



Abstract
We propose a method to produce, in a pulsed or continuous way, cold samples of highly polar molecules. Using a pulsed or continuous standard (supersonic) beam of these molecules, our idea consists of transforming the molecules into their anionic counterparts, which are decelerated to a standstill by a well-controlled external electric field and ultimately neutralized. The neutral-to-anion transformation occurs through collisions with Rydberg atoms coming from an additional atomic beam. This Rydberg electron transfer process is possible provided that the molecular species has a sufficiently strong electric dipole ( D, i.e.,
D, i.e.,  cm). Whatever the mass of the species, the deceleration stage is realized by a temporally and spatially controlled electric field within a range of less than one centimeter, which is much shorter than in current deceleration experiments of neutral molecules. Once stopped, the molecular anions are neutralized by laser photodetachment or a pulsed electric field process. The resulting molecules might be held and accumulated, for instance, in a magnetic trap.
cm). Whatever the mass of the species, the deceleration stage is realized by a temporally and spatially controlled electric field within a range of less than one centimeter, which is much shorter than in current deceleration experiments of neutral molecules. Once stopped, the molecular anions are neutralized by laser photodetachment or a pulsed electric field process. The resulting molecules might be held and accumulated, for instance, in a magnetic trap.
Export citation and abstract BibTeX RIS

Content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Although the number of methods for producing cold molecules has grown, none of them is simple and universal. The goal of this article is to present a deceleration method that should be quite universal for highly polar molecules.
The existing deceleration methods of molecular beams often use electric/magnetic or light interactions, but there are alternative propositions such as beam collision and rotating supersonic expansion [1–12]. (For a more complete list, see [13].) The scientific community has put much effort into developing efficient and universal decelerators, and consequently important progress has been realized. However, these decelerators are often cumbersome and are better adapted to light molecules, because the interaction between any kind of molecular dipole (except for high Rydberg dipoles) and an electromagnetic field leads to because forces that are too weak to enable deceleration over short distances ( m). A simple idea for getting stronger force might be to electrically charge the molecules. Thus a small electric field would be sufficient to produce an intense decelerating force.
m). A simple idea for getting stronger force might be to electrically charge the molecules. Thus a small electric field would be sufficient to produce an intense decelerating force.
The key point of our approach is to attach an extra electron to the studied molecule, which was previously formed by standard techniques such as supersonic or cryogenic expansion, in order to form a dipole-bound anion. In such an anion, the extra electron is localized at a distance so far from the positive pole of the dipole that it does not really disturb the internal degrees of freedom. This capture roughly preserves both the beam characteristics and the internal state of the molecule. The deceleration stage of the molecular anion can be carried out by a weak electric field and implemented over very small distances (within the centimeter range). Finally, the neutralization of the molecule will be performed by laser photodetachment or a fast pulse of an intense electric field. This should restore the molecule to its initial internal state, but it would henceforth be stopped in the laboratory frame of reference. This technique should enable the production of a wide class of molecules that could be accumulated in a trap.
The present proposal only focuses on decelerating highly polar molecules cooled by supersonic expansion (1 K). This method could be applicable to any diatomic or polyatomic polar molecules since the only required condition is a dipole electric moment greater than 2.5 Debye (D). In addition to the wide range of applicability, the method shows another advantage: such a decelerator (pulsed or continuous) would be much shorter than the actual apparatus based on standard electromagnetic deceleration, and it could end up with a trap to accumulate molecules.
2. Anion M− formation
Since the main idea is to decelerate charged molecules and subsequently neutralize them in order to form well controlled cold molecules, two main possibilities exist: molecules may be transformed into either cations, M+, or anions, M−. There are some arguments in favor of the anion choice, which seems surprising at first. Indeed, although the formation step is simpler for cations than for anions, the neutralization of cations without heating molecules is much more difficult than for anions. Indeed, among the possible techniques to neutralize cations, we can cite the three-body recombination,  , the charge transfer through impact with an atom, A:
, the charge transfer through impact with an atom, A:  , or the radiative recombination,
, or the radiative recombination,  . All these techniques produce several rovibrational states that would be difficult to control. Even nastier phenomena such as dissociative recombination can play a dominant role. However, the control of the final state can be achieved with a laser inducing the recombination
. All these techniques produce several rovibrational states that would be difficult to control. Even nastier phenomena such as dissociative recombination can play a dominant role. However, the control of the final state can be achieved with a laser inducing the recombination  . A high rate of recombination would require an intense flux of very cold electrons [14], and the laser wavelength should be changed for each molecular species.
. A high rate of recombination would require an intense flux of very cold electrons [14], and the laser wavelength should be changed for each molecular species.
We have thus turned our attention to cold anion sources (if possible below 1 K) to be decelerated to a standstill. Low temperature is a strong limitation that would also arise for the cation case, because most of the anion sources, such as plasma sources, dissociative electron attachment followed by fragmentation, chemical reaction using another source of anions, laser ablation techniques, and spray techniques, have an energy dispersion in the eV range (i.e., at best, in the 1000 K region [15]). Experimental work in this area has been proposed [16]. Other interesting methods that produce anions at energy as low as 1 meV are laser photoelectron attachment in a molecular supersonic beam or irradiation of neutral molecules by synchronizing laser and electron pulses, as considered in [17]. Laser photoelectron attachment succeeds in producing anions with a very efficient formation cross section of about 10−17 m2, but it is limited to specific molecules such as SF6 [18].
