
Abstract
Capacitive storage (e.g., double layer capacitance and pseudocapacitance) with Na+ stored mainly at the surface or interface of the active materials rather than inserted into the bulk crystal is an effective approach to achieve high rate capability and long cycle life in sodium-ion batteries (SIBs). Herein, atomically thin Co3O4 nanosheets are successfully synthesized and grown directly on the stainless steel mesh as an anode material for SIBs. This anode delivers a high average capacity of 509.2 mAh g−1 for the initial 20 cycles (excluding the first cycle) at 50 mA g−1, presents excellent rate capability with an average capacity of 427.0 mAh g−1 at 500 mA g−1, and exhibits high cycling stability, which significantly outperforms the electrode prepared from conventional Co3O4 nanostructures, the electrode prepared by conventional casting method, and previously reported Co3O4 electrodes. The superior electrochemical performance is mainly attributable to the atomic thickness of the Co3O4 nanosheets and the direct growth method in electrode processing, which lead to remarkably enhanced surface redox pseudocapacitance and interfacial double layer capacitance. This Na+ capacitive storage mechanism provides a promising strategy for the development of electrode materials with high energy and power densities and ultralong cycle life for SIBs.
Export citation and abstract BibTeX RIS
1. Introduction
Wide-scale implementation of energy from nature has stimulated intensive studies on effective energy storage systems, such as rechargeable batteries and supercapacitors [1–5]. Among them, lithium-ion batteries (LIBs) with high power/energy density, long cycle life, and environmental friendliness have experienced rapid development over the past few decades and have been widely applied in smart and portable devices (e.g., personal computers and cameras), electric and hybrid electric vehicles [6–8]. Nonetheless, with the ever-growing popularity of and demand for LIBs, the high cost (over US$20 000/ton for battery grade), scarcity, and potential exhaustibility of lithium resources have started to drive up their prices and impede their further development. In view of this, sodium-ion batteries (SIBs) have come again into the researchers' line of sight after decades of dreariness and are attracting more and more attention lately due to the cheap (about US$1000/ton for battery grade), abundant, and ubiquitous sodium resources, as well as the suitable redox potential of sodium (−2.71 V versus standard hydrogen electrode) [9–14]. To date, the reported anode materials for SIBs still suffer from low specific capacity, bad rate capability, and short cycle life compared with their LIB counterparts, which significantly impedes the commercial application of SIBs [15, 16]. The poor electrochemical properties are mainly ascribed to the slow Na+ diffusion, the sluggish Na+ insertion/deinsertion kinetics, and the high volume expansion of the host materials caused by the large radius (1.02 Å) of Na+ [17–19]. Therefore, developing effective host materials for fast and reversible Na+ storage is highly desirable but also extremely challenging.
Recently, capacitive charge storage with facile reaction kinetics has been investigated for energy storage in battery systems [20–24]. This storage mode is distinguished from the conventional diffusion-controlled mode due to a number of desirable properties such as short charging time, high power density, and long-term cycling stability [25]. It mainly occurs at the surface or interface of the active materials through electrostatic adsorption, redox reaction, or intercalation [26–31], and the slow ion diffusion within the crystal lattice does not occur, which thus leads to facile reaction kinetics [32, 33]. Considering the huge volume expansion and sluggish insertion/deinsertion kinetics caused by the large radius of Na+ ion, developing a material that has the potential to show considerable capacitance, with the charge stored mainly at the surface and interface of the active materials rather than inserted into the bulk crystals, should be of theoretical benefit for the performance of SIBs.
Based on the above introduction, one way to increase the capacitive Na+ storage in SIBs is to increase the specific surface area of the active material. Recently, two-dimensional (2D) inorganic graphene analogues with atomic thickness have triggered considerable research interest following the discovery of graphene due to their fascinating properties, including extremely large surface area, improved electrical conductivity, and high structural stability and flexibility [34–40]. These properties can dramatically facilitate the highly reversible surface electrostatic adsorption and redox reactions, which consequently contribute to the surface capacitive storage. To further increase the capacitive Na+ storage, one would like to develop an electrode with large interfacial areas for facile Na+ intercalation/de-intercalation. Growing the active material directly on the surface of the current collector should be an effective approach, since it not only improves the electrical conductivity, but also facilitates the charge separation via the 'job-sharing' mechanism, which greatly increases the interfacial capacitive storage [24, 29]. Herein, as a representative example, atomically thin Co3O4 nanosheets were successfully synthesized through a bottom-up self-assembly approach and grown directly on a stainless steel mesh as an anode material (ATCSM) for SIBs. As expected, the ATCSM delivers a high average discharge capacity of 509.2 mAh g−1 for the initial 20 cycles (excluding the first cycle) at 50 mA g−1, presents excellent rate capability with an average discharge capacity of 427.0 mAh g−1 at 500 mA g−1, and also exhibits high cycling stability, significantly outperforming the electrode prepared from conventional Co3O4 nanostructures (CWM) and the electrode prepared by conventional casting method (ATCSC).
