

Abstract
The development of electrocatalysts for both oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) with high-activity and atability still remain great challenges for rechargeable Zn-air batteries. Herein, a new type of Co-doped Ce–N–C bifunctional electrocatalyst has been synthesized through a simple two-step method, which realizes the high dispersion of Co3O4 on the CeO2 carbon frame and stabilizes its specific surface area. Benefiting from the synergistic interaction between Co3O4 and CeO2, the conductivity of the electrocatalyst is improved and the oxygen reduction reaction/oxygen storage properties are promoted. The resultant Co3O4-CeO2@N–C catalyst shows remarkable ORR activity with the high initial potential (E0 = 0.8 V), the large limiting current density (jL = 6 mA cm−2), and a low Tafel slope (81 mV dec−1). In full cell tests, Co3O4-CeO2@NC as the oxygen electrode exhibites superior charge/discharge capacity and excellent cycle stability. The assembled Zn-air battery achieves a maximum power density of 110 mW cm−2 at a current density of 180 mA cm−2, and a high specific capacity of 780 mAh g−1 at a discharge current density of 10 mA cm−2.
Export citation and abstract BibTeX RIS
1. Introduction
Rechargeable metal-air batteries have captured great attention as green advanced energy devices due to their high efficiency, environmental friendliness and safety [1, 2]. However, the cathode half-reaction, oxygen reduction reaction (ORR), is the major bottleneck for large-scale applications of the rechargeable metal-air batteries [3–5]. Hence, it is urgent to develop efficient and durable electrocatalysts toward ORR. At present, Pt-based materials are currently considered state-of-the-art catalysts for ORR, but many obstacles such as high price, earth scarcity, unfavorable stability and poor methanol tolerance limit their large-scale commercialization [6–8]. Furthermore, the catalytic activity of Pt-based catalysts towards oxygen evolution reaction (OER) is relatively poor. Similarly, RuO2 is highly active for OER, while its catalytic activity for ORR needs to be enhanced [9]. Therefore, it is essential to search for a low-cost and effective bifunctional electrocatalysts for ORR and OER [10–13].
In recent years, various low-cost and highly active materials have been widely developed to replace Pt-based catalysts, including heteroatoms (such as N, P, S) -doped carbon [14–16], earth-rich transition metal oxides or carbides [17–19], Fe/Ce–N–C materials [20–22], and so on. Among them, as an important reducing oxide carrier, CeO2 has an extraordinary redox couple (Ce3+/Ce4+), which leads good redox performance and high oxygen storage capacity [23, 24], thus it has been widely used as ORR and OER electrocatalysts [25–28]. According to pioneering studies, various CeO2 nanocrystals with different shapes have been successfully synthesized, such as cubes, octahedrons, rods, and nanowires and its catalytic activities have been characterized [29–31]. For instance, Liu et al [32] found that the mainly exposing crystal planes of CeO2 nanorods have a relatively high concentration of oxygen vacancies, and showed better reducibility and catalytic activity. Unfortunately, CeO2 is rarely used as catalyst alone due to its poor structural stability and high resistivity [33]. Because the specific surface area of CeO2 decreases rapidly at high temperature, meanwhile, it might lose its redox reaction catalytic performance and oxygen storage capacity. To solve this problem, CeO2 could be modified by introducing various ions to improve its high-temperature stability, therefore enhance its catalytic activity [34–36].
Inspired by the above considerations, through Co modification, we synthesized CeO2 carbon nanorods with Co3O4 anchored on the surface (Co3O4-CeO2@N–C). As expected, Co modification stabilizes the specific surface area of CeO2, and increases the redox performance and oxygen storage performance via a synergistic effect. The prepared Co3O4-CeO2@N–C catalyst shows outstanding ORR activity with a positive initial potential (E0 = 0.8 V), a large limiting current density (jL = 6 mA cm−2) and a low Tafel slope (81 mV dec−1), as well as good long-term stability, outperforming the benchmark Pt/C. In full cell tests, Co3O4-CeO2@N–C catalyst also reveals a superior charge/discharge capacity and excellent cycle stability. This method provides a new strategy for the development of high-efficiency bifunctional electrocatalysts through CeO2 modification and the outstanding performance of Co3O4-CeO2@N–C catalyst is suggestive of potential application in rechargeable Zn-air batteries.
