
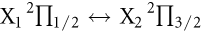

Abstract
The highly diagonal Frank–Condon factors (FCFs) are necessary for the laser cooling scheme, which means that only the molecules with the appropriate ground and excited electronic states can become candidates. Here, the laser cooling feasibility of the PbX (X = F, Cl, Br, and I) molecules is explored through four-component relativistic calculations. The potential energy curves and transition dipole moments of five Ω states are calculated and used to solve the Schrödinger equation of nuclear motion to obtain the rovibrational energy levels, spectroscopic parameters, the Einstein coefficients, and FCFs. Using the X12Π1/2 ↔ X22Π3/2 transition with highly diagonal FCFs, we construct optical schemes that can provide 104 ∼ 105 scattering phonons with four pumping lasers for PbX. The Doppler/recoil temperatures of PbX are 419.05 / 57.55, 9.63 / 61.20, 4.95 / 49.98, and 0.71 / 40.17 nK, respectively. Since the 10−4 s of the long spontaneous emission lifetime, the temperatures below microkelvin orders of magnitude can be achieved only if the adequate pre-cooling temperature is satisfied.
Export citation and abstract BibTeX RIS
1. Introduction
The production of ultracold molecules has received growing interest and emerged as an important research goal for experimentalists, at the same time, these molecules have broad applications in chemical dynamics [1], atom-ion sympathetic cooling [2–4], high-precision spectroscopic measurements [5–7], and quantum simulation of many-body interactions [8, 9]. In 2004, Di Rosa [10] proposed three conditions that diatomic candidates for laser cooling need to satisfy: (1) short radiative lifetimes for the upper states, (2) highly diagonal Frank–Condon factors (FCFs), and (3) no interference from intermediate states. Since the SrF molecule was experimentally cooled to the μK level using three lasers by Shuman et al [11] in 2010, YO [12], CaF [13], YbF [14], CaH [15], and BaH [16] molecules have also been successively cooled. Especially, the direct laser cooling of the BaH [16] molecule indicates the feasibility of laser cooling and trapping heavy molecules with relatively long-lived excited electronic states, despite previously unexplored this technical challenge. Theoretical investigations on laser cooling of diatomic molecules have also attracted the interest of many researchers. For example, the laser cooling optical schemes of BeF [17], ScO [18], CuH [19], AuH [20], and NH [21] were proposed. Moreover, Li et al [22] assessed the direct laser cooling feasibility of XH (X = Si, Ge, Sn, and Pb) and CS molecules, and found that SiH and CS molecules are promising for laser cooling candidates. Moussa et al [23] built the four-laser optical scheme for the CaCs molecule based on the X2Σ+ ↔ (2)2Π transition with an intermediate state of (2)2Σ+. Madi et al [24] investigated the low-lying electronic states of the BeX− (Cl, Br, F, and I) molecules and constructed a laser cooling scheme for BeF−. Osika et al [25] reported that the effective cooling scheme for the RaF molecule can be realized using only one pump laser. Very recently, El-Kork et al [26] reported a three-laser cooling scheme including intervening electronic states for LuF. Bigagli et al [27] built a cycling scheme for Doppler cooling of trapped OH+ ions using X3Σ− ↔ A3Π transition. The BeFr+ molecule was reported as a good candidate for laser cooling by Zrafi et al [28]. Moreover, a four-laser optical scheme of laser cooling was constructed for the AuAl molecule by Zhu et al [29].
The above investigations indicate that a small difference between the equilibrium bond lengths (|ΔRe
|) of the two electronic states involved in the pumping transitions can usually result in highly diagonal FCFs. For example, for BeF [17], LiRb [30], BH+ [31], and BH− [31] molecules, the |ΔRe
| for pseudo-closed transitions are 0.035, 0.085, 0.042, and 0.038 Å, the corresponding FCFs of the main transitions are 0.897, 0.872, 0.943, and 0.942, respectively. Yan et al [21] calculated the Re
s for the X3Σ− and 3Π1 states of NH molecule are 1.0363 Å and 1.0364 Å, respectively, and constructed the optical scheme based on the 0.99972 of FCF for the X3Σ− (ν'' = 0) ↔ 3Π1 (ν' = 0) transition. The A2Π1/2 ↔  transition was used for the laser cooling scheme for SrCl molecule by Xiao et al [32], in which the |ΔRe
| of the X2Σ+
1/2 and A2Π1/2 states is 0.028 Å, and the corresponding FCF of the A2Π1/2 (ν' = 0) ↔
transition was used for the laser cooling scheme for SrCl molecule by Xiao et al [32], in which the |ΔRe
| of the X2Σ+
1/2 and A2Π1/2 states is 0.028 Å, and the corresponding FCF of the A2Π1/2 (ν' = 0) ↔  (ν'' = 0) transition reaches 0.9124. The laser cooling scheme for the LuF molecule [26] was constructed using (1)3Π0+ (ν'' = 0) ↔ X1Σ0+ (ν = 0) transition with 0.021 Å of |ΔRe
| and 0.930636 of FCF. Therefore, to screen a molecule with a small ΔRe
| is a helpful way to find potential molecules for laser-cooling. So, we throw our light on the PbX (X = F, Cl, Br, and I) molecules because the spin–orbit coupling (SOC) states are expected to be small |ΔRe
|. Moreover, researchers have been interested in these molecules for a long time [33]. In 1936, the band spectra of PbF and PbCl molecules were analyzed and described experimentally by Rochester [34]. In the same year, Morgan [35] performed a vibrational analysis of the absorption band systems due to the PbF, PbCl, and PbBr molecules and estimated that the ground state would be expected to be a 2Π state with a splitting energy gap of 7000 cm−1 for PbCl. The ultraviolet band systems (B ← X) of PbCl and PbBr were obtained by Wieland et al [36] in 1952. The rotational spectra of (4, 0), (6, 0), (1, 1), and (3, 1) bands of PbCl were studied by Rao et al [37], while the emission spectrum of the A – X system of PbBr was obtained by Lal et al [38]. In 1992, Ziebarth et al [39] observed the emission spectra of the X2
2Π3/2 ↔ X1
2Π1/2 transitions for the PbX molecules in the near-infrared spectral region and obtained the harmonic vibrational constants (ωe
s), the anharmonic vibrational constants (ωe
χe
s) and the spectroscopic terms (Te
s) of the X1
2Π1/2 and X2
2Π3/2 states. The laser-induced fluorescence excitation spectrum of PbBr and PbBr was reported by Drake et al [40] in 1995. The high-resolution studies of the X2
2Π3/2 ↔ X1
2Π1/2 emission spectra for 208PbF, 207PbF, 206PbF, and 208Pb35, 37Cl molecules were investigated by Ziebarth et al [41] in 1998. Moreover, for the low-lying states of the PbX molecules, the spectroscopic properties such as Re
s, ωe
s, and Te
s were calculated theoretically [42–48]. Das et al [42] also obtained the radiative lifetimes of the X2
2Π3/2 and
(ν'' = 0) transition reaches 0.9124. The laser cooling scheme for the LuF molecule [26] was constructed using (1)3Π0+ (ν'' = 0) ↔ X1Σ0+ (ν = 0) transition with 0.021 Å of |ΔRe
| and 0.930636 of FCF. Therefore, to screen a molecule with a small ΔRe
| is a helpful way to find potential molecules for laser-cooling. So, we throw our light on the PbX (X = F, Cl, Br, and I) molecules because the spin–orbit coupling (SOC) states are expected to be small |ΔRe
|. Moreover, researchers have been interested in these molecules for a long time [33]. In 1936, the band spectra of PbF and PbCl molecules were analyzed and described experimentally by Rochester [34]. In the same year, Morgan [35] performed a vibrational analysis of the absorption band systems due to the PbF, PbCl, and PbBr molecules and estimated that the ground state would be expected to be a 2Π state with a splitting energy gap of 7000 cm−1 for PbCl. The ultraviolet band systems (B ← X) of PbCl and PbBr were obtained by Wieland et al [36] in 1952. The rotational spectra of (4, 0), (6, 0), (1, 1), and (3, 1) bands of PbCl were studied by Rao et al [37], while the emission spectrum of the A – X system of PbBr was obtained by Lal et al [38]. In 1992, Ziebarth et al [39] observed the emission spectra of the X2
2Π3/2 ↔ X1
2Π1/2 transitions for the PbX molecules in the near-infrared spectral region and obtained the harmonic vibrational constants (ωe
s), the anharmonic vibrational constants (ωe
χe
s) and the spectroscopic terms (Te
s) of the X1
2Π1/2 and X2
2Π3/2 states. The laser-induced fluorescence excitation spectrum of PbBr and PbBr was reported by Drake et al [40] in 1995. The high-resolution studies of the X2
2Π3/2 ↔ X1
2Π1/2 emission spectra for 208PbF, 207PbF, 206PbF, and 208Pb35, 37Cl molecules were investigated by Ziebarth et al [41] in 1998. Moreover, for the low-lying states of the PbX molecules, the spectroscopic properties such as Re
s, ωe
s, and Te
s were calculated theoretically [42–48]. Das et al [42] also obtained the radiative lifetimes of the X2
2Π3/2 and  states for PbF. The transition dipole moment of the X1
2Π1/2 → B2Σ1/2 transition, the radiative lifetime of the B2Σ1/2 state, and the spectroscopic constants for the B, D, E, and F states were determined for PbF by McRaven et al [49, 50]. The Franck–Condon factors of B2Σ+ ↔ X1
2Π1/2 transition PbF were calculated by Chen et al [33] in 2022. In the same year, Zhu et al [51] recorded the high-resolution laser-induced fluorescence spectra of the B2Σ+ ↔ X1
2Π1/2 transition for PbF under jet-cooled conditions. In addition, the previous research [35–48] showed that the SOC effect of the X2Π state of the PbX molecules is very significant, and the spin–orbit splitting energy difference can reach 8000 cm−1, and the |ΔRe
| of the SOC splitting states are about 0.01 ∼ 0.04 Å. Thus, the X1
2Π1/2 ↔ X2
2Π3/2 transitions of the PbX molecules are expected to achieve highly diagonal FCFs.