2. Experimental section
2.1. Preparation of ATCSM, CWM, and ATCSC
0.3 g polyethylene-oxide–polypropylene-oxide–polyethylene-oxide (PEO20-PPO70-PEO20, Pluronic P123) was first dissolved in an ethanol (5 g)–water (1.5 g) mixed solvent. 20 ml ethylene glycol (EG) was then added to assist the P123 to form inverse lamellar micelles in the solvent. After that, cobalt(II) acetate tetrahydrate (0.19 g for ATCSM and 0.38 g for CWM) and hexamethylenetetramine (0.1 g for ATCSM and 0.2 g for CWM) were added into the solution and dissolved under stirring for 0.5 h. After statically aging for 1 day, the solution was transferred into a 50 ml autoclave with several pieces of stainless steel mesh (300 mesh for ATCSM and 100 mesh for CWM) put in the bottom, and was then heated at 165 °C for 10 h. Finally, the atomically thin Co3O4 nanosheet- and Co3O4 nanowall-coated meshes were taken out and calcinated at 400 °C for 5 min, which lead to the formation of ATCSM and CWM, respectively. ATCSC was prepared through a conventional casting method. The atomically thin Co3O4 nanosheets were first blended with conductive acetylene black and polyvinylidene difluoride binder in a weight ratio of 8:1:1 in the presence of N-methyl-2-pyrrolidone. The obtained slurry was then brushed on the copper foils and dried at 120 °C for 12 h under vacuum. After that, the coated copper foils were tailored and pressed under 2 MPa. The effective loading masses of the active materials for these three electrodes were about 0.3 mg cm−2.
2.2. Characterizations
The morphology of the as-prepared electrodes was observed by scanning electron microscopy (SEM, JSM-7500FA, JEOL, Tokyo, Japan) and transmission electron microscopy (TEM, JEM-2011F, JEOL, Tokyo, Japan). The crystal structures of the Co3O4 nanosheets and the discharged/charged electrodes were confirmed by high-resolution TEM (HRTEM), fast Fourier transforms (FFT), and selected area electron diffraction (SAED) analyses. Atomic force microscopy (AFM, MPF-3D, Asylum Research, Santa Barbara, USA) was applied to evaluate the thickness of the Co3O4 nanosheets. The specific surface area and the pore size distribution were determined by the Brunauer–Emmett–Teller nitrogen adsorption technique (Micromeritics TriStar II 3020) with the powders degassed at 150 °C under vacuum overnight. X-ray diffraction patterns (XRD, MMA, GBC Scientific Equipment LLC, Hampshire, IL, USA) and Raman spectra (Lab RAM HR, Horiba Jobin Yvon SAS) were collected to evaluate the phase composition. The surface chemical states of different Co3O4 nanostructures were investigated by x-ray photoelectron spectroscopy (XPS, Phoibos 100 Analyser, Specs, Germany; Al Kα x-rays).
2.3. Electrochemical measurements
CR 2032 coin-type cells were assembled in Ar-filled glovebox (Mbraun, Unilab, Germany) with both the O2 and H2O levels less than 0.6 ppm. The prepared ATCSM, CWM, or ATCSC anode was used as the working electrode, porous polypropylene membrane as the separator, and Na foil as the counter and reference electrode. 1 M NaClO4 in a mixed solvent of ethylene carbonate (EC, 54.6 wt%), diethyl carbonate (DEC, 40.4 wt%), and fluoroethylene carbonate (FEC, 5 wt%) was chosen as the electrolyte. The assembled cells were galvanostatically discharged and charged at various current densities in the voltage range of 0.01–2.5 V (versus Na/Na+) on an automatic battery tester system (Land®, China). Cyclic voltammetry (CV) at different scan rates and electrochemical impedance spectroscopy (EIS) were conducted using a Biologic VPM3 electrochemical workstation.
