
Abstract
The dispersion-corrected density functional theory (DFT-D) has been used to study the interactions of SO2 on the pristine and defective single-walled MgO nanotubes (MgONTs). A geometric optimization was performed for MgONTs and adsorbents/MgONTs. The adsorption energies of SO2 molecules at different layers of MgONTs were considered. The adsorption property of SO2 was analyzed in terms of the adsorption energy, the electron donation (basicity), and the atomic charges on the adsorbed materials. The densities of states (DOS) have been calculated and used for examining the adsorption properties. In addition, the presence of vacancy defects increases the adsorption energies of the SO2molecules. The band gaps of the defective MgONTs are sensitive to SO2 molecules.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
The interaction of gas molecules with metal oxide surfaces has important environmental and technological significance, in addition to being intrinsically scientifically interesting [1, 2]. The basic metal oxides are commonly used for 'scrubbing' acidic gases from combustion emissions, and a particularly significant recent application of this phenomenon is the use of alkaline earth oxides for the removal of CO2, SO2 and NO2 [3, 4]. From a technological point of view, one would like to be able to tailor the properties of these metal oxide adsorbents to, for instance, release a particular adsorbate over a desired temperature range, or to exhibit a particular selectivity for one adsorbate over another. From a scientific perspective, an important first step in this rational design of metal oxide adsorbents is an understanding of the systematics of their interactions with gaseous adsorbates.
In a typical industrial process, the flue gas stream containing CO2, SO2 and NO2 is passed over an oxide (MgO, CaO, BaO, ZnO, TiO2, Al2O3, CeO2, etc) bed [5–11]. Once the oxide sorbent is saturated, it is replaced with a fresh load of the material. This approach generates waste and is not practical for continuous operations. The study of the complex chemistry of CO2, SO2 and NO2 on oxide surfaces [12–15] is of great importance because metal oxides offer a potential route for removing or destroying CO2, SO2 and NO2, which is one of the most dangerous components of atmospheric pollution. Magnesium oxide (MgO) is frequently used in industry as scrubber to reduce CO2, SO2 and NO2 emission [16–19], and we have chosen it as the substrate for the adsorption because the MgO surface has been widely studied and well-characterized both experimentally [20, 21] and computationally [22–25], including several specific studies of CO2, SO2 and NO2 adsorption [26, 27]. It has been established that the catalytic activity of MgO is due to a small number of defect sites (steps, kinks, corners, etc) with surface ions, particularly oxygen, having low coordination numbers [28]. MgO is particularly interesting in nanoparticle form. It has been possible to prepare MgO in a very high surface area (500 m2 g−1) with average crystallite sizes of about 4 nm [29]. The high surface areas and the intrinsically high surface reactivity allow these materials to be especially effective as adsorbents [30].
Kakkar [31] found several modes of adsorption for formaldehyde on the MgO nanosurfaces. Depending upon the coordination site, different adsorption products are formed and all the reactions are highly exothermic. Chen [32] had discussed the atomic and electronic structure of magnesium oxide nanotube (MgONT) units and found that the three-membered ring (MgO)3, was the most stable species over all MgONT units. Beheshtian [28] had performed DFT calculations to study the interaction between MgONT with CO and NO molecules and found that NO and CO can be strongly adsorbed on MgONT with remarkable adsorption energies. Peyghan [33] had studied the adsorption of N2O molecule on MgONT and found that the N2O molecule cannot significantly be adsorbed on the pristine MgONT. Ammar and El-Gharkawy [34] found NO molecule was strongly adsorbed on the MgONTs containing an oxygen vacancy. However, to the best of our knowledge, few computational investigations of the defective MgONT for SO2 capture have been performed. Thus, systematic computational studies are necessary for elucidating the various mechanisms of surface adsorption.
In this work, we focus on the effects of vacancy defects on the propensity of the MgONT surface to the uptake of SO2. The resulting defective MgONT surfaces can affect adsorption properties of SO2 from flue gas. With this in mind, we performed one-unit van der Waals density functional theory (DFT-D) calculations to elucidate initial adsorption mechanisms of SO2 on MgONT surfaces.