2. Experimental section
2.1. Materials
Ce(NO3)3·6H2O (≥99.95%, Aladdin), C6H3(CO2H)3 (≥99.00%, Aladdin), C2H7NO (≥99.00%, AR, Sinopharm Chemical Reagent Co., Ltd, China), Co(NO3)2·6H2O (≥99.0%, AR, Aladdin), NaBH4 (≥98% Xuzhou Tianhong Chemical Co, Ltd, China), All the chemicals were used without further purification, and the water used were deionized (DI) water (18.2 MΩ cm−2).
2.2. Synthesis of CeO2@N–C nanocrystals
Ce-MOF precursor was synthesized according to the literature [37, 38] with slightly modified. Typically, 1 mmol Ce (NO3)2·6H2O was dissolved in a mixture of 25 ml water-ethanol solvent (1:1, V/V) to obtain Solution I. 1 mmol trimesic acid (1, 3, 5-H3BTC) was also dispersed in the same 25 ml mixture solution to gain Solution II. Subsequently, Solution I was added into Solution II under stirring for 2 h in a water bath at 90 °C. The white precipitation was washed by DI water and ethanol, followed by 80 °C into a vacuum oven and dried for 6 h to collect. The Ce-MOF was heated at 900 °C for 2 h in a tube furnace (2 °C min−1) under Ar flow to get CeO2@N–C composite.
2.3. Synthesis of Co3O4-CeO2@N–C
0.4 g CeO2@N–C and 10 ml ethanolamine (C2H7NO) were evenly dispersed in 25 ml DI water with stirring at 50 °C for 3 h. Then 0.4 g Co(NO3)2·6H2O was added into the solution under stirring for 3 h until 0.4 g NaBH4 was added. After that, the solution was carefully transferred into a 100 ml Teflon-lined stainless-steel autoclave and heated at 80 °C for 6 h. After cooling to room temperature naturally, the product was washed with DI water until the pH of solvent became neutral and then dried at 60 °C for 6 h in an oven. Finally, the black powder of Co3O4-CeO2@N–C composite was obtained.
2.4. Materials characterization
The morphology, composition, and structure of as-prepared products were characterized by scanning electron microscope (SEM), (Hitachi S4800), transmission electron microscope (TEM, JEOL 2100F), spherical aberration (Cs) corrected TEM (FEI Titan Themis 60–300), powder x-ray diffraction (XRD, Rigaku Ultima IV) and x-ray photoelectron spectroscopy (XPS, PHI Quantum-2000).
2.5. Electrochemical characterization
Electrochemical measurements were conducted in a conventional three-electrode system controlled by CHI760E electrochemical station. The rotating disk electrode (RDE, diameter: 4 mm) and rotating-ring disk electrode (RRDE) experiments were performed on the RRDE-3A. Graphite rod and Hg/HgO electrode (1 M KOH solution) were used as counter electrode and reference electrode, respectively. The catalyst ink was prepared by dissolving 5 mg catalyst into 1 ml isopropanol and 5 μl 5% Nafion solution and sonicating for 30 min to form catalyst suspension. Then 30 μl of the ink was dropped on the glassy carbon electrode as the working electrodes. The linear sweep voltammetry (LSV) was performed at a scan rate of 5 mV s−1 in O2-saturated 0.1 M KOH with rotating from 400 to 2500 rpm for ORR. The electron transfer number (n) and %  were determined in combination with the RRDE technique:
were determined in combination with the RRDE technique:
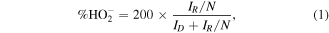
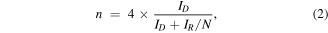
where ID and IR represent the disk and ring currents, respectively. And N is the collection efficiency of the Pt ring (N = 0.42). All the electrochemical measurements were done at room temperature.