3. Results and discussion
The atomically thin Co3O4 nanosheets were synthesized through a bottom-up self-assembly approach and grown on the stainless steel mesh in a hydrothermal reaction [41]. At the beginning of the reaction, polyethylene-oxide–polypropylene-oxide–polyethylene-oxide (Pluronic P123) surfactant formed inverse lamellar micells around the stainless steel mesh in ethylene-glycol–ethanol–water mixed solvent. After that, the hydrated cobalt oligomers were confined inside the inverse lamellar micells, and were assembled and grown along the surface of the stainless steel mesh. Finally, the ATCSM was obtained after removing the surfactant under heat treatment (experimental section). Figure 1(a) shows the SEM image of the as-prepared ATCSM. The nanosheets were uniformly coated on the surface of the steel mesh without any aggregations in mesh pores. These coated nanosheets are intertwined with each other, forming an interconnected network with numerous accessible open spaces (figure 1(b)), and in this way, most of their surfaces are exposed without overlapping each other. The structural details of these nanosheets were further studied by TEM. As can be seen in figure 1(c), there are numerous mesopores distributed uniformly throughout the 2D nanocrystals. The HRTEM image and the corresponding FFT pattern present a well-defined structure with lattice fringe spacings of 0.28 and 0.47 nm (figure 1(d)), which are associated with the (220) and (1–11) facets of cubic spinel Co3O4, respectively [27, 42]. AFM was further employed to determine the thickness of the Co3O4 nanosheets. As shown in figure 1(e), the profile along the line in the AFM image shows a height of ∼1.5 nm, which provides direct evidence for the successful synthesis of atomically thin Co3O4 nanosheets. The N2 adsorption–desorption isotherms of the Co3O4 nanosheets demonstrate a high specific surface area of ∼156 m2 g−1 and an average pore size of ∼6.8 nm (online supplementary figure S1). To confirm the advantages of the designed electrode for sodium storage performance, Co3O4 nanowalls (∼4.5 nm in thickness) grown on the stainless steel mesh (CWM, online supplementary figures S2 and S3) and an electrode produced by the conventional casting method (ATCSC, online supplementary figure S4) were also prepared for comparison (experimental section).

Figure 1. Atomically thin Co3O4 nanosheets grown on stainless steel mesh (ATCSM). (a) SEM image of the ATCSM (300 mesh). (b) High-magnification SEM image showing the coated ultrathin Co3O4 nanosheets. (c) TEM image presenting the mesoporous structure of the Co3O4 nanosheets. (d) HRTEM image of the Co3O4 nanosheets and its corresponding FFT pattern (inset). (e) AFM image and height profile (inset) along the red line of a typical Co3O4 nanosheet indicating its atomic thickness. Scale bars: (a) 300 μm, (b) 1 μm, (c) 100 nm, (d) 2 nm, (e) 1 μm.
Download figure:
Standard image High-resolution imageThe extraordinary structural features of the atomically thin Co3O4 nanosheets were studied by XRD, Raman spectroscopy, and XPS. As shown in figure 2(a), the XRD diffraction peaks of CWM can be clearly indexed to well-crystallized cubic spinel Co3O4 and austenitic stainless steel mesh. However, the Co3O4 phase is hardly to be detected in ATCSM due to the absence of long-range order in the third dimension for the atomically thin nanosheets [34]. The Raman spectrum (figure 2(b)) of the scraped atomically thin Co3O4 nanosheets from the growth substrate shows five peaks at 187, 463, 507, 605, and 665 cm−1, which are originated from the Eg, 
 and A1g vibration modes of Co3O4 [27]. These peaks are much broader and exhibit 5 cm−1 shifts to lower wavenumbers compared with those of Co3O4 nanowalls, which are attributable to the more obvious phonon confinement effect in the atomically thin Co3O4 nanosheets [43]. The surface chemical states of the atomically thin Co3O4 nanosheets were then examined by the XPS technique. As shown in figure 2(c), the Co 2p core level spectrum of the ultrathin nanosheets shows a 0.8 eV shift to lower binding energy compared with that of the nanowalls. This chemical state shifting is associated with the reduction of Co atoms caused by the lattice structural distortion in atomically thin nanocrystals [44, 45]. It was also reported that the structural distortion leads to a remarkably increased density of states near the Fermi level, which facilitates fast electron transport along the 2D conducting channels and consequently improves the electrical conductivity of the atomically thin Co3O4 nanosheets [34].