2. Computational methods
This study utilized DFT calculations to evaluate the SO2 adsorption on the MgONT surface in the one-unit slab model. Geometry optimizations and density of states (DOS) were performed using the Dmol3 code [35], which used numerical functions on an atom centered grid as its atomic basis. Double numerical basis sets including polarization functions (DNP) are performed to describe the valence orbitals of all the atoms in our calculations [36, 37]. The DNP basis set is comparable to 6–31 G** Gaussian basis sets with a better accuracy for a similar basis set size [38, 39]. Spin-polarized GGA with PBE form (GGA-PBE) is used to treat the exchange–correlation function [40]. The long range dispersion (van der Waals) contribution using the DFT-D approach of Tkatchenko and Scheffler [41] was applied to all calculations. The convergence criteria applied during geometry optimization is 1.0 × 10−6 Hartree for energy. All atoms are relaxed until the force on each atom is less than 0.002 Ha Å−1.
The pristine MgONT was represented by a quantum unit of 30 atoms, Mg15O15, consisting of five hexagonal (MgO)3 parallel layers. The defective MgONTs were represented by removing an oxygen atom to form an oxygen vacancy (F-center). Figure 1 shows the relaxed pristine and defective MgONTs used in the calculations.
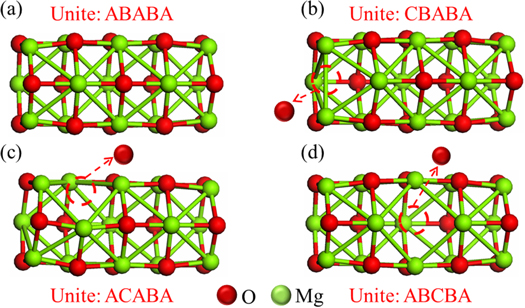
Figure 1. The optimized units of (a) the pristine Mg15O15 and the defective MgONTs for the (b) Mg15O14 FL1, (c) Mg15O14 FL2 and (d) Mg15O14 FL3.
Download figure:
Standard image High-resolution imageCoverage study determines the minimized adsorption energy between adsorbate molecules and adsorbents. It also evaluates the effect of the self-interaction of SO2 molecules in the super cell. The adsorption energy (Eads) was determined in the following way:

Where Eads is the adsorption energy, E(M/MgONT) the total energy of the promoter or SO2 molecule with the MgONT slab, EM the energy of the promoter or SO2 molecule, and EMgONT the total energy of the MgONT slab. Using the calculation of adsorption energy, the adsorption properties of adsorbates on MgONT are discussed. A Mullican electron population charge analysis was performed to determine internal charge distribution changes.
3. Results and discussions
3.1. Pristine and defective MgONTs
The optimized units of both pristine and defective MgONTs are showed in figure 1. Pipe length and pipe diameter of MgONT are 8.009 and 3.808 Å respectively. Mg15O15 is the pristine single-walled MgONT with five layers, while Mg15O14 Fx is the defective MgONTs with defect at the layer x (x = 1, 2 and 3), respectively. The average Mg–O bond lengths were optimized at 1.91 Å, 2.00 Å and 2.05 Å in the first, second and third layers of pristine MgONT, respectively, which were in good agreement with the previous [42–44]. While the nearest Mg atoms at the same layer of the F-centers were outward relaxed. In the other hand for the interlayers around the F-centers, the Mg–O bond lengths were decreased. One noticed that from table 1, the formation energy (Ef) values for the F-centers were found to be 8.51, 9.01 and 9.22 eV for the Mg15O14 FL1, Mg15O14 FL2 and Mg15O14 FL3, respectively. These values indicated that an F-center at the first layer was more stable than that at the second or the third layers. These results were in good agreement with the previous [34]. Also the Ef values were in the same range of the previous theoretical and experimental results for MgO surfaces [45–51]. It is noticed that the presence of an F-center increases the highest occupied molecular orbital (HOMO) of MgONTs. A similar behavior was noticed at MgO surfaces [45, 52].