states for PbF. The transition dipole moment of the X1
2Π1/2 → B2Σ1/2 transition, the radiative lifetime of the B2Σ1/2 state, and the spectroscopic constants for the B, D, E, and F states were determined for PbF by McRaven et al [49, 50]. The Franck–Condon factors of B2Σ+ ↔ X1
2Π1/2 transition PbF were calculated by Chen et al [33] in 2022. In the same year, Zhu et al [51] recorded the high-resolution laser-induced fluorescence spectra of the B2Σ+ ↔ X1
2Π1/2 transition for PbF under jet-cooled conditions. In addition, the previous research [35–48] showed that the SOC effect of the X2Π state of the PbX molecules is very significant, and the spin–orbit splitting energy difference can reach 8000 cm−1, and the |ΔRe
| of the SOC splitting states are about 0.01 ∼ 0.04 Å. Thus, the X1
2Π1/2 ↔ X2
2Π3/2 transitions of the PbX molecules are expected to achieve highly diagonal FCFs.
The cooling of PbF was paid attention. For example, a cold PbF molecular beam was produced by laser-ablating a Pb rod under jet-cooled conditions, followed by the reaction with SF6 [51]. The Ne buffer gas instead of He can induce the molecular beam to undergo significant translational cooling, but little or no rotational cooling [52]. Bichsel et al [53] experimentally assess the two-state Stark shifts for NO and recommend that the two-state model be used for lased cooling except that some special treats are desired. These quantitative assessments along with their two-state equations for the Stark shifts in conjunction with the equations for the rovibronic energies and for the splitting of the Λ doublet can constitute a reliable resource for cold-molecule experiments involving 2Π molecules with an odd number of electrons (e.g, LiO, OH, ClO, BeH, SH, PbF, YbF, and CH), which suggests that we can constructed optical scheme of laser cooling for these molecules with the two-state model. Inspired by these findings, we determined to explore the laser cooling feasibility with the X1 2Π1/2 ↔ X2 2Π3/2 transition of the PbX molecules. The potential energy curves (PECs) are obtained by four-component relativistic calculations with the multireference configurations interaction (MRCI) [54, 55]. By solving the Schrödinger equation of nuclear motion with the calculated PECs, we have obtained the rovibrational energy levels and the spectroscopic parameters. For the X1 2Π1/2 ↔ X2 2Π3/2 transition, the FCFs of the PbX molecules are close to 1.0. Thus, we constructed optical schemes for direct laser cooling for these molecules and found that the schemes can produce 105 scattering photons for PbF and 104 ones for PbCl, PbBr, and PbI before leaking. The Doppler/recoil temperatures are 19.05 / 57.55, 9.63 / 61.20, 4.95 / 49.98, and 0.71 / 40.17 nK for the four molecules, indicating these laser cooling schemes are expected to reach temperatures below microkelvin orders of magnitude.
2. Computational details
The quantum chemical calculations on molecules containing heavy elements have to include relativistic effects to obtain high-accuracy results [56]. DIRAC22 [57] is a program system for one-, two-, and four-component relativistic molecular calculations at the level of Hartree–Fock, Kohn–Sham (including range-separated theory), multiconfigurational self-consistent-field, multireference configuration interaction, electron propagator, and various flavors of coupled cluster theory [58]. For heavy molecules, DIRAC22 is computationally less efficient, especially for the all-electron basis sets. The lowest-order relativistic effect on atomic and molecular energies is the spin-free relativistic effect (also known as scalar relativistic effect), which consists of single-electron relativistic effects. For light atoms, this effect is relatively easy to evaluate using mass-velocity and Darwin operators or direct perturbation theory. In addition, many standard non-relativistic basis sets are available for use with lighter elements in DIRAC22. These kinds of calculations need smaller computational consumption in comparison with the all-relativistic effect method for the heavy-element-containing molecules.
By using the DIRAC22 program package [57], we calculate the PECs and the transition dipole moments (TDMs) of the PbX molecules. For each molecule, 9 Ω states are considered although the low-lying five of them are presented. The Dyall-3zp basis sets [59–61] are employed, which correspond to the large component 30s26p17d11f for Pb, 14s8p2d for F, 18s11p3d for Cl, 23s16p10d2f for Br, and 28s21p15d2f for I atom. Based on the molecular orbitals produced by the four-component Hartree–Fock method, the MRCI calculations in the Kramers-restricted relativistic large-scale configuration interaction module (KRCI) [62–64] are used to generate the convincing data of the PECs and TDMs for the PbX molecules. A Generalized Active Space (GAS) approach [65] was employed to account for electronic correlations of the MRCI calculations, and we use a 3-GAS space (8, 10/ 5), (19, 21/ 8), (21, 21/ 16). The distributions of 11 among 8 Kramers pairs in a complete active space (GAS II). Up to two holes were allowed in the 5d shell of the heavy elements (GAS I) to account for core-valence and core-core correlations. Further, the electronic dynamic correlation was considered through the inclusion of single and double excitations from the GAS I and GAS II spaces into the virtual spinor space (GAS III). For the PbF, PbCl, PbBr, and PbI molecules, the remaining 35, 39, 48, and 57 electrons were kept frozen and occupied during the configuration interaction procedure, and the calculated PECs span internuclear distance ranges of 1.60 ∼ 3.80, 1.85 ∼ 3.85, 2.00 ∼ 4.00, and 2.15 ∼ 3.95 Å with a step size of 0.05Å, respectively.