and A1g vibration modes of Co3O4 [27]. These peaks are much broader and exhibit 5 cm−1 shifts to lower wavenumbers compared with those of Co3O4 nanowalls, which are attributable to the more obvious phonon confinement effect in the atomically thin Co3O4 nanosheets [43]. The surface chemical states of the atomically thin Co3O4 nanosheets were then examined by the XPS technique. As shown in figure 2(c), the Co 2p core level spectrum of the ultrathin nanosheets shows a 0.8 eV shift to lower binding energy compared with that of the nanowalls. This chemical state shifting is associated with the reduction of Co atoms caused by the lattice structural distortion in atomically thin nanocrystals [44, 45]. It was also reported that the structural distortion leads to a remarkably increased density of states near the Fermi level, which facilitates fast electron transport along the 2D conducting channels and consequently improves the electrical conductivity of the atomically thin Co3O4 nanosheets [34].

Figure 2. Characterizations of the atomically thin Co3O4 nanosheets in ATCSM and the Co3O4 nanowalls in CWM. (a) XRD patterns showing that the diffraction peaks of ATCSM are barely visible compared with those of CWM. (b) Raman spectra with the inset showing the details of the peak broadening and shift. (c) XPS survey spectra with the inset presenting the binding energy shift of Co 2 P.
Download figure:
Standard image High-resolution imageThe electrochemical performance of ATCSM as anode material for SIBs was evaluated using CR 2032 coin-type cells with Na foil as the counter/reference electrode (experimental section). Figure 3(a) presents the cyclic voltammograms (CVs) of ATCSM in the voltage range of 0.01–2.5 V at a scan rate of 0.1 mV s–1. The first cathodic scan shows a weak peak at around 1.20 V and a strong and broad peak at around 0.58 V. According to a study on the conversion reaction of Co3O4 with Li in LIBs, the weak peak corresponds to the reduction of Co3O4 to the intermediate of NaxCo3O4, and the strong and broad peak is mainly attributable to contributions from the reduction of NaxCo3O4 to Co–Na–O clusters and finally to metallic Co, as well as the formation of the solid electrolyte interphase (SEI) film [46–49]. It is worth emphasizing that Na2O2 forms at the fully discharged state (0.01 V) as observed by the ex situ HRTEM and SAED pattern analyses (online supplementary figure S5), which is contrary to the proposed Na2O in the literature. Our result is consistent with the storage mechanism of SnS2 for SIBs studied by Meng's group [50]. They confirmed that Na2S2 forms instead of the assumed Na2S at the fully discharged state. Therefore, the reaction mechanism in Li-ion system could not be simply applied to Na-ion system due to the differences between Li–O/S and Na–O/S alloys. For the first anodic scan of the CV curve, the peak at 0.85 V represents the oxidation of Co to Co-rich intermediates and the decomposition of Na2O2, while the peaks at 1.12 and 1.54 V are ascribed to the stepwise oxidation of Co-rich intermediates to Co3O4 and the release of Na+. Further ex situ HRTEM and SAED pattern studies show that Co and Na2O2 still remain at the fully chraged state (2.5 V) (online supplementary figure S6), indicating the limited reversibility in SIBs. Generally, the electrochemical Na+ insertion/deinsertion process within spinel Co3O4 is based on the conversion mechanism of Co3O4 + 4Na+ + 4e− ↔ 2Na2O2 + 3Co. This conversion process was also confirmed by the discharge/charge voltage profiles cycled at a current density of 50 mA g−1 (online supplementary figure S7). In subsequent CV cycles, the reduction and oxidation peak intensities decrease more significantly compared with those in the Li-storage reaction, further suggesting the more irreversible conversion reaction caused by the larger size of Na+. Figure 3(b) shows the discharge/charge voltage profiles of ATCSM at different current densities. As can be seen, the discharge/charge plateaus are increasingly shortened and inclined, and the separation between them is gradually increased with increasing current