Zinc-air battery (ZAB) tests were performed in a home-made battery. The catalyst ink was drop-casted onto a Teflon-coated carbon cloth (W0S1002) with a catalyst loading amount of 1.2 mg cm−2 as the gas diffusion electrode (GDE). The achieved GDE was dried under vacuum at 85 °C. A control sample is also prepared by using 0.24 mg Pt/C. For battery tests, the GDE layer was used as the air cathode, a polished Zn foil as anode and 6.0 M KOH with 0.2 M Zn(CH3COO)2 as the electrolyte. The discharge/charge curves were recorded in ambient on a Neware battery test instrument (Shenzhen, China).
3. Results and discussion
3.1. Characterization of the electrocatalysts
The synthetic route of the catalyst is shown in figure 1. First, based on previous literature reports, the Ce-MOF900 precursor was slightly modified to prepare a bundled Ce-MOF900 (Ce3+/Ce4+) carbon–nitrogen nanorod precursor template with mixed valence. Then the amine group of ethanolamine chelated the metal cobalt ion, and the hydroxyl group at the other end was adsorbed and anchored on the carbon–nitrogen nanorod precursor template. Secondly, cobalt ions were reduced to Co3O4 nanoparticles by NaBH4 through low temperature hydrothermal method, which are highly uniform and dense anchored on the CeO2 carbon–nitrogen framework, avoiding the agglomeration. Finally, Co3O4-CeO2@N–C catalyst with mixed valence state (Ce3+/Ce4+) is prepared successfully.
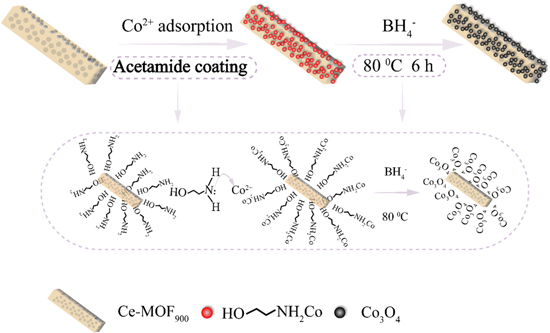
Figure 1. Schematic diagram of catalyst synthesis.
Download figure:
Standard image High-resolution imageThe crystal structure and surface chemical composition were investigated by XRD and XPS, respectively. Figure 2 is the XRD pattern including the two samples. The CeO2/NC (i) shows diffraction peaks at 2θ angles of 28.58°, 33.16°, 47.52°, 56.46°, 59.39°, 69.50°, 76.74° and 79.09°, which belong to the (111), (200), (220), (311), (222), (400), (331) and (420) planes of CeO2 (PDF#89–8436), respectively. After Co doping, Co3O4-CeO2@N–C (ii) exhibits the new diffraction peaks at 19.68° and 38.50°. Both the peaks are corresponding to the (111) and (222) planes of Co3O4 (PDF#43–1003), indicating the Co element was successfully doped. Furthermore, the strength of peaks is weak because it is covered by the CeO2@N–C, which can be further proved by the results of TEM. In addition, after comparison, the peaks width of Co3O4-CeO2@N–C is larger than CeO2@N–C. Thus, the grain size of Co3O4-CeO2@N–C (∼65 nm), computed by the Debye–Scherrer equation, is smaller than those of CeO2@N–C (∼80 nm). Smaller particle size is beneficial to increase the specific surface area and further increase the active sites to increase the catalytic performance.