By solving the Schrödinger equation of nuclear motion based on the calculated PECs, we have calculated the wavefunctions of each electronic state, and the rovibrational energy levels. The spectroscopic parameters are obtained by fitting the vibrational-rotational energy levels with Dunham expansion [66]. The Einstein coefficients (Aν'ν'' ) and FCFs (fν'ν'' ) of the X1 2Π1/2 (ν'' ) ↔ X2 2Π3/2 (ν' ) transitions are calculated to understand the laser cooling feasibility of the PbX molecules. These calculations are completed by the LEVEL8.0 program [67] that can give the width of the quasi-binding energy level and the inertial rotation constants and centrifugal distortion constants of diatomic molecules at a given energy level. It should be noted that the S (J'; J'') expression is only valid for the transition between singlet and singlet states, but can be generalized to other situations. For example, it can treat the 2Π ↔ 2Δ transition because the spectral structure is the same as that of 1Π ↔ 1Δ transition when the splitting constants of the electronic states are small.
3. Results and discussions
3.1. Potential energy curves and spectroscopic parameters
The PECs of the PbX molecules of the five low-lying states by DIRAC22 [57] with Dyall-3zp basis sets are shown in figure 1. They are attributed to the X1 2Π1/2/ X2 2Π3/2/ A2Σ1/2/ B2Σ1/2/ C1 2Π3/2 states for the PbF, PbCl, and PbBr molecules, and the X1 2Π1/2/ X2 2Π3/2/ A2Σ1/2/B1 2Π3/2/ C2Σ1/2 states for the PbI one. Based on the calculated PECs, the rovibrational energy levels are obtained by solving the Schrödinger equation of nuclear motion. There are 51/ 49 vibrational energy levels for the X1 2Π1/2/ X2 2Π3/2 states of the PbF molecule, and 51/ 51 ones for those of the PbCl, PbBr, and PbI molecules. For the sake of brevity, the vibrational energy levels over 8 are omitted in tables 1 and 2.
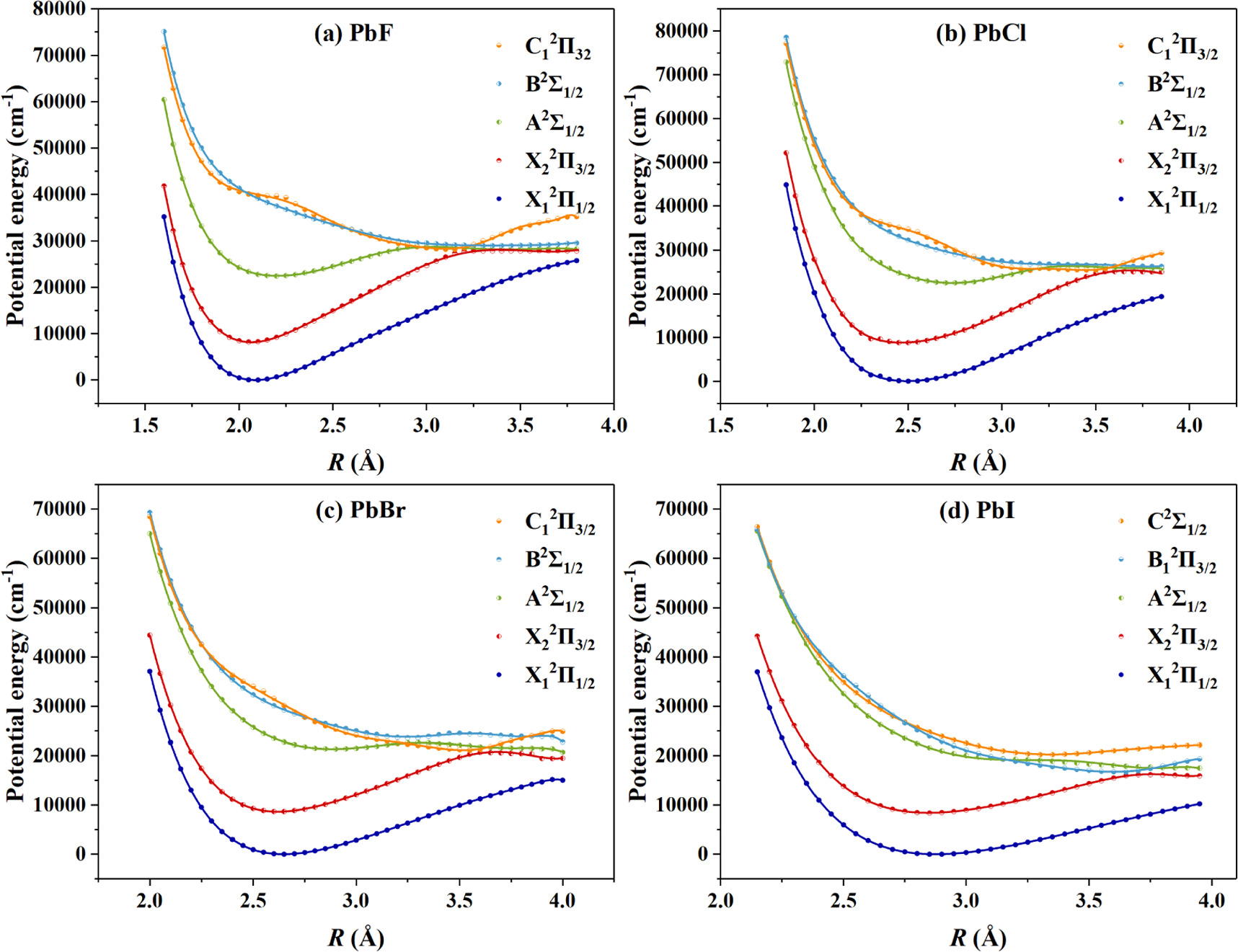
Figure 1. The calculated PECs for (a) the X1 2Π1/2, X2 2Π3/2, A2Σ1/2, B2Σ1/2, and C1 2Π3/2 states of PbF; (b) the X1 2Π1/2, X2 2Π3/2, A2Σ1/2, B2Σ1/2, and C1 2Π3/2 states of PbCl; (c) the X1 2Π1/2, X2 2Π3/2, A2Σ1/2, B2Σ1/2, and C1 2Π3/2 states of PbBr; (d) the X1 2Π1/2, X2 2Π3/2, A2Σ1/2, B1 2Π3/2, and C2Σ1/2 states of PbI.
Download figure:
Standard image High-resolution imageTable 1. The vibrational energy levels of the PbF and PbCl molecules (in cm−1).
| PbF | PbCl | |||
|---|---|---|---|---|
| X1 2∏1/2 | X2 2∏3/2 | X1 2∏1/2 | X2 2∏3/2 | |
| ν = 0 | 228.1557 | 240.9362 | 147.8020 | 135.6125 |
| ν = 1 | 707.5739 | 742.2173 | 441.9110 | 444.9914 |
| ν = 2 | 1182.7644 | 1239.3312 | 736.4642 | 754.8542 |
| ν = 3 | 1653.7346 | 1732.3179 | 1031.2126 | 1025.6471 |
| ν = 4 | 2120.4831 | 2221.1571 | 1308.6427 | 1254.2053 |
| ν = 5 | 2582.9888 | 2705.8179 | 1554.8247 | 1522.6548 |
| ν = 6 | 3041.2235 | 3186.2818 | 1796.5914 | 1813.8198 |
| ν = 7 | 3495.1515 | 3662.5439 | 2060.2448 | 2104.3958 |
| ν = 8 | 3944.7368 | 4134.5976 | 2333.9940 | 2393.6734 |
Table 2. The vibrational energy levels of the PbBr and PbI molecules (in cm−1).
