

Abstract
The structures, stability mechanical properties and electronic properties of X (X=Nb, Ge, Ni) doped Al2Cu were studied by first-principles. The results show that the stable structures can be formed are Al64Cu32, Al63Cu32Nb, Al64Cu31Nb, Al63Cu32Ge, Al63Cu32Ni, Al64Cu31Ni. Doping X improved the stiffness of the Al2Cu. Al64Cu31Ni has the largest stiffness and Al64Cu31Nb has the smallest stiffness. However, the ductility of Al2Cu is reduced after doping with X. Doping X can greatly reduce the anisotropy of the whole system and the {100}, {010} surface. Among them, Al63Cu32Ge has the lowest degree of anisotropy, and Al64Cu31Ni has the highest degree of anisotropy. Doping Nb cause strong orbital hybridization between Al (3s) and Nb (4d), doping Ni causes strong orbital hybridization between Al (3s) and Ni (3d). The bonding strength and stability of Al2Cu will be significantly changed by these two elements.
Export citation and abstract BibTeX RIS
1. Introduction
In recent years, 7xxx series aluminium alloy with high specific strength, high specific stiffness and low density have achieved great success in industrial development, such as 7020, 7050 and 7075 [1–4]. Applications are involved in aerospace, automobile and other fields. However, lower corrosion resistance, fracture toughness and other problems greatly restrict its application [5, 6]. In order to make 7xxx series aluminium alloy obtain considerable application, researchers usually improve its mechanical properties by adjusting the content of main alloying elements, reducing the content of impurities and adding trace amounts of alloying elements [7]. Among them, the technology of adding trace amounts of alloying elements has been deeply studied [8–10]. Lin et al [11] found that hydrogen embrittlement of 7xxx series aluminium alloy can be inhibited by Ti, Zr, Cr and Mn as early as 1988. And Chen and He et al [12, 13] found that Zr, Cr and Pr significantly inhibits the recrystallization of 7xxx series aluminum alloy, thus enhances the resistance to stress corrosion and exfoliation corrosion. Zhao et al [14] studied the strength and toughness of 1.1 wt% Li-Containing 7xxx aluminum alloy. The results of Lai et al [15] show that Ce can remarkably refine the as-cast grains and eutectic microstructure. Some researchers have tried to add Nb, Ge and Ni, and found that the mechanical properties of 7xxx series aluminium alloys would also be improved. Zhang et al [16] found that Nb can refine the grain of as cast structures in 7xxx aluminum alloy, improve the stress corrosion cracking resistance and mechanical properties. Wen et al [17] found that Ge can significantly refine the as-cast micro-structures of 7xxx aluminum alloy and improve the peak age hardening and strengthening effects of the alloy. Wu et al [18] found that the addition of Ni makes the 7xxx aluminum alloy show high strength and good stress corrosion resistance.
The extensive demand and development of 7xxx series aluminium alloy have prompted people to study the composition design [19, 20], intermetallic compounds [21, 22] and phase transformation [23] of alloy. Optimizing the intermetallics has become one of the most important goals of developing alloys.[24]. Mechanical or thermal problems such as fracture toughness, stress corrosion resistance, fatigue strength and heat resistance of 7xxx series aluminium alloy can be controlled by optimizing the intermetallics [25]. The most common and important phases in 7xxx aluminum alloy are η (MgZn2), S (Al2CuMg), θ (Al2Cu) and Τ (Al2Mg3Zn3) [24, 26]. It has been reported that η (MgZn2) can improve the mechanical properties of 7xxx series aluminium alloys or magnesium alloys. As an aging strengthening phase, θ (Al2Cu) is a plastic phase. It is reported that the fracture toughness, stress corrosion resistance and fatigue strength of 7xxx aluminum alloy can also be controlled by the intermetallic phases θ (Al2Cu) [25, 27]. Yang et al [28] studied the correlation between mechanical behaviors and electronic structures of Al2Cu by first-principles. Gu et al [29] studied the phase structure and stability of Fe, Si, Mn, Ti and Zr doped Al2Cu, based on density functional theory (DFT). Therefore, it can be predicted that the lattice constants and bonding properties of Al2Cu are changed by solid solution or refine Al2Cu grain by dispersion will have an important influence on the mechanical properties of 7xxx aluminum alloy. What changes will the doping of Nb, Ge and Ni bring to the structures and mechanical properties of Al2Cu? Is the improvement of mechanical properties of 7xxx series aluminium alloys due to the dissolution of Nb, Ge, and Ni into Al2Cu? At present, there are few studies on Nb, Ge, Ni doped Al2Cu whether in experiments or in theoretical calculations, which have not been reported to the best of our knowledge. Since the lattice constant of the cell is usually only a few angstroms, it is very difficult to predict the effect of doping elements on the structures and properties of Al2Cu by experiments. For the study of micro-structures such as cell, the first-principles calculation is an effective method to study the doped alloys with a trace of elements [30–32].