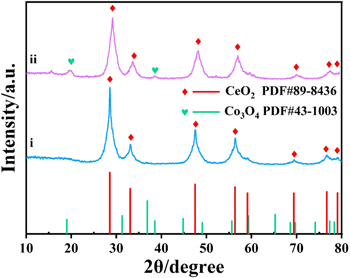
Figure 2. XRD pattern of (i) CeO2@N–C, (ii) Co3O4-CeO2@N–C.
Download figure:
Standard image High-resolution imageTo explore the effect of Co doping on the surface chemical composition and valence state of Co3O4-CeO2@N–C and CeO2@N–C, XPS measurement was also employed. Figure 3(a) shows the XPS survey spectrum of Co3O4-CeO2@N–C. Typical peaks of Ce3d (924–878 eV), Co2p (805–770 eV), C1s (280–295 eV), N1s (393–410 eV) and O1s (526–536 eV) is discovered in corresponding energy range, which can illustrate the co-existence of Ce, Co, C, N and O elements. Peak fitting of the elements core-level is all obtained after deduction of Shirley background and is shown in figures 3(b)–(f). In the high-resolution Ce3d spectrum (figure 3(b)), it can be deconvolved into eight peaks. The peaks are denoted as μ and ν to identify the spin–orbit composition of Ce 3d5/2 and Ce 3d3/2, respectively. Two peaks which labeled as μ' and ν' located at 903.16 eV and 885.12 eV are assigned to the Ce3+. The remaining six peaks of μ''' (916.82 eV), μ'' (907.62 eV), μ° (901.09 eV), ν''' (898.49 eV), ν'' (888.89 eV) and ν° (882.53 eV) are attributed to the characteristic peaks of Ce4+ [23, 39, 40]. Therefore, Ce3+ and Ce4+ are co-existence in the Co3O4-CeO2@N–C and the mainly oxidation states were tetravalent [23, 40]. The electronic state of Ce3+ is 3d104f1, so there is no doubt about the exist of lone electron (4f1 orbital). Lone electron is beneficial to enhance the interaction between ceria and the surrounding Co atoms. In the other hand, the percentage contents of Ce 3+ (μ' + ν') and Ce 4+ (μ''' + μ'' + μ° + ν''' + ν'' + ν°) can get by calculate the fitted area that under the corresponding peaks. Co3O4-CeO2@N–C (0.194) has the larger ratio value than CeO2@N–C (0.188), proving more defects in Co3O4-CeO2@N–C. Moreover, the exist of Ce3+ can not only donate charge compensation, but also help to form the unsaturated chemical bonds in the CeO2 crystal. Both of them are in favor of generating oxygen vacancies and promoting the oxygen adsorption on the catalyst surface subsequently [39, 41].
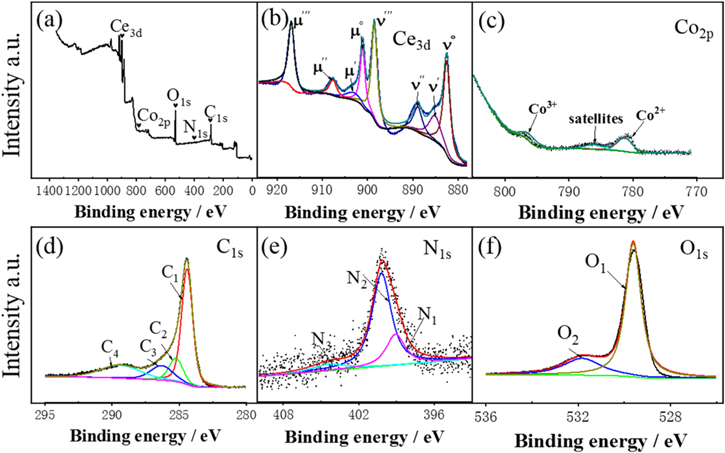
Figure 3. The XPS spectra of (a) the full survey spectra, (b) the Ce3d peaks, (c) the Co2p peaks, (d) the C1s peaks, (e) the N1s peaks, (f) the O1s peaks of Co3O4-CeO2@N–C.
