

Abstract
Self-assembling peptides are attractive materials for tissue engineering applications because of their functionality including high biocompatibility and biodegradability. Modification of self-assembling peptides with functional motifs, such as the cell-adhesive tripeptide sequence RGD leads to functional artificial extracellular matrices (ECMs). In this study, we developed an artificial self-assembling ECM protein tethered with a growth factor via heterotrimer triple-helix (helix A/B/C) formation. The helix A and helix C peptides, which are capable of forming a heterodimer coiled-coil structure, were fused to both ends of a matrix protein composed of the elastin-derived structural unit (APGVGV)12 with an RGD motif. The helix B peptide, which constituents the third helix of the triple-helix structure, was fused with basic fibroblast growth factor (bFGF) for tethering to the artificial ECM proteins. Each recombinant protein exhibited cell adhesion and cell proliferation activities similar to the original, while the designed bFGF-tethered ECM protein exhibited superior cell proliferation activity. These results demonstrate that the approach of creating growth factor-tethered self-assembling proteins via triple-helix formation can be applied to develop functional ECMs for tissue engineering applications.
Export citation and abstract BibTeX RIS
1. Introduction
Development of functional artificial extracellular matrices (ECMs) is desirable for applications such as regeneration of damaged tissues and construction of human tissues in vitro, which are goals of regenerative medicine and tissue engineering [1–3]. The ECM provides a microenvironment that supports the mechanical strength and induction of biological signals of cells, allowing for the control of cellular functions, such as cell adhesion, proliferation, and differentiation [4, 5].
Efforts aimed at creating artificial ECMs composed of synthesized and naturally derived materials with functions similar to native ECMs have been undertaken by various groups [6–11]. Previously, we designed and genetically synthesized artificial ECM proteins [12–18]. Our approach to the design of high-functional ECM proteins was based on combining structural peptides and bioactive peptides that control cellular activities. One such ECM protein named ERE consists of 12 repeats of the APGVGV peptide motif derived from elastin as the stable structural unit (E12) and the RGD peptide motif known to cell adhesive sequence as the bioactive unit [12]. Moreover, multi-functional ECM proteins were developed for controlling cellular functions in our genetic engineering-based designs. The ECM proteins were composed of not only bioactive peptide motifs derived from native ECMs such as fibronectin, laminin, and neural cell adhesion molecule but also of signal factors and transcriptional factors [14, 16–18]. For tethering such signal factors, heterodimer coiled-coil interactions were used. Two such helix peptides useful for noncovalent coupling of two proteins for the construction of functional biomaterials were developed by O'Shea et al, namely ACID-p1 and BASE-p1 [19], which form a favorable coiled-coil structure via precise electrostatic stabilization.
Our genetically designed growth/transcriptional factor-tethered artificial ECM proteins demonstrated applicability as functional ECMs. However, the formation of nanofibers requires supramolecular assembly via precise interactions. Nanofibers composed of self-assembling peptides are attractive candidates because the fibrillary structures mimic the microenvironment of native ECMs that enhance cellular activities, and also exhibit high biocompatibility and biodegradability [20–25]. To this end, self-assembling peptides have been designed utilizing protein secondary structures, such as α-helix [26–28], β-sheet [29–31], and β-hairpin [32, 33], which generate synergetic effects of precise array control and multi-point recognition of weak noncovalent interactions.
Herein, we focused on three artificial α-helices, namely helix A, B, and C (we call them hA, hB, and hC, respectively, in this paper). They form a heterotrimeric coiled-coil stabilized by hydrophobic isoleucine residues' core and specific electrostatic interactions between lysine and glutamic acid residues [34, 35]. The α-helices work as the sticky end of interest fusion proteins and link them. We demonstrated the triple helix can be used as self-assembly domains without inhibiting coiled-coil formation even though fusing 66 kDa proteins [36]. Thus, self-assembling proteins fibers composed of naturally derived or existing artificial ECM proteins frame can be constructed using the α-helices. Meanwhile, other self-assembling peptides, such as β-sheet forming peptides, could not apply to such application as the sticky end of ECM proteins. Therefore, high functional ECM proteins that mimic microenvironment, not only the physical but also the biological environment, would be developed using a triple helix. Moreover, the triple helix can be had another function, such as immobilization of signal factors to the ECM proteins. The dual functions of triple helix could not be obtained in other peptides including double helix coiled-coil.