In this work, we have calculated the structures, stability, mechanical properties and electronic properties of X (X=Nb, Ge, Ni) doped Al2Cu by first-principles. Al2Cu is used as matrix, X (X=Nb, Ge, Ni) is used as doped elements. The micro-mechanism of Al2Cu doped with X (X=Nb, Ge, Ni) was revealed, which provides a theoretical basis for further related experiments.
2. Computational details
Al2Cu belongs to the tetragonal system, the space group is I4/mcm, the lattice constants: a = b = 6.067 (Å), c = 4.877 (Å), α = β = γ = 90°, the sites of Al and Cu atoms are equivalent to each atom in the unit cell. In this study, a 2 × 2 × 2 super-cell was constructed to show the largest distortion from the perfect lattice. There are 64 Al atoms and 32 Cu atoms in the super-cell. In this work, We use an X atom to replace the Al and Cu sites respectively to form two substitution doping ( substitutional solid solutions: Al63Cu32X and Al64Cu31X ). The atomic concentration of each doping atom equals to 1.04% in the Al-Cu-X system [29]. The cell's structures before X doping and after X doping are shown in figure 1.

Figure 1. Geometries of Al64Cu32, Al63Cu32X, Al64Cu31X.
Download figure:
Standard image High-resolution imageThe calculations are carried out by using CASTEP software package based on density functional theory (DFT). All energy calculations are performed by using the pseudo-potential plane wave method [33], and the cut-off energy is 550 eV. The generalized gradient approximation (GGA) with the PBE functional is adopted to describe the exchange and correlation interaction [34, 35]. Potential function using the ultra-soft pseudo-potentials (USPP). A high symmetrical K point method with Monkhorst-Pack in Brillouin zone integral. The k-point mesh are 2 × 2 × 3. In the self-consistent field (SCF) calculation, the accuracy of the self-consistent convergence energy is 1 × 10−6 eV. Geometric optimization of each cell to obtain the most stable structures.
3. Results and discussion
3.1. Structures
The optimized unit lattice constants are shown in table 1. The lattice constants of Al2Cu are in good agreement with the literature value (the errors are 1%) [36, 37], which shows that the method of calculation is correct and feasible. When X doped into the Al64Cu32 cells, the lattice constants and cell volume have changed. The reason is that the radius of the atoms are different, the lattices are distorted.
Table 1. The optimized lattice parameters (Å) and cell's volume (Å3) for X (X = Nb, Ge, Ni) doped Al2Cu (Al64Cu32).
