (b) Cs2AgBiCl6 (c) Cs2AgBiBr6 (d) Cs2AgBiI6.
Download figure:
Standard image High-resolution image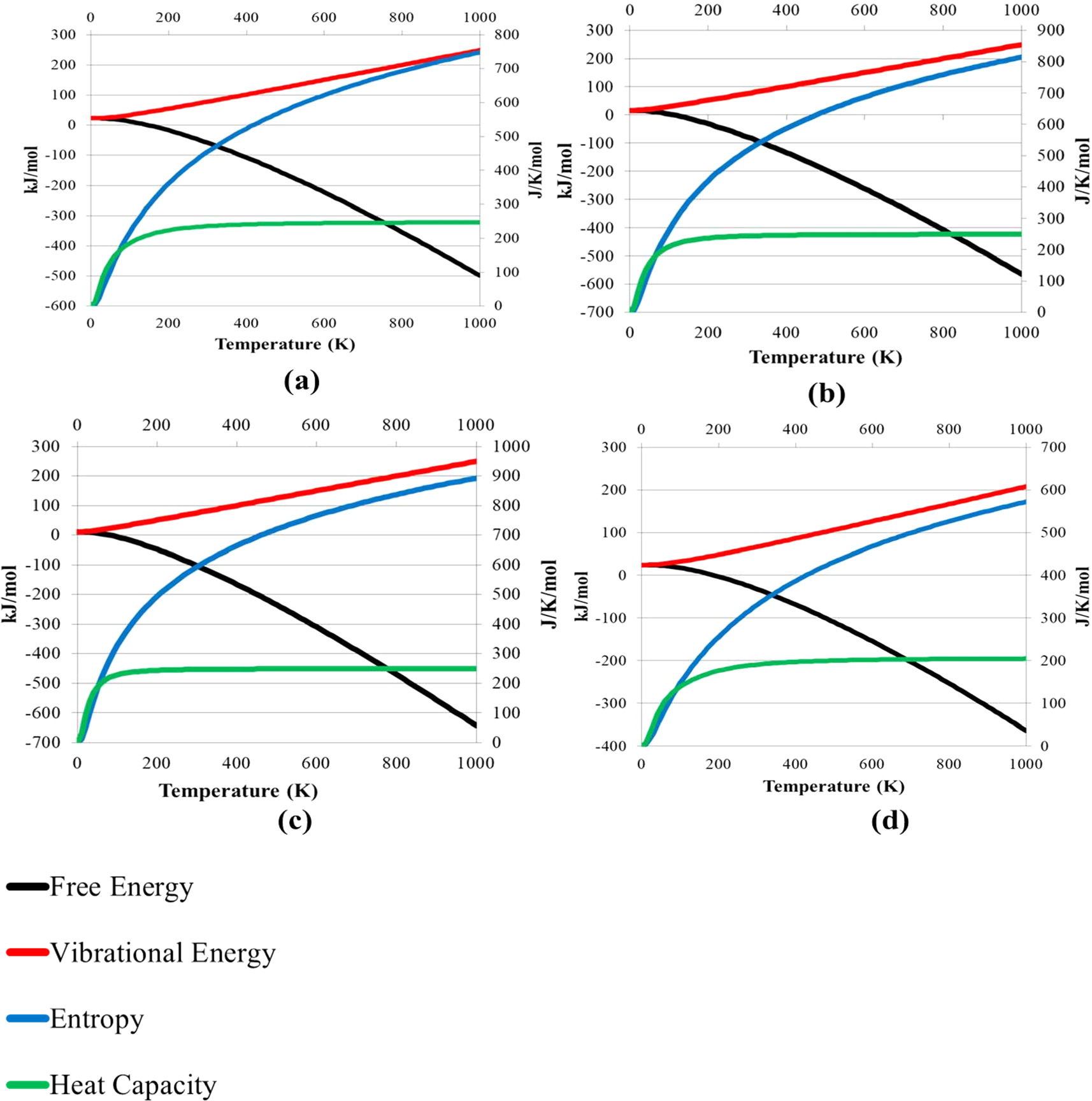
Figure 13. Computed thermodynamic parameters Free energy, Entropy, heat capacity (CV), and Vibrational energy of tetragonal phases (a) Cs2AgBiF6 (b) Cs2AgBiCl6 (c) Cs2AgBiBr6 (d) Cs2AgBiI6.