Therefore, we suggest a more general technique to produce a bright and cold anion beam based on the charge exchange process
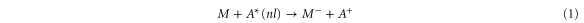
that can be controlled by the proper choice of the Rydberg energy level, n (usually of an atom, A). Using a bright and cold source of neutral molecules, the idea is to use such a Rydberg electron-transfer process to form a bright and cold anion beam. In the following, it is assumed that our source of neutral molecules is a supersonic beam providing typically, on a given rovibrational level, more than 105 molecules per pulse of tens of microsecond, or 1010 molecules per second in a continuous beam. The schematic of the experimental setup is given in figure 1 with Cs as example for the atom, A.

Figure 1. Schematic of the experimental setup. A pulsed or continuous supersonic molecular beam is produced. A (cesium) atomic beam laser excited to the Rydberg state crosses this beam, and through electron transfer, transforms the neutral molecular beam in an anionic beam. The deceleration stage is realized by a pulsed electric field. Once stopped, molecular anions are neutralized by laser photodetachment or by another pulsed electric field process.
Download figure:
Standard image High-resolution image2.1. Dipole-bound anion, M−
To understand how to extract the anion beam properties from the neutral beam properties, we will now study in more detail the anion formation process of equation (1).
The anion formed is usually a so-called dipole-bound anion. Its electron is bound with an electron affinity (binding energy) in the 1–100 meV range, which is much lower than the electron affinity of a valence-bound anion in the 0.1–1 eV range. Indeed, a dipole-bound anion exists simply because the neutral molecule dipole moment is large enough to bind an electron through the electron-dipole potential. Roughly speaking, when the dipole is sufficiently large, an excess electron can be bound into a very diffuse molecular orbital close to the positive pole. By studying the capture of negatively charged mesons in matter, Fermi and Teller were the first to predict that the long-range dipolar potential can bind an electron, provided that the dipole moment is larger than 1.625 D [19]. Various molecular effects, such as the length of the dipole, the presence of a short-range repulsive potential in the molecular core of the anion, and the rotational degrees of freedom of the anion turned out to increase this cutoff value. It is now commonly accepted that any molecule with a dipole moment above 2.5 D can form a dipole-bound anion [20]. In addition, there is conclusive evidence for long-range binding to quadrupolar molecules [21]. Thus the number of molecules that can be formed in this way ranges from simple molecules (LiH, LiF, SrF, BaF, HCN, SiO, BaO, AgCl, BrK ...) to molecules of chemical interest (HCN,CH3CHO, H2SiO,) to big polymers or large biological molecules and even DNA bases. Indeed, for big molecules individual dipoles add up, and the final one can easily be bigger that 2.5 D. Obviously, some small but interesting molecules (NH, CH, OH, CN, CO, SO, NO, ...) have lower dipole moments, but we can combine them to form molecules (such as CH3CN or C2H2N2O) that have dipole moments larger than 2.5 D, which, once decelerated, can be dissociated at threshold to form cold fragments [22]. However, this last method is far from being universal and we will not consider it here.
2.2. Formation via Rydberg charge exchange
Weakly bound anions are best produced by the Rydberg electron transfer technique described by equation (1) [23]. The formation of dipole-bound anions via Rydberg electron transfer typically occurs over a narrow range of effective principal quantum numbers, n, of the Rydberg atom, and the excess electron-binding energy, Eb (electron affinity (EA)), of a dipole-bound molecular anion can be estimated using the empirical relationship
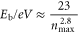
where  is the effective principal quantum number of the Rydberg that produces the maximum yield of dipole-bound anions (
is the effective principal quantum number of the Rydberg that produces the maximum yield of dipole-bound anions ( being the quantum defect) [24].
being the quantum defect) [24].
The most surprising fact in this relationship is that the charge-exchange process is strongly nonresonant since the cross section reaches its maximum value when the binding energy of the Rydberg atom is much larger than that of the created negative ion ( in atomic units).
in atomic units).
The physical explanation of this phenomenon was given by [24] and has been recently studied in detail in [25–29]. We now recall the main results. The process, sketched in figures 2 and 3, describes the ionic interaction between  and
and  and the covalent interaction between M and the Rydberg atom, A*. In the center of mass frame, at relative velocity v, the neutral molecule M crosses the electron orbits of A
and the covalent interaction between M and the Rydberg atom, A*. In the center of mass frame, at relative velocity v, the neutral molecule M crosses the electron orbits of A
 at the crossing distance
at the crossing distance  , such that
, such that  . A (small) Landau–Zener nonadiabatic transfer probability,
. A (small) Landau–Zener nonadiabatic transfer probability,  , exists because there is an avoided crossing between the ionic and covalent curves caused by the exchange interaction (
, exists because there is an avoided crossing between the ionic and covalent curves caused by the exchange interaction ( play the same role as
play the same role as  in the H2 molecule).
in the H2 molecule).

Figure 2. Interaction potential curves,  , between
, between  and
and  , and
, and  between M and A*.
between M and A*.  is the electronic affinity and I* is the ionization potential of the atom.
is the electronic affinity and I* is the ionization potential of the atom.
Download figure:
Standard image High-resolution image
Figure 3. Schematic of the charge exchange process,  . The exchange mainly occurs at the second crossing because the survival probability is smaller after the first crossing.