| Phase | Lattice constants (Å) | Cell volume (Å3) | References | ||
|---|---|---|---|---|---|
| Al2Cu | a = 6.041 | c = 4.929 | V = 179.85 | This study | |
| a = 6.04 | c = 4.89 | V = 178.40 | [38] | ||
| a = 6.067 | c = 4.877 | V = 179.52 | [36] | ||
| [39] | |||||
| a = 6.063 | c = 4.872 | V = 179.09 | [37] | ||
| a = 6.064 | c = 4.885 | V = 179.63 | [28] | ||
| a = 6.059 | c = 4.889 | V = 179.48 | [29] | ||
| a = 6.063 | c = 4.874 | V = 179.17 | [40] | ||
| Al64Cu32 | a = 12.089 | c = 9.851 | V = 1439.82 | ||
| Al63Cu32Nb | a = 12.101 | b = 12.100 | c = 9.836 | V = 1440.15 | |
| Al64Cu31Nb | a = 12.131 | c = 9.877 | V = 1453.60 | ||
| Al63Cu32Ge | a = 12.084 | c = 9.866 | V = 1440.79 | ||
| Al64Cu31Ge | a = 12.100 | b = 12.104 | c = 9.892 | V = 1448.72 | |
| Al63Cu32Ni | a = 12.085 | c = 9.806 | V = 1432.14 | ||
| Al64Cu31Ni | a = 12.084 | c = 9.837 | V = 1436.43 | ||
3.2. Stability
3.2.1. Thermodynamic stability
The formation energy of the reaction is equal to the total energy of the reaction product minus the total energy of the reactant. It can measure the degree of difficulty in the formation of intermetallic compounds, determine whether a stable structure can be formed. When the formation energy is negative, the smaller the formation energy, the easier the formation of intermetallic compounds. The formula for the reaction of X doped Al2Cu are as follows:
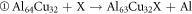
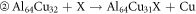
It can be seen from figure 2 that the except for the positive value of Al64Cu32Ge, the formation energies of other structures are negative, indicating that all structures can exist stably except Al64Cu32Ge. For stable structures, the formation energies in accordance with the order from low to high is Al63Cu32Nb < Al64Cu31Nb < Al64Cu31Ni < Al63Cu32Ni < Al63Cu32Ge. So, Al63Cu32Nb is easiest to form, and Al63Cu32Ge is most difficult to form. Moreover, However, for the three elements, the formation energies of Nb-doped solid solution are the smallest, whereas that of Ge-doped solid solution are the largest, regardless of where X replaces Al or Cu. This indicates that Al-Cu-Nb doped with Nb are the easiest to form, while Al-Cu-Ge compounds doped with Ge are the most difficult to form.

Figure 2. Formation energies of Al63Cu32Nb, Al64Cu31Nb, Al63Cu32Ge, Al64Cu31Ge, Al63Cu32Ni, Al64Cu31Ni.
Download figure:
Standard image High-resolution imageThe cohesive energy of a crystal is the energy released by free atoms combine into a crystal. In other words, the work done by the outside world when a crystal resolved into single atoms. The structural stability of the crystal is closely related to the cohesive energy, the greater the absolute value of cohesive energy, the more stable the crystal formed. The formula of cohesive energy is given by reference [41].
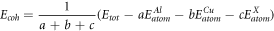
Where 

 are the energies of the free atoms of Al, Cu and X, respectively. The results are shown in figure 3. Observing all the cohesive energies, the absolute values are in turn: Al63Cu32Nb > Al64Cu31Nb > Al64Cu31Ni > Al63Cu32Ni > Al63Cu32Ge, indicating that the structure of Al63Cu32Nb is the most stable, and the structure of Al63Cu32Ge is the most unstable. For the three elements, the absolute value of binding energies after Nb doping are the largest, while that after Ge doping are the smallest. This indicates that the Nb is more stable in the form of Al-Cu-X, whereas Ge is just the contrary. Al-Cu-Nb system is the most stable, while Al-Cu-Ge system is the most instable. This corresponds to the case of formation energies.
are the energies of the free atoms of Al, Cu and X, respectively. The results are shown in figure 3. Observing all the cohesive energies, the absolute values are in turn: Al63Cu32Nb > Al64Cu31Nb > Al64Cu31Ni > Al63Cu32Ni > Al63Cu32Ge, indicating that the structure of Al63Cu32Nb is the most stable, and the structure of Al63Cu32Ge is the most unstable. For the three elements, the absolute value of binding energies after Nb doping are the largest, while that after Ge doping are the smallest. This indicates that the Nb is more stable in the form of Al-Cu-X, whereas Ge is just the contrary. Al-Cu-Nb system is the most stable, while Al-Cu-Ge system is the most instable. This corresponds to the case of formation energies.

Figure 3. Absolute value of cohesive energies of Al63Cu32Nb, Al64Cu31Nb, Al63Cu32Ge, Al63Cu32Ni, Al64Cu31Ni.