Download figure:
Standard image High-resolution imageFigure 3(c) shows the XPS survey spectrum of Co2p. The peak at 781.30 eV belongs to Co2+, while the peak at 796.81 eV is ascribed to Co3+. Generally, Co3+ plays an important role in the electrocatalytic performance toward ORR [42]. More details with C1s is depicted in figure 3(d) and it can be divided into four peaks (C1–C4). Two main peaks located at 284.38 eV (C1) and 286.22eV (C3) are attributed to C–C/C–N and C–O [43, 44], which are generated from the organic ligands. In addition, there are the other two weak peaks at 285.08 eV (C2) and 289.38 (C4), which is responsible for the characteristic of C–O and π–π* [39]. The high resolution XPS spectrum of N1s shown in figure 3(e) are fitted by three parts at 399.08 eV (N1), 400.19 eV (N2) and 404.41 eV (N3), corresponding to pyrrolic-N, graphitic-N and oxidized-N[43, 45]. Based on previous reports in open literatures, the graphitic-N is generally considered to greatly improve the ORR catalytic performance of N-doped carbon materials, especially the limiting current density on ORR[45, 46].
The O1s XPS spectrum is shown in figure 3(f), where the peak at 529.61 eV (O1) stands for the lattice oxygen (Olatt), while the peak at 531.82 eV (O2) represents the surface active oxygen (Osur). Surface active oxygen consist of superoxide species (O2-) and peroxide species (O-), both of them are activated oxygen species which comes from the oxygen defects [47]. The ratios of Osur/Olatt of Co3O4-CeO2@N–C900 (0.31) are higher than CeO2@N–C900 (0.19), which is meets the catalytic activities of the samples.
The morphology information of the Co3O4-CeO2@N–C were obtained by SEM and TEM. From the low magnification SEM images (figure 4(a)), the Co3O4-CeO2@N–C are composed by bundles of nanorods. Moreover, the high magnification SEM image shown in figure 4(b) indicates that each nanorod with three dimensions is about 2 μm height, 100 nm length and width. Compared with Co3O4-CeO2@N–C, CeO2@N–C exhibited fewer distributed nanorods (figure S1 (available online at stacks.iop.org/NANO/33/415404/mmedia)). What is more, at higher resolutions, CeO2 nanoparticles agglomerate on the surface of nanorods, while distribute uniformly after Co modification. Such morphology could inevitably lead to lower specific surface area and less available active sites, affecting their catalytic performance seriously. In TEM image (figure 4(c)), the structure of nanorods bundles of Co3O4-CeO2@N–C can be further confirmed, and some of CeO2 grain which closed to hexagon also can be observed on the nanorod (figure 4(d)). As shown in high-resolution TEM image of figure 4(e), Co3O4 and CeO2 nanoparticles distribute on the carbon–nitrogen nanorods uniformly, and the close contact between them was confirmed by lattice analysis. As a result, the lattice fringes of 0.32 nm, 0.25 nm and 0.28 nm correspond to the (112) plane of CeO2 and the (311), (220) plane of Co3O4, respectively. Obviously, Co3O4 nanoparticles anchored on the surface are expected to cooperate with CeO2 to promote the ORR/oxygen storage performance. The select area electron diffraction (SAED) pattern (figure 4(f)) shows well-defined diffraction rings, which can be well indexed to the (111), (200), (220), (422), (311) and (422) planes. All the planes are origin from CeO2 except (422) plane is attributed to the Co3O4. In addition, the elements distribution in Co3O4-CeO2@N–C is measured by mapping (figure 4(g)) and EDS (figure 4(h)), which further verified the co-existence of C, N, O, Ce, and Co, and their uniform distribution on the entire carbon support.