density due to the slow Na+ diffusion in active material and the increase in ohmic resistance [51]. These variations are much less obvious, however, when compared with those of CWM and ATCSC (online supplementary figure S8), indicating less ohmic polarization for ATCSM. Having studied the Na+ storage behavior in ATCSM, the rate performance was then investigated at stepwise current densities from 50 to 5000 mA g−1 (figure 3(c)). After the capacity fading in the initial 15 cycles, the average discharge capacities (and the capacity ratios) for ATCSM at 50, 100, 200, 500, 1000, 2000, and 5000 mA g−1 are 418.8 (averaged from the 16th cycle to the 20th cycle), 344.2 (82.2%), 278.1 (66.4%), 216.5 (51.7%), 176.7 (42.2%), 145.3 (34.7%), and 116.9 (27.9%) mAh g−1, respectively. In contrast, they are only 202.5/190.4, 166.4 (82.1%)/108.5 (57.0%), 133.2 (65.8%)/49.5 (26.0%), 99.9 (49.3%)/23.6 (12.4%), 75.0 (37.0%)/15.4 (8.1%), 56.7 (28%)/12.6 (6.6%), and 49.7 (24.5%)/9.7 (5.1%) mAh g−1 for CWM and ATCSC, respectively. These results indicate the superior rate capability of ATCSM and also signify the importance of the atomic thickness for Co3O4 nanocrystals and the direct growth method in electrode processing for improving Na+ storage on the surface of the nanosheets and facilitating charge separation at the interface between the Co3O4 nanosheets and the stainless steel mesh.

Figure 3. Electrochemical performances of ATCSM, CWM, and ATCSC in SIBs. (a) CV curves for the first five cycles of ATCSM at a scan rate of 0.1 mV s–1. (b) Discharge–charge voltage profiles of ATCSM at different current densities. (c) Rate capabilities and capacity ratios (inset) of ATCSM, CWM, and ATCSC at stepwise current densities from 50 to 5000 mA g–1. (d) Cycling performances and CEs of ATCSM, CWM, and ATCSC at a high current density of 500 mA g–1.
Download figure:
Standard image High-resolution imageThe cycling performance was further studied at a high current density of 500 mA g–1. As shown in figure 3(d), ATCSM delivers a very high initial discharge capacity of 1239.7 mA g–1, which is comparable with that in Li+ storage. Nevertheless, the Coulombic efficiency (CE) of the first cycle is only 46.8%. By contrast, CWM and ATCSC deliver much lower initial discharge capacities of 661.4 and 425.3 mA g–1, but higher CEs of 51.2% and 67.2%, respectively. The higher initial discharge capacity and lower initial CE of ATCSM are mainly attributable to the atomic thickness of the Co3O4 nanosheets and the direct growth method in electrode processing, which provide an ultrahigh ion-accessible surface area and consequently leads to the formation of large-area SEI films for irreversible Na+ storage. The ATCSM delivers an average discharge capacity of 427.0 mAh g−1 in the initial 20 cycles at 500 mA g–1 (exclude the first cycle), which is 83.9% of the average capacity (509.2 mAh g−1) obtained at 50 mA g–1 (figure 3(c)). This further confirms the superior rate capability of ATCSM with fast and facile Na+ storage. In the following cycles, the discharge capacities of these three electrodes decrease while the CEs increase gradually. That is because the Na+ ions could not be completely extracted from the interior of Co3O4 nanocrystals due to the sluggish insertion/deinsertion kinetics, which consequently leads to the decrease of diffusion-controlled storage [24]. After 100 cycles, the discharge capacity of ATCSM still has 172.9 mAh g–1, while those of CWM and ATCSC remain only 62.5 and 56.4 mAh g–1, respectively, indicating the better reversibility and cycling stability of ATCSM. The better cycling stability of ATCSM could also be verified by its well-maintained graphene-like nanostructure after 100 cycles (online supplementary figure S9). The electrochemical performance of ATCSM not only stands out from those of CWM and ATCSC, but also outperforms previously reported results obtained at relatively low current densities of 25–178 mA g–1 (online supplementary figure S10), further suggesting the advantages of the atomic thickness of Co3O4 nanosheets and the direct growth method in electrode processing.