Download figure:
Standard image High-resolution image3.2.2. Mechanical stability
The elastic constant is an important index to reflect the external stress of a crystal. In order to test the mechanical stability of the cell after doping, we calculated the elastic constants of the six structures, as shown in table 2. The mechanical stability criterion of tetragonal system are as follows: [42, 43].
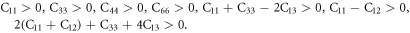
Table 2. The elastic constants of the calculated structures.
| Elastic stiffness constants Cij (GPa) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Phase | C11 | C12 | C13 | C22 | C23 | C33 | C44 | C55 | C66 |
| Al64Cu32 | 151.76 | 97.68 | 46.34 | 151.76 | 46.34 | 187.56 | 20.48 | 20.48 | 50.23 |
| Al63Cu32Nb | 188.25 | 51.43 | 45.19 | 178.66 | 65.22 | 189.61 | 47.61 | 53.82 | 45.88 |
| Al64Cu31Nb | 156.39 | 78.60 | 59.84 | 156.39 | 59.84 | 193.62 | 35.43 | 35.43 | 56.65 |
| Al63Cu32Ge | 197.54 | 50.01 | 56.40 | 173.68 | 69.10 | 169.12 | 66.73 | 66.92 | 68.33 |
| Al63Cu32Ni | 218.85 | 36.80 | 35.81 | 201.66 | 49.69 | 204.17 | 56.04 | 50.22 | 54.47 |
| Al64Cu31Ni | 172.93 | 92.97 | 27.77 | 172.93 | 27.77 | 246.32 | 55.83 | 55.83 | 86.31 |
It is shown in table 2 that all structures satisfy the criterion. This shows that all structures are stable at the mechanical level. The C11, C22 and C33 of these six structures (Al64Cu32, Al63Cu32Nb, Al64Cu31Nb, Al63Cu32Ge, Al63Cu32Ni, Al64Cu31Ni) are larger than other elastic constants, which indicates that the ability of resisting deformation under normal stress is stronger than the ability of resisting deformation under shearing stress in these six structures.
3.3. Mechanical properties
3.3.1. Elastic modulus
We calculated the bulk modulus (K), shear modulus (G), Young's modulus (E), the ratio of K/G and Poisson's ratio (u) for Al64Cu32, Al63Cu32Nb, Al64Cu31Nb, Al63Cu32Ge, Al63Cu32Ni, Al64Cu31Ni. For K, G, E, the Voigt, Reuss, and Hill (VRH) approximations are used for estimation, and the related formulas are presented by reference [44–46]. The results of elastic modulus are shown in figure 4.
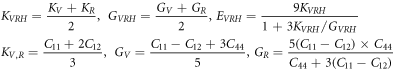
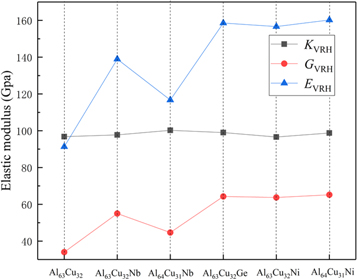
Figure 4. The elastic modulus of Al64Cu32, Al63Cu32Nb, Al64Cu31Nb, Al63Cu32Ge, Al63Cu32Ni, Al64Cu31Ni.
Download figure:
Standard image High-resolution imageThe Young's modulus (E) reflects the stiffness of the materials. The smaller the Young's modulus, the smaller the stiffness of the material, the better the ductility. From figure 4, we know that the values of Young's modulus (E) from low to high is Al64Cu32 < Al64Cu31Nb < Al63Cu32Nb < Al63Cu32Ni < Al63Cu32Ge < Al64Cu31Ni, which shows the stiffness of Al64Cu32 is the smallest, the stiffness of Al64Cu31Ni is the largest, Al64Cu32 has the best ductility. With the addition of Nb, Ge and Ni, the stiffness of Al64Cu32 became larger, and the ability to resist the elastic deformation are stronger. It can be seen from figure 4 that doping does not significantly change the bulk modulus of Al64Cu32, but affects the shear modulus, which leads to a significant change in Young's modulus. The ratio of bulk modulus (K) and shear modulus (G) can be used to evaluate the brittleness and ductility of crystals. When K/G < 1.75, the materials are brittle, conversely the materials behave as toughness. The values of K/G are shown in table 3. As shown in table 3, the values of Al64Cu32, Al63Cu32Nb and Al64Cu31Nb are greater than 1.75, so they are ductile materials, and the K/G values of Al63Cu32Ge, Al63Cu32Ni, Al64Cu31Ni are less than 1.75, so Al63Cu32Ge, Al63Cu32Ni, Al64Cu31Ni are brittle. This indicates that the doping of Ge and Ni causes a transition from plasticity to brittleness in Al64Cu32. Poisson's ratio (u) is used to evaluate the shear stability of the structure. The bigger the value of Poisson's ratio, the better the ductility of the material. The results of Poisson's ratio (u) are also shown in table 3. From table 3, we observed that the Poisson's ratios of Al64Cu32 are the largest, indicated the ductility are the best, followed are Doping of Nb, Ge and Ni, which are consistent with the results of ductility obtained by Young's modulus (E).