Figure 4. The SEM (a), (b) and TEM (c), (d) image of Co3O4-CeO2@N–C. (e) The HRTEM image of Co3O4-CeO2@N–C. (f) The SAED image of Co3O4-CeO2@N–C. (g) The corresponding mapping images of Co3O4-CeO2@N–C. (h) The EDS image of Co3O4-CeO2@N–C.
Download figure:
Standard image High-resolution image3.2. Electrochemical performance of electrocatalysts
Figure 5(a) shows the RRDE curves of CeO2@N–C, Co3O4-CeO2@N–C and commercial Pt/C (20 wt%) toward ORR at 1600 rpm. Evidently, the ORR activity of CeO2@N–C is very low due to the most negative RRDE curve. After the introduction of Co3O4, the Co3O4-CeO2@N–C catalyst shows more positive E0 (0.80 V), half-wave potential (0.75 V) and the Pt-like limiting current density (6.0 mA cm−2), indicating that the Co3O4-CeO2@N–C catalyst possesses more active sites and the catalytic performance toward ORR is enhanced. Through the RRDE measurement, we also calculated the yield of H2O2 and the electron transfer number (n) to obtain selectivity and electron transfer pathway during ORR, as shown in figure 5(a) and (c). Apparently, the disk current density corresponding to ORR is significantly larger than the ring current density arisen from the H2O2 oxidation, implying the predominant product of OH‒. The H2O2 yield observed on Co3O4-CeO2@N–C remains below 12% in the potential range of 0.25–0.6 V and the n is approximately 4, indicating an ideal four-electron transfer pathway proceeded on Co3O4-CeO2@N–C. Particularly, the Co3O4-CeO2@N–C catalyst also showed excellent electrocatalytic activity for OER (figure 5(d)). To obtain the current density of 10 mA cm−2, the potential required by Co3O4-CeO2@N–C and Pt/C was 1.65V and 1.82V, respectively, that is, Co3O4-CeO2@N–C shows better OER performance. What is more, according to previous literatures [48, 49], a smaller potential gap between ORR and OER (ΔE = EJ=10-E1/2) indicates better catalytic activity. Compared with Pt/C (ΔE = 0.98 V), Co3O4-CeO2@N–C has a smaller ΔE of 0.92 V, indicating that Co3O4-CeO2@N–C has better ORR and OER bifunctional catalytic activity. The Tafel slope corresponding to Co3O4-CeO2@N–C is 81 mV dec−1 (figure 5(e)), which is smaller than that of Pt/C (121 mV dec−1) and CeO2@N–C (141 mV dec−1), illustrating that Co3O4-CeO2@N–C is conducive to generating better dynamic processes. Moreover, the long-term stability of the Co3O4-CeO2@N–C was investigated under 0.7 V in O2-saturated 0.1 M KOH solution (figure 5(f)). After 40 000 s of continuous test, the current density of Pt/C and CeO2@N–C only retained 66.1% and 42.3%, respectively. While those of Co3O4-CeO2@N–C could reach 81.1%. This result confirms that Co3O4-CeO2@N–C has superior ORR stability than those of Pt/C and CeO2@N–C catalysts.