| PbBr | PbI | |||
|---|---|---|---|---|
| X1 2∏1/2 | X2 2∏3/2 | X1 2∏1/2 | X2 2∏3/2 | |
| ν = 0 | 101.0078 | 88.3004 | 59.5426 | 78.2719 |
| ν = 1 | 303.1707 | 299.5909 | 209.5371 | 234.4211 |
| ν = 2 | 504.4429 | 509.9825 | 358.9903 | 390.0046 |
| ν = 3 | 704.8271 | 719.477 | 507.9032 | 545.0222 |
| ν = 4 | 904.3269 | 928.0753 | 656.2762 | 699.4746 |
| ν = 5 | 1102.9435 | 1135.782 | 804.111 | 853.3602 |
| ν = 6 | 1300.6829 | 1342.5952 | 951.4077 | 1006.6806 |
| ν = 7 | 1497.5474 | 1548.5192 | 1098.1661 | 1159.4354 |
| ν = 8 | 1693.5376 | 1753.5577 | 1244.3901 | 1311.6227 |
The spectroscopic parameters for the lowest two Ω states are fitted from the obtained energy levels with Dunham expansion [66], as shown in table 3. In fact, we have tested the small basis sets and the larger Dyall-4zp basis sets [59, 60, 68] for the PbF molecule and found the remarkable increase in computational consumption has not substantially improved the results in comparison with the experimental data. As shown in table 3, the results with Dyall-4zp basis sets only a little improvement than those with Dyall-3zp basis sets, and the latter are also in good agreement with the experimental data [39, 69]. Generally, the larger active space can get more sufficient electron correlation energy and should give accurate results, but the computational consumption also increases rapidly. The valence electrons of 6s26p2 of Pb and 2s22p5 of F in the active space, together with the 5d10 of Pb core electron as the reference space, need ∼107 determinants per double group irrep for the present core-valence project with Dyall-3zp basis sets. The calculations with larger reference space of inner shells (All 14 electrons of the f shell of Pb should be included if they are considered.) are difficult to perform because the huge memory is needed (Of course, only 1s2 of F is included, the calculational consume increase is not remarkable, but the impact is also small). Considering the results of the present core-valence project are in agreement with the available experimental values, the projects with larger reference space have not been explored. For the PbF molecule, the spectroscopic term Te of 8144 cm−1 for the X2 2Π3/2 state is only 1.430% and 1.452% smaller than the experimental values of 8262.11 [39] and 8264 cm−1 [69] respectively, and consistent with the other theoretical ones [42, 43, 47, 48]. Meanwhile, for the PbCl, PbBr, and PbI molecules, the experimental Te s of 8272.78, 8197.92, and 7969.15 cm−1 by Ziebarth et al [39] and the theoretical ones [44–47] in the literature are close to the present 8830, 8636, and 8404 cm−1, respectively. The equilibrium positions of 2.086/ 2.065 Å for the X1 2Π1/2/ X2 2Π3/2 states of the PbF molecule well correspond to the experimental [69] and theoretical [42, 43, 47, 48] values available in the literature. At the same time, the calculated Re s of the X1 2Π1/2/ X2 2Π3/2 states for the PbCl, PbBr, and PbI molecules are 2.493/ 2.464, 2.647/ 2.619, and 2.875/ 2.846 Å, respectively, which deviate from the theoretical values [44–47] with percent differences from 0.244% to 4.483%. The absolute differences between the Re s for the X1 2Π1/2 and X2 2Π3/2 states are 0.021, 0.029, 0.028, and 0.029 Å for the PbF, PbCl, PbBr, and PbI molecule, respectively. These close Re s indicate that the highly diagonal FCFs for the X1 2Π1/2 ↔ X2 2Π3/2 transition of the PbX molecules are feasible, which is necessary for laser cooling. Moreover, the calculated ωe s for the X1 2Π1/2 and X2 2Π3/2 states in the present work of the PbX molecules are also close to the previous experimental [39, 69] and theoretical [42–48] values. For example, the present ωe s of the PbBr molecule for the X1 2Π1/2 and X2 2Π3/2 states deviate from the experimental values of Ziebarth et al [39] by 1.804% and 2.834%, respectively, which are better than the other theoretical result reported by Benavides-Garcia et al [46]. The present results of the ωe χe values of the X1 2Π1/2 and X2 2Π3/2 states for the PbX molecules also match the previous experimental results by Ziebarth et al [39].
Table 3. The calculated spectroscopic parameters of the X1 2Π1/2/ X2 2Π3/2 states for PbX (X = F, Cl, Br, and I) (Re in Å, the other variables are in cm−1).
| Molecules | States | Methods | Te | Re | ωe | ωe χe |
|---|---|---|---|---|---|---|
| PbF | X1 2∏1/2 | This work a | 0 | 2.086 | 484.892 | 2.547 |
| This work b | 0 | 2.076 | 489.837 | 2.391 | ||
| Expt. c | 0 | 506.97 | 2.200 | |||
| Expt. d | 0 | 2.0575 | 503 | |||
| Calc. e | 0 | 2.050 | 521 | |||
| Calc. f | 0 | 2.084 | 530 | |||
| Calc. g | 0 | 2.08 | 540 | |||
| Calc. h | 0 | 2.08 | 494 | |||
| X2 2∏3/2 | This work a | 8144 | 2.065 | 505.508 | 2.235 | |
| This work b | 8359 | 2.051 | 510.809 | 2.269 | ||
| This work i | 5068 | 1.892 | ||||
| Expt. c | 8262.11 | 533.12 | 2.200 | |||
| Expt. d | 8264 | 2.0342 | 529 | |||
| Calc. e | 7417 | 2.038 | 533 | |||
| Calc. f | 7805 | 2.066 | 542 | |||
| Calc. g | 7895 | 2.06 | 547 | |||
| Calc. h | 8259 | 2.07 | 522 | |||
| PbCl | X1 2∏1/2 | This work a | 0 | 2.493 | 262.121 | 0.807 |
| Expt. c | 0 | 304.17 | 0.904 | |||
| Calc. e | 0 | 2.475 | 310 | |||
| Calc. j | 0 | 2.61 | 304 | |||
| X2 2∏3/2 | This work a | 8830 | 2.464 | 277.775 | 0.835 | |
| Expt. c | 8272.78 | 322.52 | 0.878 | |||
| Calc. e | 7526 | 2.458 | 317 | |||
| Calc. j | 8473 | 2.58 | 313 | |||
| PbBr | X1 2∏1/2 | This work a | 0 | 2.647 | 203.039 | 0.444 |
| Expt. c | 0 | 206.77 | 0.446 | |||
| Calc. e | 0 | 2.603 | ||||
| Calc. k | 0 | 2.712 | 195 | |||
| X2 2∏3/2 | This work a | 8636 | 2.619 | 212.162 | 0.445 | |
| Expt. c | 8197.92 | 218.35 | 0.433 | |||
| Calc. e | 7577 | 2.588 | ||||
| Calc. k | 7220 | 2.674 | 205 | |||
| PbI | X1 2∏1/2 | This work a | 0 | 2.875 | 150.535 | 0.271 |
| Expt. c | 0 | 161.02 | 0.287 | |||
| Calc. e | 0 | 2.772 | 168 | |||
| Calc. l | 0 | 2.885 | 153 | |||
| X2 2∏3/2 | This work a | 8404 | 2.846 | 156.564 | 0.257 | |
| Expt. c | 7969.15 | 169.28 | 0.323 | |||
| Calc. e | 7659 | 2.760 | 171 | |||
| Calc. l | 8255 | 2.859 | 162 |
a Based on the PECs with the Dyall-3zp basis sets. b Based on the PECs with the Dyall-4zp basis sets. c Ref. [39]. d Ref. [69]. e Ref. [47]. f Ref. [42]. g Ref. [43]. h Ref. [48]. i Based on the PECs with the MRCI method and x2c-TZVPPall-2c basis sets free of relativistic effects. j Ref. [44]. k Ref. [46]. l Ref. [45].
Meanwhile, we performed test calculations for PbF with non-relativistic effects and found the results for the equilibrium position and spectroscopic term (1.8916 Å and 5068.30 cm−1 at MRCI/x2c-TZVPPall-2c [70]) are apparently different from the experimental values, while the ones with the relativistic effects are substantially improved, as shown in table 3. Overall, the calculations with the relativistic four-component method can give accurate spectroscopic parameters in comparison with the previous experimental and theoretical values.