Table 3. The calculated K/G ratio, Poisson's ratio (u), elastic anisotropy (AU) and Debye temperature (ΘD) for the Al64Cu32, Al63Cu32Nb, Al64Cu31Nb, Al63Cu32Ge, Al63Cu32Ni, Al64Cu31Ni.
| Al64Cu32 | Al63Cu32Nb | Al64Cu31Nb | Al63Cu32Ge | Al63Cu32Ni | Al64Cu31Ni | |
|---|---|---|---|---|---|---|
| K/G | 2.85 | 1.78 | 2.24 | 1.54 | 1.51 | 1.52 |
| u | 0.34 | 0.26 | 0.31 | 0.22 | 0.23 | 0.23 |
| AU | 1.45 | 0.14 | 0.30 | 0.05 | 0.26 | 0.65 |
| ΘD (K) | 3.12 | 3.21 | 3.17 | 3.03 | 3.25 | 3.26 |
3.3.2. Elastic anisotropy
Cracks in materials are an important cause of material failure. Cracks expand and merge slowly through the initial micro-cracks. Ultimately, the strength of the material decreases markedly, even breaks. This has seriously affected the active life of materials and will bring incalculable consequences to the project. For this reason, we have calculated the elastic anisotropy (AU) of the materials, which can give us an understanding of the cause of induce micro-cracks in materials. The value of AU indicates the extent of the anisotropy. When AU is 0, the material is isotropic, and the more the AU deviates from the 0, the higher the degree of anisotropy of the materials. The formula for AU are given by references [47, 48].
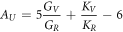
Table 3 shows the values of the anisotropy of the five structures. It can be seen that all structures are anisotropic. The value of Al64Cu32 is the largest, the degree of anisotropy is the highest, the value of Al63Cu32Ge is minimum, indicated the degree of anisotropy is lowest. The results show the periodicity, density and physicochemical properties of the Al64Cu32 are most different from each other in the different directions. Under the same conditions, cracking caused by external factors is the easiest. But Al63Cu32Ge is just the opposite of Al64Cu32.
In most cases, the cracks initiate from the surface of the materials. The difficulty of surface cracking directly affect the occurrence of micro-cracks. In order to determine the surface where cracks are most likely to occur, the analysis of shear anisotropy of materials is an effective method. The shear anisotropy is an important criterion for judging the formation of micro-cracks on surface. The shear anisotropic factor reflects the degree of anisotropy of the bonding between atoms in different planes, which explains the reason of the cracking on the surface of the materials. For the {100} shear planes between 〈011〉 and 〈010〉 directions, the {010} shear planes between 〈101〉 and 〈001〉 directions, and the {001} shear planes between 〈110〉 and 〈010〉 directions, the shear anisotropic factors are given by reference [49].
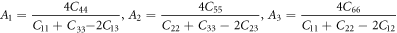
For isotropic crystals, the values of A1, A2, and A3 must be 1, while any values less than or greater than 1 are the measures of elastic anisotropy, and the more the deviation between the value and 1, the higher the degree of anisotropy [50]. The results of A1, A2, and A3 are shown in figure 5.