. The exchange mainly occurs at the second crossing because the survival probability is smaller after the first crossing.
Download figure:
Standard image High-resolution imageHowever, once the crossing is overcome, the anion must survive all the other crossings, and mainly the first ones that occur with higher potential covalent curves linked to  (see figure 2). The product of all these probabilities can be performed using the integral approximation with a probability,
(see figure 2). The product of all these probabilities can be performed using the integral approximation with a probability,  [25]. The total cross section is given by
[25]. The total cross section is given by  , where b is the impact parameter. Typically, the value of the total cross section is about
, where b is the impact parameter. Typically, the value of the total cross section is about  cm2. More precisely, the experimental formation rate,
cm2. More precisely, the experimental formation rate,  , ranges from 10−7 cm3 s−1 for the Butanal (C4H8O with a dipole μ of 2.7 D,
, ranges from 10−7 cm3 s−1 for the Butanal (C4H8O with a dipole μ of 2.7 D,  meV and
meV and  ) to 10−8 cm3 s−1 for the Acetonitrile (CH3CN with a dipole of 3.9 D,
) to 10−8 cm3 s−1 for the Acetonitrile (CH3CN with a dipole of 3.9 D,  meV and
meV and  ) [24, 26, 27]. The total cross section slightly depends on the relative velocity of the reactants, which is shown in figure 4 using the formula from [25].
) [24, 26, 27]. The total cross section slightly depends on the relative velocity of the reactants, which is shown in figure 4 using the formula from [25].
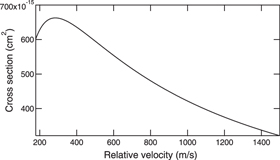
Figure 4. Total cross section of the BaF− formation through Rydberg electron transfer according to the relative velocity, v. For BaF seeded in Xe, Ar, and He supersonic beams with respective forward velocity 308 m s−1, 559 m s−1, 1770 m s−1 in the frame of laboratory, the relative velocities with a beam of Rydberg cesium typically moving at 250 m s−1 correspond to the horizontal coordinates, v = 282, 445, 1313 m s−1.
Download figure:
Standard image High-resolution imageAnother important feature of this reaction is the attraction between the products  and
and  under the effect of the Coulomb potential,
under the effect of the Coulomb potential,  . This leads to a conical trajectory in the center of mass, and we can extract anion beam properties from the scattering angle
. This leads to a conical trajectory in the center of mass, and we can extract anion beam properties from the scattering angle
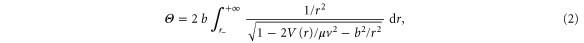
where  is the distance of smallest approach. From this relation we can extract the differential cross section given by
is the distance of smallest approach. From this relation we can extract the differential cross section given by  . Figure 5 illustrates the behavior of the differential cross section calculated for different molecular velocities.
. Figure 5 illustrates the behavior of the differential cross section calculated for different molecular velocities.
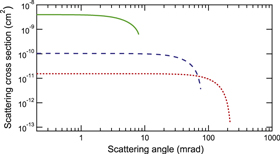
Figure 5. Example of differential cross section for the formation through charge exchange of a Cs Rydberg collisional velocity,  m s−1, and three different BaF forward velocities, vM, corresponding to the supersonic beam speed of He (green solid line, 1770 m s−1 forward velocity), Ar (blue dashed line, 559 m s−1 forward velocity), and Xe (red dotted line, 308 m s−1 forward velocity). A forward BaF velocity,
m s−1, and three different BaF forward velocities, vM, corresponding to the supersonic beam speed of He (green solid line, 1770 m s−1 forward velocity), Ar (blue dashed line, 559 m s−1 forward velocity), and Xe (red dotted line, 308 m s−1 forward velocity). A forward BaF velocity,  m s−1.
m s−1.
Download figure:
Standard image High-resolution imageThe formula for the cross section used here, provided by [25], is based on an s-orbital anion wave function, dipole approximation, and a zero-range pseudopotential model for the interaction between the excess electron and the anion core. It is sufficient for our purposes and enough to provide a qualitative description of the ion-pair formation. We simply point out that it significantly underestimates, by almost a factor of 10, the absolute values of the cross sections [26, 27]. The model does not account for the rovibrational state of the molecule. However, it is expected that the dipole-bound negative ion formation is a relatively nonperturbative process, and the produced anions must have exactly the same nuclear configurations as their neutral parents [30]. Only in very specific cases, such as for some floppy molecules like the water dimer, the molecule configuration can be strongly modified [31].
The coupling between the electron motion and the molecular rovibration can limit the lifetime of the anion through, for instance, an autodetachment process:  . However, because of the conserved nuclear configurations in the formation process, which can be written as
. However, because of the conserved nuclear configurations in the formation process, which can be written as  , the autodetachment has to be energetically allowed to occur—that is, the rovibrational internal energy has to be larger than the binding energy of the anion. Thus for molecules with a ground vibrational state and low rotational quantum numbers, long-lived anions must be formed, as confirmed by [32, 33]. For instance, in the case of CH3CN, the data indicate that the creation of long-lived ions requires that target molecules be in states with rotational quantum numbers
, the autodetachment has to be energetically allowed to occur—that is, the rovibrational internal energy has to be larger than the binding energy of the anion. Thus for molecules with a ground vibrational state and low rotational quantum numbers, long-lived anions must be formed, as confirmed by [32, 33]. For instance, in the case of CH3CN, the data indicate that the creation of long-lived ions requires that target molecules be in states with rotational quantum numbers  . Further measurements demonstrate that the longest anion lifetime (
. Further measurements demonstrate that the longest anion lifetime ( s) is limited by black body radiation-induced photodetachment.
s) is limited by black body radiation-induced photodetachment.