In this study, the helix peptides served as self-assembling domains and growth factor-tethering domains. Specifically, we developed growth factor tethered self-assembling ECM proteins (figure 1). For promoting supramolecular assembly and tethering growth factors, hA and hC were fused to both ends of the ERE matrix protein; the resulting proteins were designated hA-ERE-hA (ARA) and hC-ERE-hC (CRC), respectively. hB was fused to the N-terminus of basic fibroblast growth factor (bFGF), which exhibits various bioactivities, including cell proliferation and differentiation, for tethering to ARA and CRC assemblies (ARA–CRC). These helix-fused proteins were genetically produced, and the cellular behavior of the artificially constructed functional ECM was investigated.

Figure 1. Schematic illustration of a functional nanofiber comprised of bFGF-tethered self-assembling ECM proteins via coiled-coil triple-helix formation. (a) ECM proteins and bFGF were non-covalently associated via coiled-coil triple helix formation between the interaction of helix peptide motifs fused to each protein. (b) Control of the cellular functions with bFGF-tethered self-assembling ECM proteins on the plate surface.
Download figure:
Standard image High-resolution image2. Materials and methods
2.1. Materials
The pBluescript SKII(-) plasmid was purchased from Toyobo (Japan). The pUC18, pET28b, pET32c plasmids and E. coli BL21 (DE3) were obtained from Novagen. Escherichia coli KRX was purchased from Promega. Synthesized oligo DNA were obtained from FASMAC (Japan). Restriction enzymes and ligase were purchased from Takara Bio (Japan). All other chemicals were analytical grade.
2.2. Plasmid construction
Three plasmids were constructed to conduct following experiments described in this paper: pET28b-hA-ERE-hA-CHis, pET28b-hC-ERE-hC-CHis, and pET32c-NHis-hB-(GGGS)8-bFGF. The pET32c-NHis-ERE, pET28b-hA-CHis, pET28b-hB-CHis, pET28b-hC-CHis, pBS-(GGGS)8, and pET32c-NHis-bFGF plasmids were previously constructed in our lab [14, 36].
To obtain the ERE fragment, pET32c-NHis- ERE was digested with BamHI and BglII. The resultant fragment was ligated into a pUC18 vector, which included the adapter sequence BamHI-BglII-BspHI-EcoRI, digested with BglII. pUC18-ERE was digested with BamHI and EcoRI and ligated to pET28b-hA-CHis or pET28b-hC-CHis using the same enzymes. The resultant pET28b-hA-ERE-CHis or pET28b-hC-ERE-CHis sequences were digested with XbaI and BspHI and inserted into the XbaI and NcoI digested sites of the pET28b-hA-CHis or pET28b-hC-CHis plasmids, respectively. The resulting plasmids were named pET28b-hA-ERE-hA-CHis and pET28b-hC-ERE-hC-CHis, respectively.
To obtain the (GGGS)8 linker fragment, pBS-(GGGS)8 was digested with BglII and EcoRI. The resulting fragment was ligated to pET28b- hB-CHis digested with BamHI and EcoRI. The resultant pET28b-hB-(GGGS)8-CHis sequence was digested with NcoI and EcoRI and ligated to the pET32c-NHis-bFGF plasmid, which included the same restriction enzymes sites. The resulting plasmid was named pET32c-NHis-hB-(GGGS)8-bFGF.
2.3. Protein expression and purification
For expression of hA-ERE-hA (ARA) and hC-ERE-hC (CRC), E. coli KRX competent cells were transformed with pET28b-hA-ERE-hA-CHis and pET28b-hC-ERE-hC-CHis plasmids, respectively. Transformed E. coli cells were cultured at 37 °C in a Luria−Bertani (LB) medium with 20 μg ml−1 kanamycin at an optical density of 0.6 at 660 nm. Protein expression was induced by addition of 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) and 0.1% rhamnose. Cells were cultured for 5 h at 30 °C and harvested by centrifugation. Collected cells were suspended in Bug Buster Protein Extraction Reagent (Novagen) with Benzonase Nuclease (Novagen), followed by rotation at room temperature for 30 min after pelleting by centrifugation (8000 g, 4 °C) for 15 min, supernatants were collected. The objective fusion proteins were purified by His-select Nickel Affinity Gel (Sigma-Aldrich) using a Poly-Prep column (Bio-Rad) from the soluble fractions. After 2 h incubation at 4 °C, the column was washed five times with the wash buffer (16 mM Na2HPO4, 4 mM NaH2PO4, 500 mM NaCl, pH 8.0) of three times volume of resin. Then, the column was washed three times with the same buffer containing 5 and 10 mM imidazole each (six times in total) of three times volume of resin. The fusion proteins were eluted three times with the elution buffer (16 mM Na2HPO4, 4 mM NaH2PO4, 500 mM NaCl, 500 mM imidazole, pH 7.4) of an equivalent volume of resin. The fusion proteins were then dialyzed in a phosphate-buffered saline (PBS) and concentrated using an Amicon Ultra filter (Merck Millipore).