3.2. Franck–Condon factors and laser cooling schemes
The FCFs (fν'ν''
) for the X1
2Π1/2 ( '' ≤ 4) ↔ X2
2Π3/2 (ν' ≤ 4) transitions of the PbX molecules are calculated based on the vibrational wavefunctions evaluated by solving the nuclear motion Schrödinger equation with the obtained PECs. Figure 2 shows that the FCFs of the X1
2Π1/2 (ν'' ≤ 4) ↔ X2
2Π3/2 (ν' ≤ 4) transitions of the PbX molecules are highly diagonal. For example, the f00 values of the X1
2Π1/2 (ν'' = 0) ↔ X2
2Π3/2 (ν' = 0) are respectively 0.939125, 0.914541, 0.892669, and 0.885371. Therefore, these transitions are pseudo-closed transitions and suitable for constructing laser cooling schemes for the PbX molecules.
'' ≤ 4) ↔ X2
2Π3/2 (ν' ≤ 4) transitions of the PbX molecules are calculated based on the vibrational wavefunctions evaluated by solving the nuclear motion Schrödinger equation with the obtained PECs. Figure 2 shows that the FCFs of the X1
2Π1/2 (ν'' ≤ 4) ↔ X2
2Π3/2 (ν' ≤ 4) transitions of the PbX molecules are highly diagonal. For example, the f00 values of the X1
2Π1/2 (ν'' = 0) ↔ X2
2Π3/2 (ν' = 0) are respectively 0.939125, 0.914541, 0.892669, and 0.885371. Therefore, these transitions are pseudo-closed transitions and suitable for constructing laser cooling schemes for the PbX molecules.
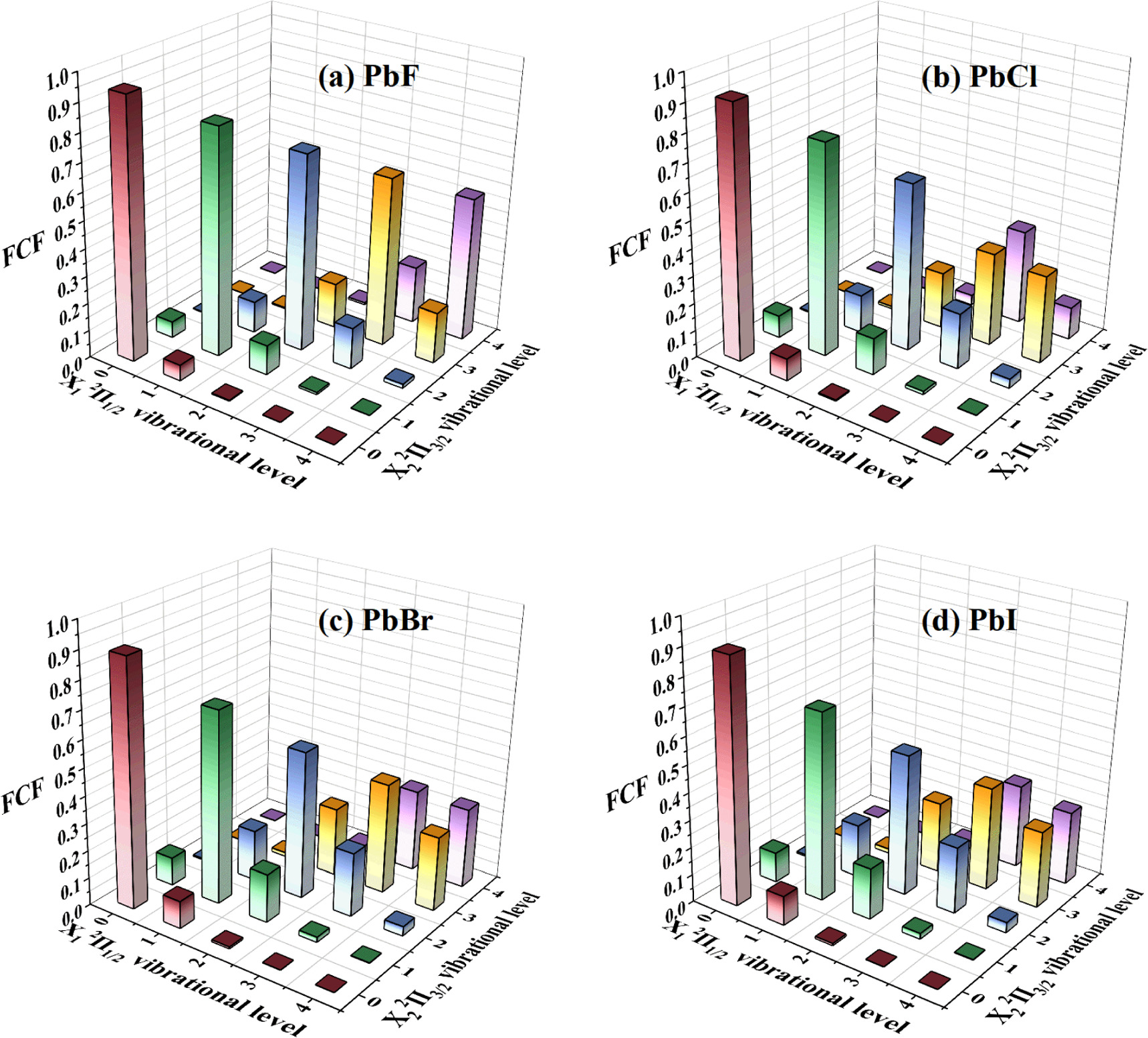
Figure 2. The FCFs for the X1 2Π1/2 (ν'' ≤ 4) ↔ X2 2Π3/2 (ν' ≤ 4) transitions. (a) PbF, (b) PbCl, (c) PbBr, and (d) PbI.
Download figure:
Standard image High-resolution imageThe laser cooling scheme is constructed for the PbF molecule according to the obtained spectroscopic properties, as shown in figure 3(a), and the actual values of fν'ν''s
are presented in table 4. A laser with the wavelength of λ00 = 1213.16 nm is used to pump the main X1
2Π1/2 (ν'' = 0) ↔ X2
2Π3/2 (ν' = 0) transition. Considering that there are small probabilities of decaying from the X2
2Π3/2 (ν' = 0, 1, 2) states to the X1
2Π1/2 states (ν'' = 1, 2, 3), we added three pump lasers with the wavelengths of 1209.95, 1206.75, and 1279.47 nm to ensure the pseudo-closed loop transition more efficiently. As a result, the accumulated FCFs for PbF can reach 0.999994 which can provide 1.779 × 105 scattered photons before leaking. These scattered photons are enough to exert an obvious cooling effect. In the present work, the accumulated FCFs are calculated with the sum of f00, f01, f02, and f03, and number of the scattered photons is evaluated by 

Figure 3. The optical schemes for laser cooling the PbX molecules with the X1
2Π1/2 (ν'') ↔ X2
2Π3/2 (ν') transitions. The solid red lines represent the laser-driven transition wavelengths. Here,  00/
00/ 11/
11/ 22/
22/ 23 are 1213.16/1209.95/1296.75/1279.47 nm for PbF, 1135.87/1133.68/1131.62/1171.04 nm for PbCl, 1155.44/1154.22/1153.01/1180.28 nm for PbBr, and 1186.88/1186.02/1185.15/1206.45 nm for PbI, respectively. The dotted lines represent the spontaneous decay from the (ν' = 0, 1, 2) states. fν'ν''
represents FCFs and the actual values are presented in table 4.