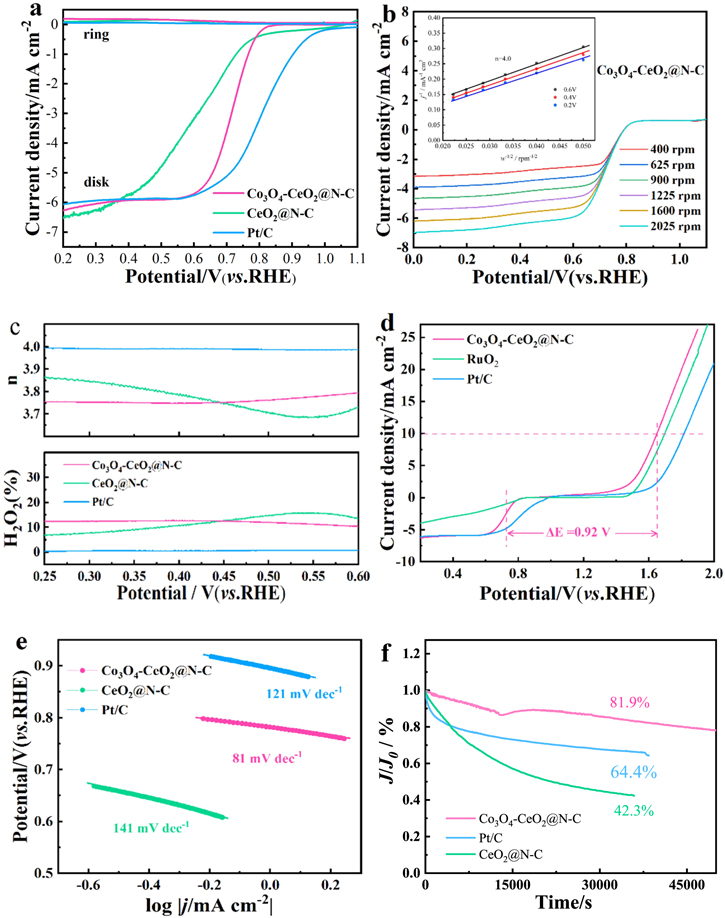
Figure 5. (a) RRDE curves for different catalysts at 1600 rpm; (b) LSV curves of Co3O4-CeO2@N–C at different rotation rates from 400 to 2050 rpm (inset: electron transfer number (n)); (c) electron transfer number (n) and H2O2 yield of various catalysts at 1600 rpm; (d) ORR/OER bifunctional LSV curves of various catalysts at 1600 rpm; (e) Tafel slopes of of various catalysts; (f) chronoamperometric curves of various catalysts.
Download figure:
Standard image High-resolution image3.3. Zn-air battery test
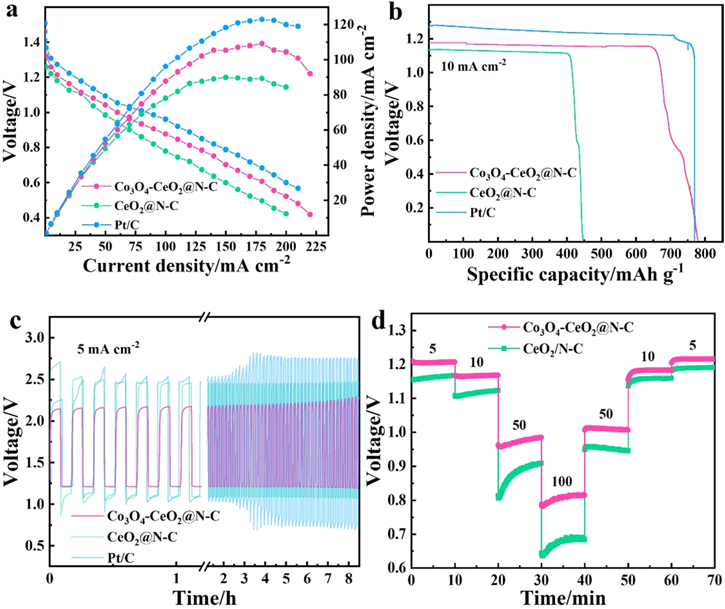
To further evaluate the practical application potential of the catalyst in Zn-air batteries, Teflon carbon cloth coated with CeO2@N–C, Co3O4-CeO2@N–C and commercial Pt/C (20 wt%) catalysts were used as the air cathode to assemble ZABs. As shown in figure 6(a), the open-circuit voltage of the ZAB assembled by the Co3O4-CeO2@N–C is 1.46 V, which is higher than that with CeO2@N–C cathode and only 50 mV smaller than that with Pt/C, suggesting its good catalytic performance. The maximum power density for ZAB assembled by the Co3O4-CeO2@N–C reaches about 110 mW cm−2 at around 180 mA cm–2 (figure 6(a)), which outperforms CeO2@N–C cathode (90 mW cm–2) and up to 90% of Pt/C. In addition, at the constant discharge current density of 10 mA cm−2, ZAB assembled by the Co3O4-CeO2@N–C has the highest specific capacity of 780 mAh g−1 (based on the mass of consumed Zn, figure 6(b)). This is superior to CeO2@N–C (447 mAh g−1) and commercial Pt/C (770 mAh g−1). Moreover, the rechargeability of the air electrode at a current density of 5 mA cm−2 was evaluated (10 min per cycle and 200 cycles) and the results are shown in figure 6(c). Co3O4-CeO2@N–C shows excellent cycle durability and has the lowest initial charge/discharge voltage gap (0.89 V) in the initial dozens of cycles than those of CeO2@N–C (1.42 V) and Pt/C (1.1 V). Besides, for Co3O4-CeO2@N–C, the voltage gap of ZAB does not increase significantly within 40 h, which proved that it had high round-trip efficiency, while the gap of ZAB with Pt/C catalyst increases obviously. Finally, by testing at different discharge rates, the rate performance of Co3O4-CeO2@N–C based ZABs was evaluated (figure 6(d)). Under different