Download figure:
Standard image High-resolution image3.10. Band edge positions
The lead-free halide components could be of interest in the field of photocatalytic water splitting. For the first time, Wang et al used Cs2AgBiBr6 to produce H2 under visible light irradiation [66]. The band edge positions of the semiconductor play a vital role. The optimal water splitting materials are selected where the conduction band edge (CBM) of the semiconductor must be more negative than the H2O/H2 level of water (0 eV) and its valence band edge (VBM) must be more positive than the H2O/O2 level of water (1.23 eV) [67]. Thus, we determine the CBM and VBM of halides and mixed halide compounds relative to the normal hydrogen electrode potential (NHE). The resulting VBM and CBM are present in figures 14 and 15 where the VBMs and CBMs correspond to their eigenvalues obtained from the DOS calculations under HSE06 approximation (table S8-S11). The visible region between 2 to 2.75 eV was considered [68]. In figure 14, Under UV irradiation, Cs2AgBiF6 shows the ability to lead the redox process as the CBM had more negative values (−0.317 eV and −0.707 eV) than the reduction level of the water (0 eV) and more positive values (3.303 eV and 2.341 eV) than the oxidation level of the water (1.23 eV) for cubic and tetragonal phases respectively. The tetragonal fluoride compound shows strong reduction power whereas the cubic phase shows strong oxidation power. Cs2AgBiCl6 shows the oxidation ability under UV irradiation as its VBM levels are more positive values (1.935 eV and 1.917 eV) than the oxidation level of water in cubic and tetragonal phases respectively. It also has a good reduction ability the CBM level is 0.103 eV than the reduction level of water. We could expect higher reduction power in the tetragonal phase of the Fluoride compound and higher oxidation power in the cubic phase of Cs2AgBiF6 among the halide compounds of both phases. Significantly under visible light irradiation the Cs2AgBiBr6 compound shows photocatalytic ability in both oxidation and reduction for both phases. The tetragonal phase of Cs2AgBiBr6 shows more reduction ability than the cubic phase. Under visible light irradiation, both the phases of Cs2AgBiI6 show the ability of oxidation than the Bromide compound. We could expect more oxidation power in the iodide compounds under visible light irradiation and also the best visible-light photocatalytic material will be the tetragonal phase of Cs2AgBiBr6 among our halide compounds.

Figure 14. The computed band edge positions in HSE06 approximation for the Cs2AgBiX6 (X = F, Cl, Br, and I) for both the cubic and tetragonal phases, denoted in black and red colors, respectively.