In conclusion, the high formation rates indicate that a supersonic or cryogenic beam of highly polar molecules lying in their low rovibrational states can be efficiently transformed into anions, thanks to a charge exchange process.
2.3. Choice of the Rydberg-molecule collisional velocity
Since the final aim is the production of a cold sample of neutral molecules, it is crucial to follow the velocity properties of the molecular anions' M− beam. Indeed, the charge transfer is expected to modify the initial trajectory of the neutral parent and lead to heating and deviation of the anion beam. Here, we calculate these mechanical effects in order to optimize the experimental configuration. First, it is worth noting that for a too-small relative velocity,  , between the Rydberg atoms, A, and the molecule M, M− and A+ do not have enough energy to be separated and will form a bound ionic pair, M− - A+. On the other hand, M− and A+ are dissociated if the relative velocity is equal to or greater than the critical velocity,
, between the Rydberg atoms, A, and the molecule M, M− and A+ do not have enough energy to be separated and will form a bound ionic pair, M− - A+. On the other hand, M− and A+ are dissociated if the relative velocity is equal to or greater than the critical velocity,  where
where  is the internal (electronic + rovibrational) energy of the products (with similar notation for the reactants). Such a threshold is visible in figure 4. As the reaction (1) is endothermic, there is no reaction at zero relative velocity, which has strong experimental implications. In particulars, this means that A cannot flow within the supersonic beam in which M is seeded—by exciting the carrier gas in Rydberg state, for instance—because of the small relative velocity between the reactants.
is the internal (electronic + rovibrational) energy of the products (with similar notation for the reactants). Such a threshold is visible in figure 4. As the reaction (1) is endothermic, there is no reaction at zero relative velocity, which has strong experimental implications. In particulars, this means that A cannot flow within the supersonic beam in which M is seeded—by exciting the carrier gas in Rydberg state, for instance—because of the small relative velocity between the reactants.
Therefore, the Rydberg atoms must come from a different source, such as another beam or a vapor cell. The choice of the Rydberg system will strongly affect the efficiency of the process and the final beam properties. For instance, the anion formation rate,  , decreases with decreasing Rydberg atom velocity [34] (see also figure 4). This effect is also enhanced by the post-attachment electrostatic interactions between the produced ions.
, decreases with decreasing Rydberg atom velocity [34] (see also figure 4). This effect is also enhanced by the post-attachment electrostatic interactions between the produced ions.
To get more insight about the process, we calculated the trajectories of an ideal monocinetic Rydberg beam orthogonal to a monocinetic molecular beam, a situation that is summarized in figure 6. Our results are given for BaF, a molecule that has a dipole moment of 3.17 D, seeded in a supersonic beam whose mean velocity differs by the use of the carrier gas. The calculation itself, performed in the center of mass, gives the scattering angle and the relative velocity of the products for a specific choice of the reactant velocities and impact parameter b. For a same relative velocity, and provided that the electron exchange is possible because the condition  is fulfilled, note that the scattering angle increases with the impact parameter. It is found that there is a maximal scattering angle when
is fulfilled, note that the scattering angle increases with the impact parameter. It is found that there is a maximal scattering angle when  and no deviation when
and no deviation when  . As this system has an axial symmetry, all the velocity vectors corresponding to a single value of b describe a cone, as illustrated in figure 6. Therefore, in the frame of the laboratory, the reaction leads to a redistribution of velocities (see figure 7; i.e., a global deviation and heating of the anion beam with respect to the molecular beam). Quantitatively the deviation is given by the change of direction of
. As this system has an axial symmetry, all the velocity vectors corresponding to a single value of b describe a cone, as illustrated in figure 6. Therefore, in the frame of the laboratory, the reaction leads to a redistribution of velocities (see figure 7; i.e., a global deviation and heating of the anion beam with respect to the molecular beam). Quantitatively the deviation is given by the change of direction of  corresponding to
corresponding to  . Figure 8 shows that the deviation of BaF− is always small for a fast supersonic beam. The heating process is evaluated by the maximal extents of transverse velocity (
. Figure 8 shows that the deviation of BaF− is always small for a fast supersonic beam. The heating process is evaluated by the maximal extents of transverse velocity ( ) and longitudinal velocity (
) and longitudinal velocity ( ) of the molecular beam with respect to the deviated axis of the anion beam. As shown in figure 9, there is a global trend of little heating for a fast supersonic beam. It is noteworthy that the anion velocity tends to be less altered when the relative velocity is large because it is not much affected by the Coulomb effect. Anyway, our calculation for BaF unambiguously indicates that the use of a fast molecular beam, as can be provided by He as a carrier gas, should lead to the smallest increase in temperature.
) of the molecular beam with respect to the deviated axis of the anion beam. As shown in figure 9, there is a global trend of little heating for a fast supersonic beam. It is noteworthy that the anion velocity tends to be less altered when the relative velocity is large because it is not much affected by the Coulomb effect. Anyway, our calculation for BaF unambiguously indicates that the use of a fast molecular beam, as can be provided by He as a carrier gas, should lead to the smallest increase in temperature.