23 are 1213.16/1209.95/1296.75/1279.47 nm for PbF, 1135.87/1133.68/1131.62/1171.04 nm for PbCl, 1155.44/1154.22/1153.01/1180.28 nm for PbBr, and 1186.88/1186.02/1185.15/1206.45 nm for PbI, respectively. The dotted lines represent the spontaneous decay from the (ν' = 0, 1, 2) states. fν'ν''
represents FCFs and the actual values are presented in table 4.
Download figure:
Standard image High-resolution imageTable 4. The FCFs (fν'ν''s ) of the X1 2Π1/2 (ν'' ≤ 4) ↔ X2 2Π3/2 (ν' ≤ 2) transitions for the PbF, PbCl, PbBr, and PbI molecules, respectively.
| PbF | PbCl | PbBr | PbI | |
|---|---|---|---|---|
| f00 | 0.939125 | 0.914541 | 0.892669 | 0.885371 |
| f01 | 0.058087 | 0.081338 | 0.100140 | 0.106555 |
| f02 | 0.002667 | 0.003962 | 0.006803 | 0.007627 |
| f03 | 1.17 × 10−4 | 1.45 × 10−4 | 3.69 × 10−4 | 4.25 × 10−4 |
| f04 | 5.28 × 10−6 | 8.23 × 10−6 | 1.79 × 10−5 | 2.05 × 10−5 |
| f10 | 0.059678 | 0.082538 | 0.102390 | 0.108891 |
| f11 | 0.823645 | 0.769645 | 0.700327 | 0.682352 |
| f12 | 0.108539 | 0.135067 | 0.176987 | 0.186194 |
| f13 | 0.007658 | 0.011698 | 0.018813 | 0.020870 |
| f14 | 4.53 × 10−4 | 9.84 × 10−4 | 0.001393 | 0.001589 |
| f20 | 0.001188 | 0.002863 | 0.004817 | 0.005581 |
| f21 | 0.114667 | 0.139731 | 0.185295 | 0.194763 |
| f22 | 0.716662 | 0.614080 | 0.538306 | 0.514772 |
| f23 | 0.151666 | 0.205450 | 0.233447 | 0.242896 |
| f24 | 0.014641 | 0.033863 | 0.034592 | 0.037989 |
Similarly, the optical schemes pumping with four lasers are built for the PbCl, PbBr, and PbI molecules with the X1 2Π1/2 (ν'' = 0) ↔ X2 2Π3/2 (ν' = 0) as the main transition, which is shown in figures 3(b)–(d), respectively, and table 4 shows the corresponding values of fν'ν''s . For the PbCl molecule, the pumping laser wavelength for the main transition is 1135.87 nm, and those of the additional ones are 1133.68 (λ11), 1131.62 (λ22), and 1171.04 nm (λ23), respectively, inducing 6.975 × 104 of the scattered photons. For the PbBr and PbI molecules, the driven lasers with the wavelengths of 1155.44 and 1186.88 nm to pump the main X1 2Π1/2 (ν'' = 0) ↔ X2 2Π3/2 (ν' = 0) transition respectively, in addition, those of the additional wavelengths are 1154.22, 1153.01, 1180.28 for the PbBr and 1186.02, 1185.15, and 1206.45 nm for the PbI molecules to re-pump the photons falling on the auxiliary vibrational energy levels (ν' = 1, 2, 3). As a result, the accumulated FCFs are 0.999982 and 0.999978, and the correspondingly scattered photons are 5.424 × 104 and 4.568 × 104 for the PbBr and PbI molecules, respectively. The scattered photons are enough to perform remarkable cooling effects for the three molecules. Meanwhile, all required laser-cooling wavelengths are in the infrared region which the continuous wave laser can easily achieve.
3.3. The validities of the constructed optical schemes
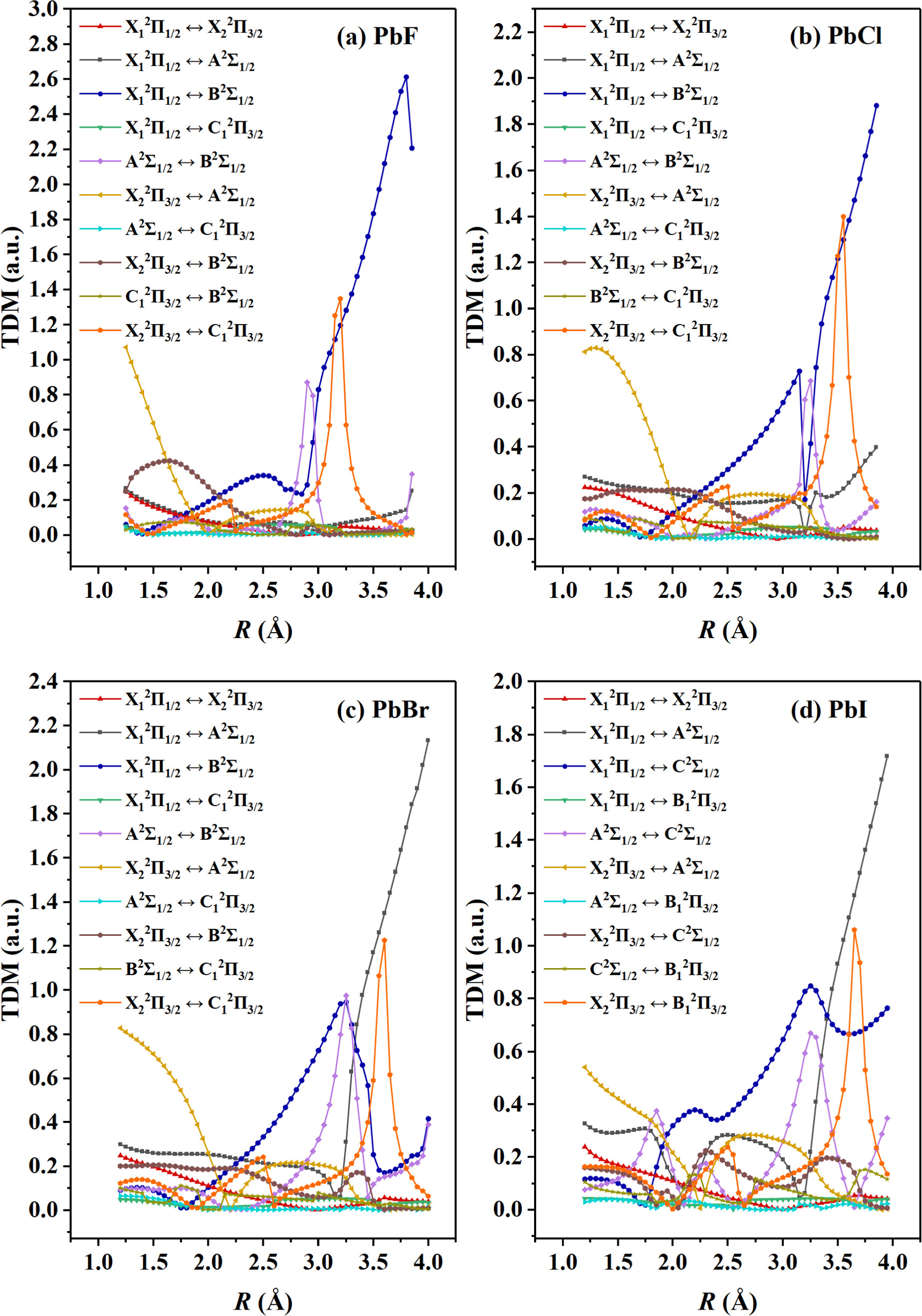
The TDMs of all possible transitions for the 5 lowest-lying Ω states of the PbX molecules are calculated, the results are shown in figure 4. Based on the obtained TDMs, we calculated the Einstein coefficients (Aν'ν'' ) and radiative lifetimes (τs) for the X1 2Π1/2 (ν'' = 0 ∼ 9) ↔ X2 2Π3/2 (ν' = 0, 1, 2) transitions involving in the optical schemes for laser cooling the PbX molecules, as shown in tables 5–8. For the X1 2Π1/2 (ν'' = 0 ∼ 9) ↔ X2 2Π3/2 (ν' = 0) transitions, the sums of the Aν'ν''s are the order of magnitude of 103 for the PbF, PbCl, and PbBr molecules while the corresponding τs reach 10−4 s, and the sums of the Aν'ν''s and the calculated τs for the PbI molecule are respectively the order of magnitude of 102 and 10−3. The smaller Aν'ν''s and longer τs indicate that the driving forces of the optical schemes for the PbX molecules are limited, and a lower pre-cooling temperature for each molecule is required to achieve good cooling effects. The vibrational branch ratios (Rν'ν''s ) are calculated for the X1 2Π1/2 (ν'' ≤ 4) ↔ X2 2Π3/2 (ν' ≤ 2) transitions by [71]:
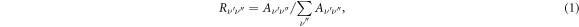
{kind=link}
{kind=link}
{kind=link}
Figure 4. The curves of TDMs for the X1 2Π1/2 ↔ X2 2Π3/2/ A2Σ1/2/ B2Σ1/2 / C1 2Π3/2, X2 2Π3/2 ↔ A2Σ1/2/ B2Σ1/2 / C1 2Π3/2, A2Σ1/2 ↔ B2Σ1/2/ C1 2Π3/2, and C1 2Π3/2 ↔ B2Σ1/2 transitions of PbF, PbCl and PbBr; and the X1 2Π1/2 ↔ X2 2Π3/2/ A2Σ1/2/ C2Σ1/2 / B1 2Π3/2, X2 2Π3/2 ↔ A2Σ1/2/ C2Σ1/2 / B1 2Π3/2, A2Σ1/2 ↔ C2Σ1/2/ B1 2Π3/2, and C2Σ1/2 ↔ B1 2Π3/2 transitions of PbI.