For expression of hB-bFGF, E. coli BL21 (DE3) competent cells were transformed with pET32c-NHis-hB-(GGGS)8-bFGF plasmid. Transformed E. coli cells were cultured at 37 °C in LB medium with 50 μg ml−1 ampicillin at an optical density of 0.6 at 660 nm. Protein expression was induced by addition of 1 mM IPTG. Cells were cultured overnight at 16 °C. The protein was purified by the procedure described above for ARA and CRC.
The purity of all fusion proteins were evaluated by 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The concentrations of the purified fusion proteins were examined using the bicinchoninic acid (BCA) Protein Assay Kit (Pierce).
2.4. Transmission electron microscopy (TEM)
ARA and CRC proteins were dissolved in 3-morpholinopropanesulfonic acid (MOPS), pH 7.0, at a final concentration of 50 μM. ARA was mixed with an equivalent volume of CRC, and the mixture was incubated at 4 °C for 1 h to promote self-assembly. The sample was adsorbed onto a hydrophilically treated specimen grid covered with a carbon film (Nisshin EM) for 1 min. After washing with ultrapure water, the grid was stained with 0.25% phosphotungstic acid (PTA) for 1 min. After washing with ultrapure water again, the stained sample was dried on filter paper at room temperature for 2 h or more. TEM images were taken with a transmission electron microscope H-7500 (Hitachi) at 120 kV.
2.5. Cell culture
Human umbilical vein endothelial cells (HUVECs) were obtained from Kurabo (Japan). These cells were maintained at 37 °C under 5% CO2 condition in HuMedia-EG2 supplemented with 2% fetal bovine serum, 10 ng ml−1 human EGF, 5 ng ml−1 human bFGF, 1.34 μg ml−1 hydrocortisone, 10 μg ml−1 heparin, 50 μg ml−1 gentamicin, and 50 ng ml−1 amphotericin B.
2.6. Cell adhesion activity
The helix-fused ERE proteins (ARA and CRC) were prepared in PBS at a final concentration of 100 nM. These protein solutions were added to a 96-well suspension culture plate (Sumilon MS-8096R) to coat the surface. A fibronectin-coated surface was prepared for as a positive control. Uncoated plates and plates coated with the helix-combined linker proteins hA-(GGGS)8-hA (ALA) and hC-(GGGS)8- hC (CLC) were prepared for as negative controls. After 1 h incubation at 37 °C, the protein-coated wells were washed with PBS. HUVECs were seeded in each plate at a density of 104 cells/well. After 3 h incubation at 37 °C, plate surfaces were washed with PBS. Attached cells on the plates were analyzed by Cell Counting Kit (CCK-8) (Dojindo) using water-soluble tetrazolium salt, WST-8. The activities were measured at a wavelength of 450 nm as described in the CCK-8. The cells were concomitantly stained with Calcein-AM (Dojindo) and observed using a fluorescence microscopy.
2.7. Cell proliferation activity of hB-bFGF
HUVECs were utilized to evaluate the cell proliferation activity of hB-bFGF. HUVECs were seeded in a 96-well tissue culture plate (Costar 3595) at a density of 2000 cells/well. Then hB-bFGF was supplied with the HuMedia-EG2 without bFGF. The cells were cultured at 37 °C under 5% CO2 condition. The culture media were changed on the first and fourth day. The day on which hB-FGF was added was defined as day 0. The activity was evaluated at an absorbance of 450 nm as described in the CCK-8 on days 1 and 4.