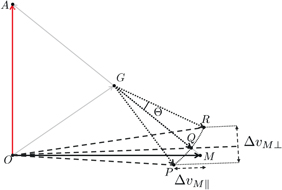
Figure 6. Newton diagram of the charge exchange reaction. The velocities of the Rydberg atom,  , and the neutral molecule,
, and the neutral molecule,  , are initially orthogonal, and the velocity of the center of mass is
, are initially orthogonal, and the velocity of the center of mass is  . In the frame of the center of mass, the molecule with a velocity
. In the frame of the center of mass, the molecule with a velocity  is transformed into an anion with a smaller velocity,
is transformed into an anion with a smaller velocity,  whose deviation, Θ, with respect to
whose deviation, Θ, with respect to  is given by equation (2). For a given impact parameter and given velocity,
is given by equation (2). For a given impact parameter and given velocity,  belongs to a cone of apex, G;
belongs to a cone of apex, G;  ,
,  ,
,  are three possible resulting velocities. The cone of velocities with the largest possible base corresponds to a distribution,
are three possible resulting velocities. The cone of velocities with the largest possible base corresponds to a distribution,  ,
,  ,
,  :
:  and
and  are respectively the maximal extents of the transverse and longitudinal velocities. In the frame of the laboratory, the deviation is given by the angle between
are respectively the maximal extents of the transverse and longitudinal velocities. In the frame of the laboratory, the deviation is given by the angle between  and
and  .
.
Download figure:
Standard image High-resolution image
Figure 7. Maximal, minimal, and average anion velocity for the anion beam, after the formation through charge exchange, for different Rydberg collisional velocities, vA. Results are given for an initial ideal molecular beam of BaF at a single forward, vM (temperature of 0 K), respectively corresponding to supersonic beams of He (green solid line, 1770 m s−1 forward velocity), Ar (blue dashed line, 559 m s−1 forward velocity), and Xe (red dotted line, 308 m s−1 forward velocity).
Download figure:
Standard image High-resolution image
Figure 8. Deviation angle of an anion beam for different Rydberg collisional velocities, vA. The line styles correspond to caption description in figure 7.
Download figure:
Standard image High-resolution image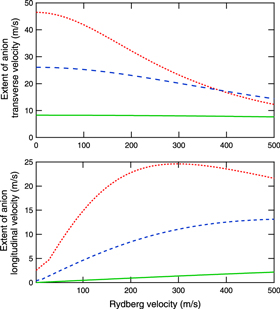
Figure 9. Extent of the transverse velocity,  (above), and longitudinal velocity,
(above), and longitudinal velocity,  (below), of the anion beam, after formation through charge exchange, for different Rydberg collisional velocities, vA. The line styles correspond to caption description in figure 7.
(below), of the anion beam, after formation through charge exchange, for different Rydberg collisional velocities, vA. The line styles correspond to caption description in figure 7.
Download figure:
Standard image High-resolution imageIn conclusion, a fast, neutral molecular beam—such as that produced using He seeded gas in a supersonic expansion—will remain almost identical especially with a beam temperature that is almost unchanged when transformed into its anionic counterpart by Rydberg electron transfer.
2.4. Choice of the Rydberg target
Ideally we would like to transform about 100% of the molecules into anions. Assuming a molecular beam with a velocity in the lab frame  m s−1, a realistic Rydberg cloud size of
m s−1, a realistic Rydberg cloud size of  cm, and a typical formation rate of
cm, and a typical formation rate of  cm3 s−1, it would require a Rydberg density
cm3 s−1, it would require a Rydberg density  cm−3, which must be produced during the necessary collisional time of
cm−3, which must be produced during the necessary collisional time of  μs. This is a strong limitation because the density, or similarly the Rydberg separation
μs. This is a strong limitation because the density, or similarly the Rydberg separation  , that can be sustained during this time is given by the equation
, that can be sustained during this time is given by the equation  μs
μs  [35, 36]. It is thus advisable to use heavy atoms like Cs. As an example, 5 μs Penning ionization time is obtained for Cs(43p) at a density of
[35, 36]. It is thus advisable to use heavy atoms like Cs. As an example, 5 μs Penning ionization time is obtained for Cs(43p) at a density of  cm3 [37]. We could reach longer Penning ionization times, and thereby a higher density, by using the Rydberg state without resonant dipole-dipole interaction, as in the van der Waals
cm3 [37]. We could reach longer Penning ionization times, and thereby a higher density, by using the Rydberg state without resonant dipole-dipole interaction, as in the van der Waals  case, or by using a repulsive force between atoms. Atoms in the s state are also advantageous because the efficiency of the ion-pair (anion) production depends not only on the principal quantum number, n, but also on the orbital angular momentum, l, of the Rydberg electron; an s electron should produce the highest production rate [26].
case, or by using a repulsive force between atoms. Atoms in the s state are also advantageous because the efficiency of the ion-pair (anion) production depends not only on the principal quantum number, n, but also on the orbital angular momentum, l, of the Rydberg electron; an s electron should produce the highest production rate [26].