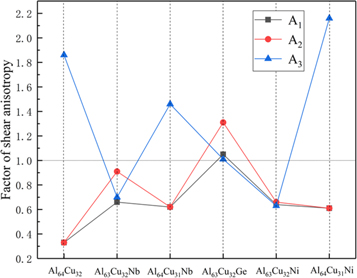
Figure 5. The factor of shear anisotropy of Al64Cu32, Al63Cu32Nb, Al64Cu31Nb, Al63Cu32Ge, Al63Cu32Ni, Al64Cu31Ni.
Download figure:
Standard image High-resolution imageAs shown in figure 5 the six structures are all anisotropic on three planes. The anisotropic values of A2 and A3 are generally larger than those of A1, implying that the degree of anisotropy in the {010} and {001} surface are higher than that in {100}. On the {001} plane, Al64Cu31Ni has the highest anisotropy, which means that the distribution of the charge on {001} plane has the highest anisotropy, and most easily induce the germination of micro-cracks. The value of Al63Cu32Ge in {001} is closest to 1, and the degree of anisotropy is the lowest in this plane, indicated it is most difficult to germinate micro-cracks. On the {100} and {010} planes, The values of A1 and A2 of the six structures are similar, which indicates that their anisotropic degrees on these two surfaces are little difference. Al64Cu32, Al64Cu31Nb, Al63Cu32Ni and Al64Cu31Ni have the closest A1 and A2 values. Overall analysis, the anisotropic value of Al63Cu32Ge is closest to 1, followed by Al63Cu32Nb. No matter what element is doped, the anisotropic value of Al64Cu32 will be closer to 1, and the anisotropy of Al64Cu32 will be reduced, except for the A3 of Al64Cu31Ni. In summary, the addition of Nb, Ge and Ni can greatly reduce the anisotropy of the {100}, {010} and the whole anisotropy, especially Nb and Ge replaced Al sites are more obvious.
3.4. Debye temperature
In solid materials, Debye temperature (ΘD) is an another important physical parameter that affects the bonding force between atoms. The higher the Debye temperature (ΘD), the better the thermal conductivity of the materials, the stronger the chemical bond between the atoms, the greater the hardness of the materials, and the better the wear resistance. Debye temperature (ΘD) can be obtained by average sound velocity. Its formula are presented by [51, 52].
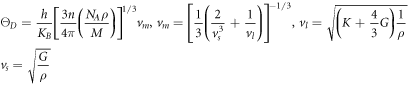
Among them, h and KB are Planck and Boltzmann constants respectively. n is the number of atoms in the compound. NA is Avogadro's number. ρ is the mass density. M is the molar mass of a compound. vm is the average velocity. vl and vs are longitudinal wave velocity and transverse wave velocity, respectively.
The results of Debye temperature (ΘD) are shown in table 3. It can be seen that the Debye temperature (ΘD) of Al64Cu31Ni is the highest, which shows the thermal conductivity of Al64Cu31Ni is the best, the chemical bond between the cell's atoms are the strongest, and the hardness is the largest. Al63Cu32Ge has the lowest Debye temperature (ΘD), the thermal conductivity is the worst in six structures, the chemical bonds between the atoms are not as strong as those of the other structers, and the hardness is also the least. The values of Debye temperature (ΘD) from high to low: Al64Cu31Ni > Al63Cu32Ni > Al63Cu32Nb > Al64Cu31Nb > Al64Cu32 > Al63Cu32Ge. It can be seen that doped Nb and Ni can increase the Debye temperature (ΘD) of Al64Cu32 and make the chemical bonds more stable, the thermal conductivity, hardness and wear resistance of Al64Cu32 will be improved. When Ge replaced the sites of Al formed Al63Cu32Ge, it will decrease the Debye temperature (ΘD) of Al64Cu32, the hardness of the materials will be decreased and make the wear resistance deteriorated.