Download figure:
Standard image High-resolution image{kind=link}
Table 5. The Einstein coefficients Aν'ν'' and radiative lifetimes τ of the X1 2Π1/2 (ν'' = 0 ∼ 9) ↔ X2 2Π3/2 (ν' = 0, 1, 2) vibrational transitions for PbF.
| ν'' (X1 2∏1/2) | ν' (X2 2∏3/2) | ||
|---|---|---|---|
| ν' = 0 | ν' = 1 | ν' = 2 | |
| ν'' = 0 | 4.55 × 103 | 5.97 × 10−2 | 1.19 × 10−3 |
| ν'' = 1 | 4.11 × 102 | 0.82 | 0.11 |
| ν'' = 2 | 22.91 | 0.11 | 0.72 |
| ν'' = 3 | 1.07 | 7.66 × 10−3 | 0.15 |
| ν'' = 4 | 5.40 × 10−2 | 4.53 × 10−4 | 1.46 × 10−2 |
| ν'' = 5 | 3.52 × 10−3 | 2.58 × 10−5 | 1.10 × 10−3 |
| ν'' = 6 | 2.64 × 10−4 | 1.50 × 10−6 | 7.57 × 10−5 |
| ν'' = 7 | 1.78 × 10−5 | 8.92 × 10−8 | 5.15 × 10−6 |
| ν'' = 8 | 9.17 × 10−7 | 5.27 × 10−9 | 3.55 × 10−7 |
| ν'' = 9 | 2.91 × 10−8 | 3.09 × 10−10 | 2.49 × 10−8 |
| ∑ Α ν'ν'' (s−1 ) | 4.98 × 103 | 4.85 × 103 | 4.73 × 103 |
| τ = 1/∑ Α ν'ν'' (s) | 2.01 × 10−4 | 2.06 × 10−4 | 2.11 × 10−4 |
Table 6. The Einstein coefficients Aν'ν'' and radiative lifetimes τ of the X1 2Π1/2 (ν'' = 0 ∼ 9) ↔ X2 2Π3/2 (ν' = 0, 1, 2) vibrational transitions for PbCl.
| ν'' (X1 2∏1/2) | ν' (X2 2∏3/2) | ||
|---|---|---|---|
| ν' = 0 | ν' = 1 | ν' = 2 | |
| ν'' = 0 | 2.17 × 103 | 83.46 | 0.77 |
| ν'' = 1 | 3.23 × 102 | 1.78 × 103 | 1.31 × 102 |
| ν'' = 2 | 23.96 | 5.30 × 102 | 1.38 × 103 |
| ν'' = 3 | 1.43 | 68.91 | 7.80 × 102 |
| ν'' = 4 | 0.10 | 7.32 | 1.85 × 102 |
| ν'' = 5 | 1.05 × 10−2 | 0.41 | 24.35 |
| ν'' = 6 | 7.60 × 10−5 | 2.51 × 10−4 | 1.22 |
| ν'' = 7 | 1.18 × 10−3 | 8.22 × 10−4 | 7.03 × 10−2 |
| ν'' = 8 | 7.96 × 10−4 | 6.32 × 10−5 | 3.04 × 10−2 |
| ν'' = 9 | 3.77 × 10−6 | 5.10 × 10−4 | 1.87 × 10−2 |
| ∑ Α ν'ν'' (s−1 ) | 2.52 × 103 | 2.47 × 103 | 2.50 × 103 |
| τ = 1/∑ Α ν'ν'' (s) | 3.97 × 10−4 | 4.05 × 10−4 | 4.00 × 10−4 |
Table 7. The Einstein coefficients Aν'ν'' and radiative lifetimes τ of the X1 2Π1/2 (ν'' = 0 ∼ 9) ↔ X2 2Π3/2 (ν' = 0, 1, 2) vibrational transitions for PbBr.
| ν'' (X1 2∏1/2) | ν' (X2 2∏3/2) | ||
|---|---|---|---|
| ν' = 0 | ν' = 1 | ν' = 2 | |
| ν'' = 0 | 1.06 × 103 | 50.14 | 0.42 |
| ν'' = 1 | 2.07 × 102 | 8.13 × 102 | 87.06 |
| ν'' = 2 | 21.32 | 3.58 × 102 | 6.09 × 102 |
| ν'' = 3 | 1.60 | 57.80 | 4.62 × 102 |
| ν'' = 4 | 0.10 | 5.93 | 1.04 × 102 |
| ν'' = 5 | 5.66 × 10−3 | 0.47 | 13.73 |
| ν'' = 6 | 3.00 × 10−4 | 3.25 × 10−2 | 1.34 |
| ν'' = 7 | 1.54 × 10−5 | 2.03 × 10−3 | 0.11 |
| ν'' = 8 | 7.79 × 10−7 | 1.20 × 10−4 | 7.83 × 10−3 |
| ν'' = 9 | 3.86 × 10−8 | 6.88 × 10−6 | 5.25 × 10−4 |
| ∑ Α ν'ν'' (s−1 ) | 1.30 × 103 | 1.29 × 103 | 1.28 × 103 |
| τ = 1/∑ Α ν'ν'' (s) | 7.72 × 10−4 | 7.77 × 10−4 | 7.83 × 10−4 |
Table 8. The Einstein coefficients Aν'ν'' and radiative lifetimes τ of the X1 2Π1/2 (ν'' = 0 ∼ 9) ↔ X2 2Π3/2 (ν' = 0, 1, 2) vibrational transitions for PbI.