However, Penning ionization always occurs [38] due to black body radiation or irregularity in the arrangement of the atoms [36]. Clearly, for molecules having a dipole moment close to the critical 2.5 D, as is required for high Rydberg states to form their anionic counterpart, it is trickier to get high efficiency in the formation process. However, for Rydberg states such that  —in other words, for molecules with a dipole moment bigger than ∼3 D—such a high Rydberg density (
—in other words, for molecules with a dipole moment bigger than ∼3 D—such a high Rydberg density ( cm−3) can be produced [39, 40].
cm−3) can be produced [39, 40].
Collisions of molecules with other molecules, ions, Rydberg or ground-state atoms might degrade the beam [41, 42]. Estimation of these effects is difficult. If some study of collisional effects with atomic negative ions exists [43], the collisional properties of dipole-bound negative ions have only been studied partially by Dunning's group [44, 45].
Interaction between  and
and  can lead to collisional destruction of the anion or simply heating of the beam. Let us assume that a 0.1 nA BaF beam (this will be limited by the space charge effect, as discussed later in the paper) with a 1-mm radius moving at 1760 m s−1 corresponds to a density of roughly 4 × 1011 m−3 and a mean interparticule separation of
can lead to collisional destruction of the anion or simply heating of the beam. Let us assume that a 0.1 nA BaF beam (this will be limited by the space charge effect, as discussed later in the paper) with a 1-mm radius moving at 1760 m s−1 corresponds to a density of roughly 4 × 1011 m−3 and a mean interparticule separation of  m. It is thus expected that going through the 1-cm cloud, the distance of minimal approach between
m. It is thus expected that going through the 1-cm cloud, the distance of minimal approach between  and
and  atoms is greater than 1 μm, a distance for which the anions see a weaker electric field than the field required to detach the electron [23]. We can conclude that, except for very low-binding-energy anions where the detachement field is especially low, no electron detachment would occur through a collision with
atoms is greater than 1 μm, a distance for which the anions see a weaker electric field than the field required to detach the electron [23]. We can conclude that, except for very low-binding-energy anions where the detachement field is especially low, no electron detachment would occur through a collision with  . However, deflection of the
. However, deflection of the  through such Coulomb interaction has to be evaluated. A simple estimation, which will be confirmed by the study of the space charge effect, is that the
through such Coulomb interaction has to be evaluated. A simple estimation, which will be confirmed by the study of the space charge effect, is that the  velocity change during the 1 ns needed to travel 1 μm at the closest distance from
velocity change during the 1 ns needed to travel 1 μm at the closest distance from  is on the order of 1 m s−1.
is on the order of 1 m s−1.
Regarding collisions between M and  , destruction of the dipole-bound anion has been seen in collisions with polar targets as a result of rotational energy transfer. The measured reaction rates are as large as the typical anion formation rate,
, destruction of the dipole-bound anion has been seen in collisions with polar targets as a result of rotational energy transfer. The measured reaction rates are as large as the typical anion formation rate,  cm3 s−1 (cross section 10−12 cm2) [44, 45]. Fortunately, the density of the neutral molecules is much smaller than the density of the Rydberg atoms, and the anion formation rate will dominate. The collisional cross section between the formed anions, M−, and other nonpolar neutral species such as the atomic gas can be evaluated to be at most given by the Langevin rate, leading to the typical rate of
cm3 s−1 (cross section 10−12 cm2) [44, 45]. Fortunately, the density of the neutral molecules is much smaller than the density of the Rydberg atoms, and the anion formation rate will dominate. The collisional cross section between the formed anions, M−, and other nonpolar neutral species such as the atomic gas can be evaluated to be at most given by the Langevin rate, leading to the typical rate of  cm3 s−1 [46], which is a factor hundreds lower than the typical anion formation rate. We mention that electron transfer in collisions between dipole-bound negative ions and the attaching targets, such as SF6, have been studied and the reaction rate is also small and limited by the geometrical size [47]. If we take the Langevin rate as an upper limit, the ratio of Rydberg atoms versus non-Rydberg atoms has to be as large as possible, and if possible greater than 1%. This value is easily achievable using a standard laser system for the Rydberg excitation [36, 48].
cm3 s−1 [46], which is a factor hundreds lower than the typical anion formation rate. We mention that electron transfer in collisions between dipole-bound negative ions and the attaching targets, such as SF6, have been studied and the reaction rate is also small and limited by the geometrical size [47]. If we take the Langevin rate as an upper limit, the ratio of Rydberg atoms versus non-Rydberg atoms has to be as large as possible, and if possible greater than 1%. This value is easily achievable using a standard laser system for the Rydberg excitation [36, 48].