To deeply understand the outstanding electrochemical performance of ATCSM, we investigated the reaction kinetics of ATCSM, CWM and ATCSC by performing CV at various scan rates from 0.1 to 100 mV s−1. As can be seen in figure 4(a) and online supplementary figure S11, both the anodic and cathodic peaks become stronger and broader as the scan rate increases. In addition, the separation between them also increases, indicating large polarization at high scan rates. Figure 4(b) presents a plot of log(cathodic peak current, i) versus log(scan rate, v) that is derived from figure 4(a) and online supplementary figure S9. According to a power law [24, 25], the relationship between them can be expressed as:
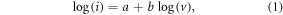
where a and b are adjustable values. Note that, the b-value of 0.5 indicates that the current is totally controlled by semi-infinite linear diffusion, while that of 1.0 represents a totally surface- and/or interface-controlled capacitive process. The slope of the log(i)–log(v) plot (b-value) for ATCSM shows an average value of 0.86 in the range of 0.1–20 mV s−1, indicating the dominant capacitive contribution with facile Na+ storage kinetics that are controlled by the surface and/or the interface. The slope decreases to 0.73–0.70, however, when the scan rate rises up to 50–100 mV s−1, suggesting an increase in ohmic polarization and the limitation of Na+ diffusion. By contrast, the diffusion-dominated Na+ storage in CWM occurs at the lower scan rate of 20 mV s−1 with a b-value of 0.73, revealing its poor rate capability as compared to ATCSM. Moreover, the Na+ storage in ATCSC is diffusion-dominated over the whole range of scan rates with an average b-value of 0.59, signifying that this conventional casting method in electrode processing cannot make full use of the surface and/or interface areas of the active materials to enhance the capacitive Na+ storage. The relationship between capacity (Q) and scan rate (v) could also be established. As shown in figure 4(c), the Q versus v−1/2 plots show that the capacity of ATCSM is more independent of the scan rate compared with that of CWM, due to its higher capacitive contribution. The capacity for ATCSC decreases linearly with decreasing v−1/2, further verifying that its Na+ storage is diffusion-dominated over the whole range of scan rates.

Figure 4. Kinetics analysis of the Na+ storage behavior in ATCSM, CWM, and ATCSC. (a) CV curves of ATCSM at various scan rates from 0.1 to 100 mV s−1. (b) Log(peak current) versus log(scan rate) plots for the determination of the capacitive-dominated region and diffusion-dominated region in ATCSM, CWM, and ATCSC. (c) Capacity versus scan rate−1/2 providing another evaluation method to identify the capacitive-dominated region and diffusion-dominated region. (d) Separation of the capacitive current from the diffusion-controlled current in ATCSM at a scan rate of 2 mV s−1. (e) Contribution ratios of the capacitive charge storage and diffusion-controlled charge storage at different scan rates in ATCSM, CWM, and ATCSC. (f) Nyquist plots of ATCSM, CWM, and ATCSC in the discharged state at 0.5 V.
Download figure:
Standard image High-resolution imageIt is possible to quantitatively separate the capacitive charge storage from the total storage capacity by distinguishing the capacitive current at a fixed voltage according to:
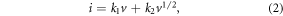
where i is the current density at a fixed potential, v is the scan rate, k1 and k2 are parameters, with k1v standing for the capacitive contribution, and k2v1/2 representing the diffusion-controlled contribution [32, 52]. Figure 4(d) shows the capacitive current (red region) separated from the total current as a function of voltage at a scan rate of 2 mV s−1. As can be seen, diffusion-controlled Na+ storage takes place mainly in the cathodic and anodic peak regions, which is associated with the redox reaction between Co3O4 and Co. The quantified capacitive contribution accounts for 68.7% of the total storage capacity. The ratios of the capacitive contribution to the diffusion-controlled contribution for ATCSM, CWM and ATCSC at scan rates from 0.1 to 5.0 mV s−1 were calculated. As shown in figure 4(e), the capacitive contribution increases with increasing scan rate. Therefore, high rates show a greater emphasis on the capacitive storage mechanism due to its facile reaction kinetics compared with those of the diffusion-controlled storage mechanism [31]. The average capacitive contribution for ATCSM in the range of 0.1–5 mV s−1 is 63.5%, much higher than the 54.9% and 25.0% for CWM and ATCSC, respectively, indicating the enhanced capacitive Na+ storage in ATCSM. The remarkable capacitive storage in ATCSM could be further confirmed by EIS performed in the discharged state at 0.5 mV (figure 4(f)). The Nyquist plots of the three electrodes present a depressed semicircle in the high-medium frequency region and a clear inclined line in the low-frequency region. The semicircle is assigned to the charge transfer resistance (Rct) and the inclined line is related to a mass (mainly Na+) transfer process [51]. The Rct-value for ATCSM is 446 Ω, which is much lower than the 582 Ω and 1035 Ω for CWM and ATCSC, respectively, suggestive of the small charge transfer resistance and the facile electrochemical reaction kinetics. Moreover, the much steeper tail in the low-frequency region for ATCSM also indicates the fast Na+ diffusion with a low energy barrier. These analyses further suggest the obvious capacitive storage behavior in ATCSM [53].