Download figure:
Standard image High-resolution image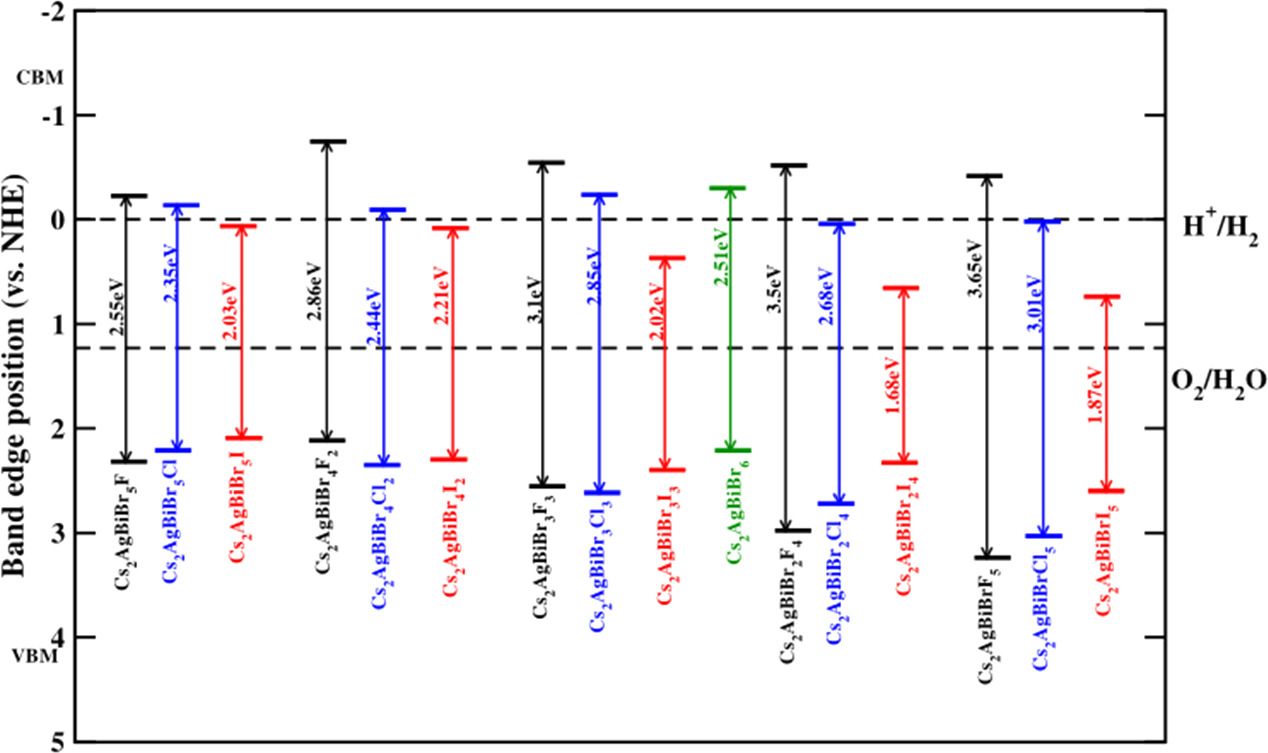
Figure 15. The computed band edge positions in HSE06 approximation for Cs2AgBiBr6−xFx, Cs2AgBiBr6−xClx, Cs2AgBiBr6−xIx x =(1,2,3,4,5).
Download figure:
Standard image High-resolution imageWe did the substitutions for the cubic phase of the Cs2AgBiBr6 as it shows the direct transitions of the electrons from the valence band to the conduction band with the 2.64 eV energy (table S4) emphasizing the good optical absorption among the halide compounds under the visible light irradiation and also shows the highest absorption coefficient among the halide compounds. We have shown the band edge positions of these mixed halides in figure 15. The gradual substitutions of the fluorine atoms to bromine atoms in Cs2AgBiBr6 show the photocatalytic ability under UV irradiation will be better for Cs2AgBiFBr5. The chloride substitutions show the visible photo-catalytic ability for the compounds of Cs2AgBi Br5Cl and Cs2AgBiBr5Cl2. The VBM of Cs2AgBiClBr5 is the same as the Cs2AgBiBr6, therefore Cs2AgBiBr5Cl and Cs2AgBiBr6 should have the same oxidation power. Among the chlorine substitutions, Cs2AgBiBr2Cl4 shows a stronger oxidation power than the pure Cs2AgBiBr6 compound under visible light irradiation. Under UV light irradiation the chloride-substituted compound, Cs2AgBiBrCl5 can be a better candidate for oxidation than the pure bromide compound. Iodine substitutions show the ability of oxidation under visible light irradiation except for the Cs2AgBiI4Br2 and Cs2AgBiBrI5 compounds that have better oxidation ability in the infrared region. Cs2AgBiBr3I3, Cs2AgBiBr4I2, Cs2AgBiBr2I4, and Cs2AgBiBrI5 show better oxidation ability than the Cs2AgBiBr6. However, the Cs2AgBiIBr5 shows a lower ability of oxidation power than the pure bromide compound. Overall our results show that the fluorine-substituted Cs2AgBiBr6 perovskites will be good photocatalytic materials under both the UV and visible regions.