Interaction between the negative ions and the Rydberg atoms is probably important. However, contrary to a collision with a cation, when a free or Rydberg electron approaches a negative atomic ion, it will experience a repulsive field. Therefore, in general we do not expect the charge-exchange process to occur even if for specific systems, some di-anionic resonances also play a role [49, 50]. Such cross sections will thus probably be orders of magnitude lower than the one found in the cation case, where it can reach the Rydberg geometrical cross section,  cm2 for (
cm2 for ( ) [51, 52]. Estimation of the cross section for all other possible processes due to interaction between
) [51, 52]. Estimation of the cross section for all other possible processes due to interaction between  and
and  is quite difficult. However, because the relative velocity,
is quite difficult. However, because the relative velocity,  m s−1, between the molecule and the Rydberg is very small compared to the orbital velocity of the Rydberg electron,
m s−1, between the molecule and the Rydberg is very small compared to the orbital velocity of the Rydberg electron,  (
( m s−1 is the atomic unit of velocity), and the impact parameter will be smaller than
m s−1 is the atomic unit of velocity), and the impact parameter will be smaller than  nm (for n = 30), we will be in the orbital adiabatic collision regime, where the Rydberg orbital electron adjusts itself adiabatically to the slow ion perturbation [53, 54], such that no energetic transition occurs. Therefore, we can estimate the n or l Stark mixing cross section being negligible. Finally, we mention that precise choice of the
nm (for n = 30), we will be in the orbital adiabatic collision regime, where the Rydberg orbital electron adjusts itself adiabatically to the slow ion perturbation [53, 54], such that no energetic transition occurs. Therefore, we can estimate the n or l Stark mixing cross section being negligible. Finally, we mention that precise choice of the  quantum state—for instance, to have a nonpolarizable Rydberg state—can also help further reduse these collisional rates.
quantum state—for instance, to have a nonpolarizable Rydberg state—can also help further reduse these collisional rates.
Even if experiments are obviously needed to clarify these aspects, we expect, except maybe in the case of very low-binding-energy anions, that no profound effect on the formation and the temperature of the anion beam will result from collisions between anions and other particles.
Several configurations for the Rydberg atoms can be suggested, including a vapor cell, a magneto-optical trap, an effusive beam, or a supersonic beam. However, the previous simulation and the constraints in terms of density and monochromaticity indicate that a Cs (slightly) supersonic beam might be a simple choice. A Mach number, M, of 5 may be enough to limit the energy dispersion of the anion beam. We recall here that  , where D is the nozzle diameter and λ is the mean-free path [55]. For example, with a nozzle diameter of D = 2 mm, a cesium temperature of 350
, where D is the nozzle diameter and λ is the mean-free path [55]. For example, with a nozzle diameter of D = 2 mm, a cesium temperature of 350  C should be sufficient. Using a recirculating oven [56–60] is probably a good choice for this because it has the great advantage that the effused element usage is minimized, so in principle, it need only be reloaded after thousands of hours of operation [61].
C should be sufficient. Using a recirculating oven [56–60] is probably a good choice for this because it has the great advantage that the effused element usage is minimized, so in principle, it need only be reloaded after thousands of hours of operation [61].
3. Deceleration
We now study the deceleration process. This should occur faster that the anion lifetime ( s) limited by black body radiation-induced photodetachment [62]. A fast deceleration condition is reinforced by the fact that a small interaction time between the anions is beneficial because it limits heating due to the Coulomb effect.
s) limited by black body radiation-induced photodetachment [62]. A fast deceleration condition is reinforced by the fact that a small interaction time between the anions is beneficial because it limits heating due to the Coulomb effect.
A possible deceleration scheme would be for the charged particle beam to climb an electric potential that will bring the beam almost to a standstill. However, such an energy deceleration process would lead to heating (increase of velocity dispersion) and is therefore probably not optimal. This can be seen as a conservation of phase space density: when decelerating, the special density increases, so the velocity dispersion (the temperature) has to increase. Conservation of energy indicates that the deceleration process leads to an increase in the temperature by a ratio,  , of twice the initial velocity to the initial velocity dispersion.
, of twice the initial velocity to the initial velocity dispersion.
We propose instead to change all velocities in the same way using a pulsed electric field, E, for a short time, T, to remove the velocity,  . For a typical molecule of 100 atomic mass, values of
. For a typical molecule of 100 atomic mass, values of  V cm−1 and
V cm−1 and  ns will stop the molecules. During this process, the molecules barely move (∼0.1 mm). We apply the field in the region where we want to produce the neutral molecules at rest. The stopping field is thus pulsed every time this region is filled again by the anionic beam: for instance, every 10 μs for a region of a centimeter size. This method produces molecules at the same temperature as the anion molecular beam and, because the Rydberg electron transfer barely modifies it, at the same temperature as the initial neutral molecular beam. If some short decelerators, such as chip-based or Rydberg decelerators, have been demonstrated [63, 64], we emphasize that our decelerator can even decelerate extremely large masses in the millimeter range and can operate in continuous regime.
ns will stop the molecules. During this process, the molecules barely move (∼0.1 mm). We apply the field in the region where we want to produce the neutral molecules at rest. The stopping field is thus pulsed every time this region is filled again by the anionic beam: for instance, every 10 μs for a region of a centimeter size. This method produces molecules at the same temperature as the anion molecular beam and, because the Rydberg electron transfer barely modifies it, at the same temperature as the initial neutral molecular beam. If some short decelerators, such as chip-based or Rydberg decelerators, have been demonstrated [63, 64], we emphasize that our decelerator can even decelerate extremely large masses in the millimeter range and can operate in continuous regime.
In principle, the space charge effect can alter the beam characteristics, as studied in [65]. Because our beam is diluted, a simple estimation of the increase in velocity dispersion can be obtained from the instantaneous Coulomb acceleration between two individual anions having (for a 0.1 nA beam) a separation of  m. Thus, after 10 μs, the velocity modification due to the Coulomb field (of 0.14 V m−1 for 100 atomic mass) is only on the order of 1 m s−1. The stray field present in the experimental chamber should then be controlled at the mV cm−1 level if we want to avoid too strong an effect on the anionic beam.
m. Thus, after 10 μs, the velocity modification due to the Coulomb field (of 0.14 V m−1 for 100 atomic mass) is only on the order of 1 m s−1. The stray field present in the experimental chamber should then be controlled at the mV cm−1 level if we want to avoid too strong an effect on the anionic beam.