2.8. Protein binding via triple-helix formation
The surfaces of a non-treated 96-well plate (Sumilon MS-8096R) were modified with 100 nM of ARA–CRC by hydrophobic interaction. After 1 h incubation at 37 °C, the surfaces were washed five times with PBS-T (PBS including 0.05% Tween 20) followed by blocking with Blocking One (Nacalai Tesque) incubated overnight at 4 °C. After washing five times with PBS-T, various concentrations (1, 10, 100, and 1000 nM) of hB-bFGF were added to the ARA–CRC modified plates. After incubation for 1 h at 37 °C to allow triple helix formation, the surfaces of the plates were washed five times with PBS-T followed by reaction with 1/5000 diluted rabbit anti-bFGF antibody (Sigma) incubated for 40 min at room temperature. After washing with PBS-T, anti-rabbit IgG antibody labeled peroxidase (Jackson ImmunoResearch) was added and incubated for 40 min at room temperature. After washing with PBS-T, 3, 3', 5, 5'-tetramethylbenzidine (TMB) horseradish peroxidase (HRP) substrate (Kirkegaard and Perry Laboratories) was reacted with protein-modified surfaces of the plates. Atter 2 min incubation, 1 N HCl was added to stop the reaction, and the absorbances at 450 nm were measured.
2.9. Cell proliferation on bFGF tethered self-assembling ECM protein
The surfaces of a 96-well hydrophobic plate (Sumilon MS-8096R) were coated with 100 nM of ARA–CRC and incubated for 1 h at 37 °C. After washing three times with PBS, 10 nM of hB-bFGF was added and incubated for 1 h at 37 °C to form coiled-coil triple helix structure. After washing three times with PBS, HUVECs suspended in HuMedia-EG2 without bFGF were seeded onto the wells at a density of 2000 cells/well. The culture media were changed on the second, fifth, and seventh day. The numbers of cells on days 2 and 7 were counted by WST-8 assay using the CCK-8.
3. Results and discussion
3.1. Protein expression and purification
The constructed helix-fused proteins are illustrated in figure 2(a). ARA and CRC were created by fusing helix peptides hA or hC to both ends of the matrix protein ERE, which is composed of two (APGVGV)12 peptides along with the RGD tripeptide motif. The RGD sequence is a major cell attachment site found in various ECMs that interact with integrin receptors [37]. On the other hand, bFGF-tethered hB peptides (hB-bFGF) were constructed by fusing hB with the linker peptide (GGGS)8 to the N-terminus of bFGF. Note that in our previous work, C-terminus modified bFGF-fusion proteins were obtained from only insoluble fraction, and showed no proliferation activity, even after refolding. The (GGGS)8 linker was also inserted between hB and bFGF to enhance the flexibility of bFGF and efficiently transmit a signal to the cell without losing activity upon immobilization of bFGF to the scaffold via coiled-coil interactions. These fusion proteins were genetically produced by E. coli KRX or BL21 (DE3) as a Histidine-tag (His-tag)-fusion protein and purified from the soluble fractions using a coordinate bond with His-select nickel affinity resins. The purities of the fusion proteins were evaluated by SDS-PAGE (figures 2(b) and (c)). The bands appeared at the expected molecular weights (ARA: 24.4 kDa; CRC: 24.4 kDa; hB-bFGF: 30.0 kDa). The purities of fusion proteins ARA, CRC, and hB-bFGF were 80.2%, 84.9%, and 60.3%, respectively, estimated by ImageJ software. The fusion proteins were considered pure enough for further experiments.

Figure 2. Construction of fusion proteins. (a) The design of helix-fused matrix proteins and growth factor. The helix A, B, and C are artificial α-helices. The amino acids sequences are indicated at the bottom. (b) and (c) SDS-PAGE analysis of purified proteins. Lane M indicates the molecular weight marker. (b) Purified helix-fused matrix proteins ARA (24.4 kDa) and CRC (24.4 kDa). (c) Purified helix-fused growth factor hB-bFGF (30.0 kDa).