4. Concluding remarks
- For the very first time in the literature, a comprehensive analysis of structural, electronic, and optical properties of Cs2AgBiX6 (X = F, Cl, Br, I) in cubic (
 ) and tetragonal phase (I4/m) are provided based on first-principles DFT analysis. In addition, we report an in-depth analysis of pure inorganic Cs2AgBiX6 (X = F, Cl, I) of the tetragonal phase (I4/m) for the first time. In the search for the best double perovskite material based on the Cs2AgBiBr6 structure, an analysis of the gradual substitution of halogen substitutions at the bromide sites for the cubic phase of Cs2AgBiBr6 was carried out. Our results lead to the following conclusions:
) and tetragonal phase (I4/m) are provided based on first-principles DFT analysis. In addition, we report an in-depth analysis of pure inorganic Cs2AgBiX6 (X = F, Cl, I) of the tetragonal phase (I4/m) for the first time. In the search for the best double perovskite material based on the Cs2AgBiBr6 structure, an analysis of the gradual substitution of halogen substitutions at the bromide sites for the cubic phase of Cs2AgBiBr6 was carried out. Our results lead to the following conclusions: - Cs2AgBiX6 possesses an indirect band gap nature and the band gap value increases when changing the halogen (X) component from F to I for both the cubic and tetragonal phases in between the ultraviolet region and the infrared region
- Cs2AgBiBr6 and Cs2AgBiCl6 materials of the tetragonal phase are dynamically stable whereas the cubic phases were dynamically unstable
- The HSE06 values of the absorption edge of the halide compounds are in the range of 2.14 eV–4.87 eV. The larger values of Cs2AgBiF6 and Cs2AgBiCl6 indicate these materials mainly absorb ultraviolet light.
- The absorption curve of Cs2AgBiBr6−x Ix is gradually red-shifted with the increasing of I– while the blue-shifting occurred for the F– and Cl–.
- TDM analysis shows that Cs2AgBiI6 has more electron transitions from VB to CB compared to the F− and Cl− substituted compounds
- Analysis of the band edge positions of the Cs2AgBiBr6 material shows that it will be a good photocatalyst due to its CBM lying more negative than the reduction level of water and VBM lying more positive than the oxidation level of water.
- Fluorine-substituted Cs2AgBiBr6 perovskites will be good photocatalytic materials under both the UV and visible regions.
- Among all the Cs2AgBiX6 (X = F, Cl, Br, I) structures, the cubic phase of Cs2AgBiBr6 was seen to have the highest absorption coefficient along with prominent electronic features that are favorable for optoelectronic applications.
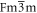
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors gratefully acknowledge the Research Council of Norway for providing the computer time (under project number NN2867k) at the Norwegian supercomputer facility.
This research was funded by the Capacity Building and Establishment of Research Consortium (CBERC), grant number LKA-3182-HRNCET, and the Higher Education and Research Collaboration on Nanomaterials for Clean Energy Technologies (HRNCET) project, Grant Number NORPART/2016/10237.
Data availability statement
All data that support the findings of this study are included within the article (and any supplementary files).
Supporting information
Band diagrams and the density of states figures for the cubic phase of Cs2AgBiX6 (X = F, Cl, Br, I). Band structures, Transition dipole moment diagrams, and the computed effective mass values for the Cs2AgBiBr6−x Fx , Cs2AgBiBr6−x Clx , Cs2AgBiBr6−x Ix (x = 1, 2, 3, 4, 5). Optical properties with their frequency-dependent linear optical calculations, computed band edge position values in detail for both the halide and mixed halide compounds.
Supplementary data (2 MB PDF)