The capacitive Na+ storage in ATCSM mainly involves the surface redox pseudocapacitance and the interfacial double-layer capacitance due to the atomic thickness of the Co3O4 nanosheets and the direct growth method in electrode processing (figure 5). On the one hand, the Co3O4 nanosheets with atomic thickness and mesoporous structure possess numerous low-coordinated surface atoms and highly accessible surface areas. Na+ ions can electrochemically adsorb onto the coordination-unsaturated surface atoms through charge-transfer processes, and therefore enhances the surface redox pseudocapacitance [28]. On the other hand, the direct growth of ultrathin Co3O4 nanosheets on the stainless steel mesh creates a large interfacial area, and the discharged products of Co and Na2O also generate very large interfacial areas between them. In these interfacial areas, the Co3O4 nanosheets (or Na2O) act as the Na+-accepting phase, while the stainless steel mesh (or metallic Co) acts as the electron-accepting phase through the 'job-sharing' mechanism to facilitate the charge separation and enhance the so-called interfacial double-layer capacitance [24, 25]. The Na+ storage behavior through these two storage modes is kinetically facile and not diffusion controlled on the time scale of interest. Therefore, both the surface redox pseudocapacitance and the interfacial double-layer capacitance contribute to the excellent electrochemical performance of ATCSM.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. Schematic illustration of the capacitive Na+ storage in ATCSM. Two mechanisms exist: the pseudocapacitance arises from two storage mechanisms: surface redox pseudocapacitive storage on the surfaces of the atomically thin Co3O4 nanosheets, and interfacial double-layer capacitive storage at the interfaces between the Co3O4 nanosheets and the stainless steel mesh, and between different phases of the electrode material.
Download figure:
Standard image High-resolution image{kind=link}
4. Conclusion
Atomically thin Co3O4 nanosheets were synthesized through a surfactant-assisted self-assembly approach and grown on stainless steel mesh as an anode material for SIBs. This novel anode could deliver a high average discharge capacity of 509.2 mAh g−1 for the initial 20 cycles (exclude the first cycle) at 50 mA g−1, presents excellent rate capability with an average discharge capacity of 427.0 mAh g−1 at 500 mA g−1, and also exhibits high cycling stability, which significantly outperforms the electrodes prepared from conventional Co3O4 nanostructures and conventional casting method, and other previously reported Co3O4 electrodes. The superior electrochemical performance is mainly attributable to the atomic thickness of the Co3O4 nanosheets and the direct growth method in electrode processing, which lead to remarkably enhanced surface and interfacial capacitive storage. The successful development of atomically thin Co3O4 nanosheets could be extended to the synthesis of other non-layered materials with ultrathin 2D nanostructures for fast and reversible Na+ storage. In addition, the active materials could also be controlled to grow on other current collectors such as graphene, carbon nanotubes, and electrical conductive porous materials for improved energy densities, and this novel design of direct growth method on the current collector provides a facile and large-scale approach for electrode processing, which could replace the conventional casting method and dramatically enhances the Na+ storage performance.
Acknowledgments
The authors are grateful for financial support from the Australian Research Council Discovery Project (DP160102627), the UOW-BUAA Joint Research Centre, an Australian Postgraduate Award (APA), and an International Postgraduate Research Scholarship (IPRS).