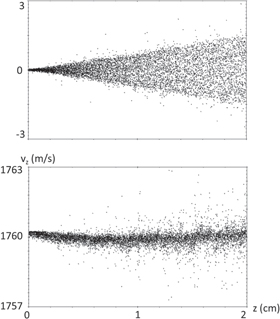
To confirm this estimation, we simulated the propagation of an ideal (meaning zero initial temperature) BaF− beam at 1760 m s−1. Evolution of 104 particles, producing a beam current of 0.1 nA, in a 1-mm Gaussian radius beam were simulated, taking into account all Coulomb interactions using General Particle Tracer software, for 20 μs. In the stopping region of the first centimeter, a steady state was reached, and figure 10 shows that even for such a high current density (which would correspond to more than 109 anions produced per second), we can propagate all the beams within a few centimeters during their lifetimes without significant transverse or longitudinal heating. The increase in velocity dispersion was on the order of 1 m s−1, corresponding to a temperature of only 10 mK.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 10. Velocity modification due to space charge effects during the propagation of a 0.1 nA BaF− beam of 1 mm radius. Upper part transverse velocity (vx). Lower part axial velocity (vz).
Download figure:
Standard image High-resolution image{kind=link}
The stopping field will thus produce molecules that are slow enough to be neutralized and eventually trapped.
The space charge effects seem to limit the beam current to slightly less than 109 (anions) molecules per second (0.1 nA), which is lower than can be produced by a standard (neutral) molecular source [66]. This relaxes our initial constraints, and all the related difficulties mentioned above, on the high Rydberg density required to ideally transform about 100% of the molecules into anions.
4. Neutralization
Once decelerated, we 'just' have to remove the electron without heating either the internal or the external degree of freedom of the molecule. Two possible schemes can be envisioned: photodetachment and electric field detachment.
4.1. Electric field
The detachment field, E, required is such that the barrier drops below the energy of the dipole (quasi-)bound state. This leads to a field of 0.1 kV cm−1 for a binding energy of 0.7 meV, and 10 kV cm−1 for a binding energy of 15 meV [67].
We can apply a pulsed electric field to a specific region when the particles have the smallest velocity. A very short but intense electric field would detach the electron without significantly affecting the velocity of the anion. The simplest way of doing this is probably to abruptly increase the value of the field producing the deceleration to a value higher that the detachment value just after the stopping is performed.
4.2. Photodetachment
Another method to neutralize the anion is to photodetach it. The photodetachement process could, in principle, even produce cooling in a similar way to one-way cooling: a proper laser spatial shaping can favor detachment of slow molecules [13].
Analytic description of dipole-bound anion photodetachment has been given in [68] based on the experimental data of [69]. Because of the very low binding energy,  , the photodetachment cross section can be quite large, but it decreases, as
, the photodetachment cross section can be quite large, but it decreases, as  , where ω is the photon frequency. From [68], we can reasonably assume a photodetachment cross section of
, where ω is the photon frequency. From [68], we can reasonably assume a photodetachment cross section of  . It is clearly advantageous to use a far infrared laser, such as a CO2 laser with a wavelength of 10.6 μm. Using a 100-watt laser focused on 3 mm leads to a photodetachment rate near 104 s−1 for a small binding energy of 1 meV, and it goes to a photodetachment rate of 106 s−1 for a binding energy of 10 meV.
. It is clearly advantageous to use a far infrared laser, such as a CO2 laser with a wavelength of 10.6 μm. Using a 100-watt laser focused on 3 mm leads to a photodetachment rate near 104 s−1 for a small binding energy of 1 meV, and it goes to a photodetachment rate of 106 s−1 for a binding energy of 10 meV.
5. Conclusions
We have shown that a supersonic molecular beam that produces very polar molecules with low vibrational states and low rotational quantum numbers can be transformed with high efficiency in anions using the charge exchange process. The formed dipole-bound negative ion is expected to have exactly the same nuclear configuration as its neutral parents. For a fat beam, the produced anionic beam should have almost identical characteristics to the initial neutral beam. Such, a charged particle beam can be of interest in and of itself for research in material science or for surface investigations such as imaging or lithography [65]. Further deceleration and neutralization can be a very simple way to produce efficiently neutral molecules at rest.
It is beyond the scope of this paper to describe in detail the experimental setup, but note that if accumulation in a trap is envisioned, it would probably need to send the anion beam off axis using deflectors and to realize the (photo-)detachement laser and stopping field in a region where the atomic and neutral molecular beam is absent in order to avoid further collisions. Furthermore, using a permanent magnet and high-permittivity material, it should be possible to construct a magnetic trap with an open access close to 1 cm3 [70–74]. However, precise shaping of the electric and magnetic field configurations would be required to properly accelerate and decelerate the anion beam in order to prevent the magnetic mirror effect.
Acknowledgments
We are indebted to C Desfrançois, M Raoult, V Kokoouline, and O Dulieu for useful discussions. The research leading to these results received funding from the European Research Council under grant agreement no. 277762 COLDNANO.