Table 1. HOMO, lowest unoccupied molecular orbital (LUMO), energy gap (Eg/eV) and formation energy (Ef/eV) of the pristine and defective MgONTs.
| HOMO | LUMO | Eg | Ef | |
|---|---|---|---|---|
| Mg15O15 | −0.19 | −0.06 | 0.13 | — |
| Mg15O14 FL1 | −0.13 | −0.06 | 0.07 | 8.51 |
| Mg15O14 FL2 | −0.12 | −0.06 | 0.06 | 9.01 |
| Mg15O14 FL3 | −0.12 | −0.05 | 0.07 | 9.22 |
3.2. SO2 adsorption on the pristine MgONTs
SO2 molecules interact in several ways with the pristine MgONTs. However, in this section, the adsorption of SO2 molecule on the anionic site of different layers was considered. The geometry structures for the adsorption of SO2 molecules on the adsorbing O2− site of MgONT were shown in figure 2. SO2 molecules approach to the O2− site at the first, second and third layers of the pristine MgONT by the S atom to form complexes denoted by adsorbent/Mg15O15 PL1, adsorbent/Mg15O15 PL2, and adsorbent/Mg15O15 PL3, respectively.
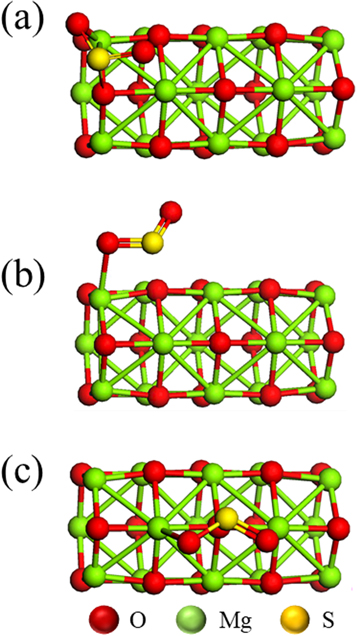
Figure 2. The optimized units of the SO2 adsorption on the pristine MgO nanotube. (a) SO2/Mg15O15 PL1, (b) SO2/Mg15O15 PL2, (c)SO2/Mg15O15 PL3.
Download figure:
Standard image High-resolution imageThe optimized parameters for the adsorption of the SO2 molecules on the adsorbing O2− site of MgONT were tabulated in table 2. One can notice that the adsorption energies (Eads) of SO2 molecule on the pristine MgONTs were in the range from−1.06 to −1.76 eV, which means that the essence of adsorption is chemisorption. It is also noticed that, the total Mulliken charge of QSO2 is approximately equal to −0.34 e. The S–O bond length is approximately equal to 1.51 Å. These values are longer than that of the free SO2 molecule (1.45 Å, the experimental value is 1.43 Å [53]). This means that there is some charge transfer between SO2 molecule and the pristine MgONT. And as a result, the interaction between SO2 and MgONTs is strong at any layer of the MgONTs. Also the Eads of SO2 molecules on the pristine MgONTs is dependent on the layer number of the adsorption site.
Table 2. HOMO, LUMO, Energy gap (Eg/eV), Mulliken charges (Q/e−) and Adsorption energy (Eads/eV) for the adsorption of SO2 on the pristine Mg15O15 nanotubes.