3.5. Density of states
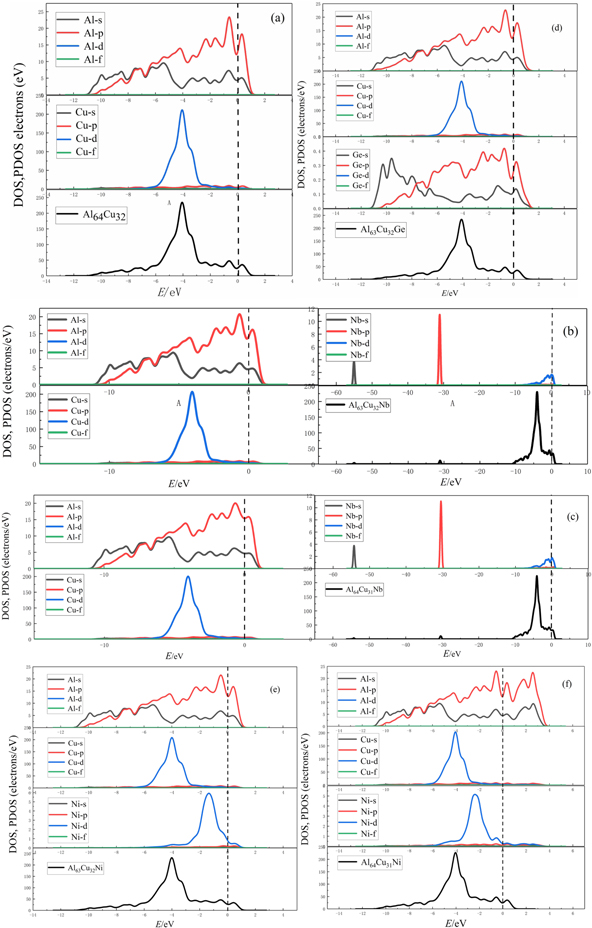
In order to understand the action mechanism, the electronic interaction, the bonding's conditions of doped atoms in structures, the total density of states (DOS) and the partial density of states (PDOS) were studied before doping and after doping. The outer electrons involved in the calculations are: Al 3s23p1, Cu 3d104s1, Nb 4p64d45s1, Ge 4s24p2, Ni 3d84s2. Figure 6 is the total density of states (DOS) and the partial density of states (PDOS).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 6. The Partial and total density of states for (a) Al64Cu32, (b) Al63Cu32Nb, (c) Al64Cu31Nb, (d) Al63Cu32Ge, (e) Al63Cu32Ni, (f) Al64Cu31Ni.
Download figure:
Standard image High-resolution image{kind=link}
In figure 6, the Fermi level is in the dashed line. Figure 6(a) is the density of states of Al64Cu32 before doping, it can be seen that the bonding electrons are mainly distributed between −11∼1 eV, mainly from the contributions of Al (3s), Al (3p) and Cu (3d). Between −6∼−2 eV, orbital hybridization exists between Al (3p) and Cu (3d), indicating that Al and Cu have bonding effects. Between −2∼1 eV, the bonding electrons mainly come from the contribution of Al (3s) and Al (3p). Figure 6(b) is the density of states of Al63Cu32Nb, the total density of states is divided into three parts. In the first part, the bonding electrons are mainly distributed between −56∼−54 eV, which are mainly contributed by Nb (5s). In the second part, the bonding electrons are mainly distributed between −32∼−30 eV, mainly contributed by Nb (4p). The third part of the bonding electrons are mainly distributed between −10∼3 eV, mainly from the contribution of Al (3s), Al (3p) and Cu (3d), a small contribution from Nb (4d). Between the −6 eV∼−3 eV, the orbital hybridization between Al (3p) and Cu (3d) are very strong, indicating that the covalent bonding effects are very powerful. Between −4∼3 eV, Al (3s), Al (3p) and Nb (4d) exist weak orbital hybridization, indicating that the bonding effects between Al and Nb are feeblish. Figure 6(c) is the density of states of Al64Cu31Nb, we can see that the case of Al64Cu31Nb is similar to that of (b) Al63Cu32Nb, it is also a strong bonding effects between Al and Cu, and a weak bonding effects between Al and Nb. Figure 6(d) is the density of states of Al63Cu32Ge the total density of states are mainly distributed between −12∼2 eV, mainly contributed by Al (3s),Al (3p), Cu (3d) and a little Ge (4s), Ge (4p). Similarly, there are strong orbital hybridization between Al (3p) and Cu (3d) from −6∼−2 eV. A weak orbital hybridization between Al (3s), Al (3p), Cu (3d), Ge (4s) and Ge (4p). It shows that there is also a weak bonding effects between Ge, Al and Cu. Figure 6(e) is the density of states of Al63Cu32Ni, and the bonding electrons are mainly distributed between −12∼1 eV, mainly from the contributions of Al (3s), Al (3p), Cu (3d) and Ni (3d). Between −6∼−2 eV, Al (3p) and Cu (3d) exist strong orbital hybridization, Al and Cu have strong bonding effects. Al (3s), Al (3p) and Cu (3d) contribute a major part of the total DOS. Between −4∼1 eV, Al (3p) and Ni (3d) exist strong orbital hybridization, Al (3s) and Ni (3d) occur slight hybridization, so Al and Ni have a bonding effects. Figure 6(f) is the density