| ν'' (X1 2∏1/2) | ν' (X2 2∏3/2) | ||
|---|---|---|---|
| ν' = 0 | ν' = 1 | ν' = 2 | |
| ν'' = 0 | 1.06 × 103 | 50.14 | 0.42 |
| ν'' = 1 | 2.07 × 102 | 8.13 × 102 | 87.06 |
| ν'' = 2 | 21.32 | 3.58 × 102 | 6.09 × 102 |
| ν'' = 3 | 1.60 | 57.80 | 4.62 × 102 |
| ν'' = 4 | 0.10 | 5.93 | 1.04 × 102 |
| ν'' = 5 | 5.66 × 10−3 | 0.47 | 13.73 |
| ν'' = 6 | 3.00 × 10−4 | 3.25 × 10−2 | 1.34 |
| ν'' = 7 | 1.54 × 10−5 | 2.03 × 10−3 | 0.11 |
| ν'' = 8 | 7.79 × 10−7 | 1.20 × 10−4 | 7.83 × 10−3 |
| ν'' = 9 | 3.86 × 10−8 | 6.88 × 10−6 | 5.25 × 10−4 |
| ∑ Α ν'ν'' (s−1 ) | 1.30 × 103 | 1.29 × 103 | 1.28 × 103 |
| τ = 1/ ∑Α ν'ν'' (s) | 7.72 × 10−4 | 7.77 × 10−4 | 7.83 × 10−4 |
The results in table 9 demonstrate that the  from X2
2Π3/2 (ν' = 0) to X1
2Π1/2 (ν' = 0) is 91.26%, 86.18%, 82.20%, and 68.57% for PbF, PbCl, PbBr, and PbI, respectively. By using four lasers to pump the X1
2Π1/2 (ν'' = 0) ↔ X2
2Π3/2 (ν' = 0), X1
2Π1/2 (ν' = 1) ↔ X2
2Π3/2 (ν' = 1) and X1
2Π1/2 (ν'' = 2, 3) ↔ X2
2Π3/2 (ν' = 2) transitions, the total
from X2
2Π3/2 (ν' = 0) to X1
2Π1/2 (ν' = 0) is 91.26%, 86.18%, 82.20%, and 68.57% for PbF, PbCl, PbBr, and PbI, respectively. By using four lasers to pump the X1
2Π1/2 (ν'' = 0) ↔ X2
2Π3/2 (ν' = 0), X1
2Π1/2 (ν' = 1) ↔ X2
2Π3/2 (ν' = 1) and X1
2Π1/2 (ν'' = 2, 3) ↔ X2
2Π3/2 (ν' = 2) transitions, the total  s of the pseudo-closed-loop transitions increase to 99.99% / 99.99% / 99.99% / 99.97% for the four molecules, which could significantly improve the cooling effect. Moreover, the scattering probabilities leaking into off-diagonal bands for the X1
2Π1/2 (ν'') ↔ X2
2Π3/2 (ν') transitions are low, which further demonstrates that these transitions of PbX are suitable for laser cooling.
s of the pseudo-closed-loop transitions increase to 99.99% / 99.99% / 99.99% / 99.97% for the four molecules, which could significantly improve the cooling effect. Moreover, the scattering probabilities leaking into off-diagonal bands for the X1
2Π1/2 (ν'') ↔ X2
2Π3/2 (ν') transitions are low, which further demonstrates that these transitions of PbX are suitable for laser cooling.
Table 9. Vibrational branch ratios (Rν'ν''s ) (in %) for the X1 2Π1/2 (ν'' ≤ 4) ↔ X2 2Π3/2 (ν' ≤ 2) vibrational transitions of PbX (X = F, Cl, Br, and I).
| ν' (X2 2∏3/2) | ||||
|---|---|---|---|---|
| ν'' (X1 2∏1/2) | ν' = 0 | ν' = 1 | ν' = 2 | |
| PbF | ν'' = 0 | 91.26 | 3.12 | 1.14 × 10−2 |
| ν'' = 1 | 8.25 | 79.87 | 5.91 | |
| ν'' = 2 | 0.46 | 15.56 | 69.32 | |
| ν'' = 3 | 2.14 × 10−2 | 1.36 | 21.87 | |
| ν'' = 4 | 1.08 × 10−3 | 8.64 × 10−2 | 2.66 | |
| PbCl | ν'' = 0 | 86.18 | 3.38 | 3.07 × 10−2 |
| ν'' = 1 | 12.8 | 72.08 | 5.23 | |
| ν'' = 2 | 0.95 | 21.44 | 55.15 | |
| ν'' = 3 | 5.66 × 10−2 | 2.79 | 31.18 | |
| ν'' = 4 | 4.15 × 10−3 | 0.3 | 7.38 | |
| PbBr | ν'' = 0 | 82.20 | 3.9 | 3.32 × 10−2 |
| ν'' = 1 | 16.02 | 63.24 | 6.81 | |
| ν'' = 2 | 1.65 | 27.87 | 47.65 | |
| ν'' = 3 | 0.12 | 4.49 | 36.16 | |
| ν'' = 4 | 7.75 × 10−3 | 0.46 | 8.15 | |
| PbI | ν'' = 0 | 68.57 | 9.04 × 10−2 | 0.24 |
| ν'' = 1 | 27.07 | 46.37 | 9.79 × 10−2 | |
| ν'' = 2 | 3.98 | 42.1 | 31.62 | |
| ν'' = 3 | 0.35 | 10.17 | 47.81 | |
| ν'' = 4 | 3.01 × 10−2 | 1.14 × 10−2 | 17.53 | |
However, the smaller Aν'ν'' will result in smaller Doppler (ΤD s) and recoil temperatures (Τr s), which can be used to obtain lower cooling temperatures. The achievable ΤD and Τr are estimated according to the following formulas [72]:
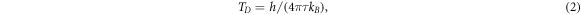
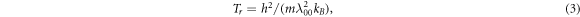
where m, h, and kB are molecular mass, Planck's constant, and Boltzmann's constant, while λ00 is the laser wavelength pumping the main transition of X1 2Π1/2 (0) ↔ X2 2Π3/2 (0). The calculated ΤD / Τr are 19.05/ 57.55, 9.63/ 61.20, 4.95/ 49.98, and 0.71/ 40.17 nK for the PbX molecules, respectively. Equation (3) shows that the recoil temperature is strongly related to molecular mass, which indicates that heavy molecules are more likely to be cooled with lower recoil temperatures. Therefore, among the molecules considered, the PbI molecule can reach a minimum recoil temperature of 40.17 nK, which implies that this molecule is promising to achieve sub-micro-Kelvin temperature by Doppler cooling. On the whole, the present schemes can achieve the sub-micro-Kelvin temperatures for the PbX molecules but need lower pre-cooling temperatures.
4. Conclusions
In summary, the PECs and TDMs are obtained for the five low-lying Ω states of the PbX molecules by using the KRCI with the molecular orbitals produced by the relativistic four-component method in DIRAC22. The reliability of the present calculational level is confirmed by the calculated spectroscopic parameters for the two low-lying Ω states (X1 2Π1/2 and X2 2Π3/2) that are in good agreement with the theoretical and experimental values available in the literature. The four-laser cooling optical schemes with the close-loop X1 2Π1/2 ↔ X2 2Π3/2 transition are constructed for the PbX molecules based on the highly diagonal Frank–Condon factors. The pumping laser wavelengths are 1131.62 ∼ 1279.47 nm, which are available for common laser devices. Remarkably, the Doppler and recoil temperatures below microkelvin orders of magnitude can be achieved for all the constructed schemes. However, the radiative lifetimes of the upper levels are in the order of 10−4 or 10−3 s, which are not short enough to produce large spontaneous scattering forces for rapid cooling. Therefore, a lower pre-cooling temperature is needed to perform the present optical schemes. These results are expected to provide a helpful guide for designing experimental laser cooling programs for the PbX molecules.
Acknowledgments
This work is supported by the National Neutral Science Foundation of China (Grant Nos.12374232). C.L. Yang thanks the Xinjiang Tianchi Talent Program (2023).
Data availability statement
All data that support the findings of this study are included within the article (and any supplementary files).
CRediT authorship contribution statement
Jing-Zhen Luan: Data curation, Investigation, Writing-original draft. Chuan-Lu Yang: Conceptualization, Methodology, Supervision, Writing-review & editing. Xiao-Hu Li: Investigation. Wen-Wang Lu:Visualization. Yu-Liang Liu: Software. Wenkai Zhao: Supervision.