| HOMO | LUMO | Eg | Eads | QO | QS | QSO2 | |
|---|---|---|---|---|---|---|---|
| SO2 | −0.30 | −0.18 | 0.12 | — | −0.23/−0.23 | 0.46 | 0 |
| SO2/Mg15O15 PL1 | −0.19 | −0.08 | 0.11 | −1.76 | −0.47/−0.40 | 0.48 | −0.39 |
| SO2/Mg15O15 PL2 | −0.20 | −0.10 | 0.10 | −1.22 | −0.44/−0.32 | 0.45 | −0.31 |
| SO2/Mg15O15 PL3 | −0.19 | −0.10 | 0.09 | −1.06 | −0.39/−0.38 | 0.44 | −0.33 |
In contrast with the previous results we observe a decrease in Eg values of the SO2/Mg15O15 PL1, SO2/Mg15O15 PL2, SO2/Mg15O15 PL3−complexes by about 0.02 eV than the bare MgONT. So we studied the influence of SO2 adsorption on the electronic properties of the complexes. The total density of states (DOS) of free SO2 molecule and projected density of states (PDOS) of SO2 for SO2/Mg15O15 PL1 are shown in figure 3. Comparing figure 3(a) with figure 3(b), one can observe that the electronic structures of the SO2 molecule and MgONT have a considerable change before and after the adsorption. So it is concluded that the decrease in Eg value of SO2/Mg15O15 PL1SO2/Mg15O15 PL2 and SO2/Mg15O15 PL3 is related to the chemisorption of SO2 molecule on the pristine Mg15O15.
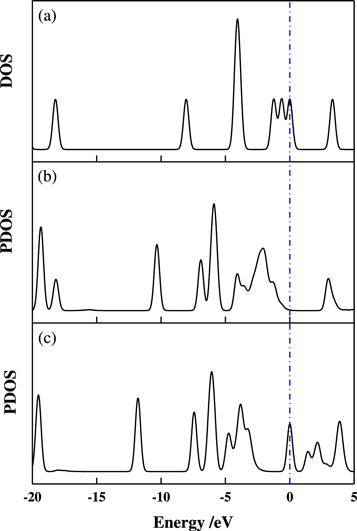
Figure 3. DOS for free SO2 molecule (a), PDOS of SO2 for SO2/Mg15O15 PL1 (b) and SO2/Mg15O14 FL1 (c).
Download figure:
Standard image High-resolution image3.3. SO2 adsorption on the defective MgONTs
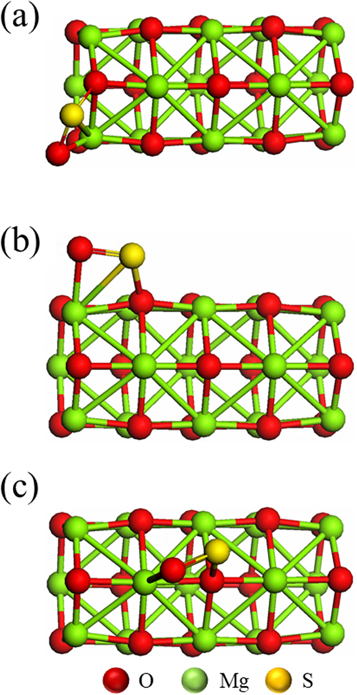
The optimized structures for the adsorption of the SO2 molecules on the defect sites F-centers were shown in figure 4. The Eads for SO2 molecules on the defective nanotubes (Mg15O14 FL1, Mg15O14 FL2 and Mg15O14 FL3), were evaluated and tabulated in table 3. Our calculations showed that all configurations for the adsorption of SO2molecules on the defective Mg15O14 FL1, Mg15O14 FL2 and Mg15O14 FL3 are exothermic interactions. The Eads for SO2 molecules on F-centers were ranged from −1.76 to −1.06 eV. This means that the adsorption of SO2 molecules on the defective MgONTs is chemisorption. The total charge on the adsorbed SO2 molecules (QSO2) on F-centers were ranged from −1.23 to −1.26e−. The strong interaction maybe due to the charge transfer from the defective MgONTs to the SO2 molecules which are emphasized from the negative charge on SO2 molecules.As a result, the S–O bond length is greater inSO2/Mg15O14 FLx configurations (figure 4) than that in SO2/Mg15O15 PLx configurations (figure 2). This is maybe referred to the higher basicity of F-centers than the pristine Mg15O15. These results were confirmed by the higher QSO2 in the case of F-centers than the pristine Mg15O15. One can conclude that the strong interaction between adsorbent molecule and the defective Mg15O14 may be explained via the charge transfer from the defective Mg15O14 to the adsorbent. The presence of an F-center, rich electronic center, increases the bascity of the MgONTs. The strong interaction between SO2 molecule and the defective Mg15O14 may be explained via spin pairing between the two single electrons of SO2 molecule and F-center.