Download figure:
Standard image High-resolution image3.2. Evaluation of the self-assembly of fusion proteins by TEM
TEM imaging was used to examine the self-assembling capacity of a mixture of ARA and CRC. Fibril formation was expected upon specific coiled-coil interactions between hA of ARA and hC of CRC. The helix peptides hA, hB, and hC form not only heterotrimer coiled-coils but also heterodimer coiled-coils comprised of each combination of two helix peptides, each of which exhibits different associative strengths (hA-hC > hB-hC > hA-hB) [34]. Representative TEM images of the ARA–CRC complex are shown in figure 3. The sample formed a fibrillar structure in which two fibers of 500 nm in width were twisted upon other. Mature fibrils appeared to be formed by lateral aggregation arising from the elastin-derived hydrophobic APGVGV repeats following the formation of nanofibers by supramolecular assembly between ARA and CRC. This result suggests that the introduced helix peptides promote self-assembly of the matrix proteins. Although some studies have addressed nanofiber formation by self-assembly of short-chain peptides, nanofibers composed of ECM proteins can be constructed by fusing helix peptides that form specific coiled-coils to both ends of the matrix proteins. In this study, we used the repeated APGVGV sequence which is one of the motifs called elastin-like polypeptides (ELPs) as a structural frame of the ECM proteins. It has been proposed that the APGVGV polypeptide forms a stable β-spiral structure [38] that provides flexibility and stability in the ECM proteins. Meanwhile, the APGVGV motif has a propensity to aggregate in high concentration due to increasing hydrophobic interaction. To inhibit aggregation and control fiber width, incorporation of the hydrophilic group could be effective. Indeed, previous experiments by our group have shown that inducing polyaspartic acid chains regulated aggregation of ELPs by the negatively charged repulsion. As a result, the polyaspartic acid fused ELPs formed size-controlled nanoparticles [39–41]. Therefore, aggregation of high-hydrophobic ECM proteins including ELPs could be controlled by introducing electrically charged amino acid chain such as polyaspartic acid.

Figure 3. Representative TEM images of the fibril formation of a mixture of ARA and CRC. The sample was negatively stained with phosphotungstic acid after the proteins were mixed and incubated for 1 h. (a) Magnification × 10 000. Scale bar = 2 μm. (b) Magnification × 60 000. Scale bar = 500 nm.
Download figure:
Standard image High-resolution image3.3. Cell adhesion activity of ARA and CRC
HUVECs were seeded onto a surface coated with ARA and CRC to evaluate the cell adhesion activity of these ECM proteins. Non-coated, hA-(GGGS)8-hA (ALA)-coated and hC-(GGGS)8-hC (CLC)-coated plates were used as negative controls, while ERE-coated and fibronectin-coated plates were used as positive controls. The repeating APGVGV sequence contained in ERE, ARA, and CRC is highly hydrophobic, allowing for the ECM proteins to adsorb hydrophobic plate surface. After 3 h of incubation, the numbers of cells on the plates were examined (figure 4(a)). HUVECs seeded onto the surfaces coated with ALA or CLC were barely attached to the plate, similar to the non-treated plate. Meanwhile, adequate cell adhesion and extension were observed on the plates coated with ARA, CRC, and their mixture (ARA–CRC), as well as ERE-coated and fibronectin-coated plates (figure 4(b)). These results suggest that cell adhesion was promoted by the RGD sequence of ARA and CRC. Thus, the introduced helix peptides do not disturb the ERE functions, and the designed ECM proteins exhibit cell adhesion activities comparable to that of fibronectin.

Figure 4. Cell adhesion activity of the designed ECM proteins. HUVECs were seeded onto the surfaces coated with designed ECM proteins. Each ECM protein was prepared at a final concentration of 100 nM. (a) Evaluation of cell numbers after 3 h incubation. The activities were evaluated using the CCK-8. Error bars represent the standard error of the mean. (b) Fluorescence images of HUVECs cultured on surfaces coated with designed ECM proteins. Cells were stained with Calcein-AM. All scale bars represent 500 μm.
Download figure:
Standard image High-resolution image3.4. Cell proliferation activity of hB-bFGF
Cell proliferation activity of hB-bFGF was investigated. The hB-bFGF was added to the HUVEC culture medium at the final concentration of 10 or 100 nM. The bFGF is an essential factor for the proliferation of HUVECs in low serum condition. The proliferation of HUVECs induced by adding hB-bFGF is shown in figure 5. HUVECs significantly proliferated in the presence of bFGF or hB-bFGF, while the same cells showed no growth without bFGF. The helix-fused protein induced cell growth at levels comparable to wild-type bFGF at each concentration. The results indicate that hB-bFGF preserved the activity as bFGF even after fusing hB-(GGGS)8.

Figure 5. Cell proliferation activity of the hB-bFGF. The proliferation of HUVECs induced by the addition of hB-bFGF were investigated on day 1 (white bars) and day 4 (gray bars). Error bars represent the standard error of the mean.