of states of Al64Cu31Ni, and the bonding electrons are mainly distributed between −12∼4 eV, similar to (d) mainly from the contributions of Al (3s), Al (3p), Cu (3d) and Ni (3d). It's just that the total density of states between −2 eV∼fermi level contributed by Al and Ni, and fermi level ∼4 eV by Al (3s) and Al (3p). Between −6∼−2 eV, Al (3p) and Cu (3d) exist strong orbital hybridization, Al and Cu have strong bonding effects. Between −4∼−2 eV, Al (3s), Al (3p) and Ni (3d) exit strong orbital hybridization, indicating that Al and Ni also have strong bonding effects. In summary, compared all density of states, we found that doped Ni causes strong orbital hybridization between Al (3p) and Ni (3d) in Al2Cu, doped Ge causes weak orbital hybridization between Al (3p), Cu (3d) and Ge (4s), Nb is in the middle of Ge and Ni. Doped Nb and Ni can greatly enhance the bonding strength of Al2Cu, and improve the stability. This is consistent with our calculated results of Debye temperature (ΘD).
4. Conclusions
We have calculated the structures, stability, mechanical properties and electronic properties of X (X=Nb, Ge, Ni) doped Al2Cu by using first-principles. The results reveal that:
- (1)When X (X=Nb, Ge, Ni) doped Al2Cu, Whether X substitutes for Al or Cu, Al-Cu-Nb compounds doped with Nb are the easiest to form, while Al-Cu-Ge compounds are the most difficult to form. Al-Cu-Nb system are the most stable, while Al-Cu-Ge system are the most instable.
- (2)The ability of resisting deformation under normal stress is stronger than the ability of resisting deformation under shearing stress in these six structures (Al64Cu32, Al63Cu32Nb, Al64Cu31Nb, Al63Cu32Ge, Al63Cu32Ni, Al64Cu31Ni). Doping Nb, Ge and Ni improved the stiffness of the Al2Cu, Al64Cu31Ni has the largest stiffness and Al64Cu31Nb has the smallest stiffness. The ability to resist elastic deformation is also increasing. Addition of Nb, Ge and Ni can greatly reduce the anisotropy of the {100}, {010} surface and the whole anisotropy, and greatly reduce the possibility of producing micro-cracks on the {100}, {010} surface. In particular, Nb and Ge replaced Al sites have the lowest degree of doping anisotropy, and the effect of doped Ge is more pronounced.
- (3)The addition of Nb and Ni can increase the Debye temperature (ΘD) of Al2Cu and make the chemical bonds more stable in the Al2Cu cells, thus improving the thermal conductivity, hardness and wear resistance of Al2Cu. This is due to the strong orbital hybridization between the Al (3s) and Nb (4d), Al (3s) and Ni(3d), resulting in strong bonding effects. The Debye temperature (ΘD) of Al2Cu will be a slight decreased when Ge is doped. This may be due to after doping, a hybridization between the Al (3s) and the Ge (4p) occurs, which weakens the strong hybridization between the Al (3s) and Cu (3d), and weakens the bonding effect between Al and Cu.
Acknowledgments
We gratefully acknowledged the financial support from the Key Fund Project (Grant No. 51374128, No. 51874172) of the National Science Foundation, People's Republic of China. Resources supporting this work were provided by High-performance Computing Platform of Renmin university of China. Finally, thanks to my ex-girlfriend Yu. Thank her for her quiet company for the past year. She is a good girl. I like her very much!