{kind=link}
{kind=link}
{kind=link}
Figure 4. The optimized units of the SO2 adsorption on the defective MgO nanotube. (a) SO2/Mg15O14 FL1, (b) SO2/Mg15O14 FL2, (c)SO2/Mg15O14 FL3.
Download figure:
Standard image High-resolution image{kind=link}
Table 3. HOMO, LUMO, energy gap (Eg/eV), adsorption energy (Eads/eV) and Mulliken charges (Q/e−) and for the adsorption of SO2 on the defective Mg15O14 nanotubes.
| HOMO | LUMO | Eg | Eads | QO | QS | QSO2 | |
|---|---|---|---|---|---|---|---|
| SO2/Mg15O14 FL1 | −0.13 | −0.08 | 0.05 | −3.74 | −0.81/−0.55 | 0.13 | −1.23 |
| SO2/Mg15O14 FL2 | −0.14 | −0.09 | 0.05 | −3.77 | −0.87/−0.51 | 0.12 | −1.26 |
| SO2/Mg15O14 FL3 | −0.14 | −0.09 | 0.05 | −3.73 | −0.88/−0.48 | 0.10 | −1.26 |
The Eads of adsorbent molecules on the defective MgONTs (F-centers) was more in the first layers than that in the second and third layers. These mean that the adsorption energies decrease by increasing of the coordination number of the defect site (F-center) where the coordination number of the defect site (F-center) is 3 at the first layer and 4 at the second and third layers. From the previous one, it can be concluded that the Eads of SO2 molecule on the defective MgONTs depends on the charge of the adsorption site (F-center), adsorption mode and the coordination number of the adsorbing site (F-center).
To emphasize the previous results in point of view the electronic structure, we studied the most stable complexes SO2/Mg15O15 FL1. The total DOS of it was shown in figure 3. One can observe that the unoccupied peaks of the free SO2 (figure 3(a)) molecule at −0.6 eV and −1.2 eV were disappeared and a new occupied one was appeared in the adsorbed SO2 molecule (figure 3(c)) at −4.8 eV, which confirm that the SO2 molecule gains a negative charge during the adsorption process. The inspection of figure 3 reveals that the SO2 electronic structure is significantly perturbed upon chemisorption, independently of the surface complex geometry. This confirms that the strong interaction between SO2 molecule and the defected Mg15O15 FL1 nanotube is due to a charge transfer process. It is therefore concluded that the adsorption of SO2 molecule can essentially increase the electrical conductance of the defected Mg15O15 FL1. Suggest that Mg15O15 FL1 has capability of generating a response to the adsorption of SO2 molecule.
4. Conclusion
DFT calculations were performed to study the interaction between the gaseous SO2 molecule with the pristine and defective MgONTs. The formation of an F-center at the terminal layers is more stable than its formation at the internal layers of MgONTs. The adsorption of SO2 molecules on O2− site of the pristine MgONTs is chemisorption with the adsorption energy range of −1.06 to −1.76 eV. Due to the charge transfer from the defective MgONTs to the SO2 molecules, the adsorption of SO2 molecules on F-centers of the defective MgONTs is stronger, up to −3.73 to 3.77 eV. Through the study, we can find that the adsorption energy of SO2 molecules on the defective MgONTs depends on the charge of the adsorption site (F-center), adsorption mode and the coordination number of the adsorbing site (F-center). The adsorption of SO2 molecules can change the electrical conductance of the defective MgONTs, suggesting that the defective MgONTs are sensitive to SO2 molecules.
Acknowledgments
This work is financially supported by National Natural Science Foundation of China with Grant No. 21676148.
Data availability statement
All data that support the findings of this study are included within the article (and any supplementary files).