Download figure:
Standard image High-resolution image3.5. Binding of hB-bFGF via triple-helix formation with ARA and CRC
ELISA analyses were carried out to examine the binding activity of hB-bFGF to ARA–CRC via heterotrimer coiled-coil interaction. Various concentrations of hB-bFGF were immobilized to the ARA– CRC modified surfaces. Anti-bFGF antibody was used to determine the amounts of hB-bFGF on the surface. As shown in figure 6(a), the signals detected with hB-bFGF increased in a concentration-dependent manner up to 1000 nM. However, there is a possibility of nonspecific binding of hB-bFGF. To elucidate that the interaction between hB-bFGF and ARA–CRC was due to the triple-helix formation between hA, hB, and hC presented each protein, competitive inhibition of the binding of hB-bFGF with hB peptide was performed. Various concentrations of hB inhibitor peptide were mixed with 100 nM of hB-bFGF solution, and the mixtures are reacted with the ARA–CRC modified surfaces. The intensities of the signal based on the amounts of hB-bFGF were evaluated as described above and shown in figure 6(b). The hB peptide inhibited the hB-bFGF binding to the surfaces modified with ARA–CRC dependent on the hB concentration. It is evident that the interaction between hB-bFGF and ARA–CRC was due to the triple-helix formation between hA, hB, and hC. Thus, the helix peptides functioned as not only self-assembling domains but also as growth factor-tethering domains for ECM proteins.
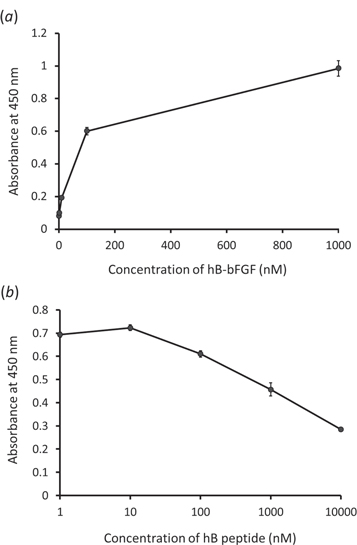
Figure 6. Binding activity of hB-bFGF to the surfaces modified with ARA–CRC ECM proteins via triple-helix formation. (a) Various concentrations of hB-bFGF were reacted with ARA–CRC modified 96-well plate surfaces. (b) Competitive inhibition of binding of hB-bFGF to ARA–CRC using the hB peptide. Various concentration of hB peptide were mixed with the hB-bFGF and the mixtures were reacted with the ARA–CRC modified plate surfaces.
Download figure:
Standard image High-resolution image3.6. Cell proliferation on bFGF tethered self-assembling ECM protein
The proliferation of HUVECs cultured on the surface of bFGF-tethered self-assembling ECM proteins was evaluated. HUVECs were grown on the hB-bFGF-presenting ARA–CRC coated plate surface, and their proliferation activities were examined (figure 7). As control experiments, HUVECs were seeded onto the ARA–CRC-coated plate with or without bFGF. In these cases, cell growth was scarcely observed or less active than when utilizing hB-bFGF. It is presumed that bFGF was washed out at the time of medium change on the second, fifth, and seventh day. Meanwhile, hB-bFGF tethered self-assembling ECM protein-absorbed surface induced significant cell proliferation. These results indicated that hB-bFGF exhibits intact bFGF activity without impairment and sustained its activity for a longer period by immobilizing to the ECM proteins via triple-helix formation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 7. Activation of cell proliferation by bFGF-tethered ARA–CRC. HUVECs were cultured on the hB-bFGF-presenting ARA–CRC coated plate surface. The cell growth activities were investigated on day 2 (white bars) and day 7 (gray bars). Error bars represent the standard error of the mean.
Download figure:
Standard image High-resolution image{kind=link}
4. Conclusion
In this study, bFGF-tethered self-assembling ECM proteins were designed and genetically synthesized to enhance cell adhesion and proliferation. Introducing helix peptides that exhibited the ability to form heterotrimer coiled-coil structures promoted not only self-assembly of the ECM proteins but also tethering of a growth factor to the ECM proteins. This novel method could be applied to various targeted cell studies by changing signal factors, and functional peptides derived from ECM and used as artificial functional ECMs for three-dimensional cell culture and tissue engineering.
Acknowledgments
This work was financially supported by Grants-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan. We are also grateful to Ms. Keiko Ikeda from the Technical Department of the Tokyo Institute of Technology for technical support with the transmission electron microscope imaging.