current densities (5 –100 mA cm−2), the discharge potential of ZABs remained basically stable. Especially after high-rate discharge at high current density, the discharge potential could recover at 5 mA cm−2, which proved the remarkable reversibility for Co3O4-CeO2@N–C. All the above results indicate that the ZABs with Co3O4-CeO2@N–C catalysts exhibit a superior charge–discharge capacity and stability due to their excellent bifunctional ORR/OER electrocatalytic activity and stability, which outperforms most of other recently reported low-cost bifunctional oxygen electrocatalysts (table 1).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 6. Performance of Zn-air battery with different catalysts: (a) polarization curve and power density diagram; (b) discharge specific capacity curve at 10 mA cm−2 discharge current density; (c) discharge–charge curve under 5 mA cm−2 current density; (d) rate performance of Co3O4-CeO2@N–C -based ZABs.
Download figure:
Standard image High-resolution image{kind=link}
Table 1. Cycle performance of rechargeable Zn-air batteries with various catalysts.
| Catalyst | Peak power density (mW cm−2) | Conditions (mA cm−2) | Cycling performance | Reference |
|---|---|---|---|---|
| Fe0.5Co0.5Ox /NrGO | 86 | 10 | 2 h per cycle for 60 cycles (120 h) | [50] |
| Co3FeS1.5(OH)6 | 113.1 | 2 | 20 min per cycle for 108 cycles (36 h) | [51] |
| Co3O4/N-CNTAs | — | 5 | 10 min per cycle for100 cycles (16.7 h) | [52] |
| C-MOF-C2-900 | 105 | 10 | 20 min per cycle for 90 cycles (30 h) | [13] |
| NPMC-1000 | 55 | 2 | 10 min per cycle for 180 cycles (30 h) | [53] |
| CeO2@N–C | 90 | 5 | 10 min per cycle for 200 cycles (33 h) | This work |
| Co3O4-CeO2@N–C | 110 | 5 | 10 min per cycle for 200 cycles (33 h) | This work |
4. Conclusions
In summary, we developed a new Co-doped bifunctional electrocatalyst (Co3O4-CeO2@N–C) through a simple two-step method. XPS surface analysis shows that there are Ce3+/Ce4+ redox centers, which will facilitate the high-concentration oxygen vacancy catalysis of oxygen. High-resolution SEM and TEM imaging analysis, the optimized Co3O4-CeO2@N–C sample contains abundant Ce atoms, Co atoms and N atom sites. In an alkaline medium, the initial potential of the catalyst is 0.80 V, and the reversible oxygen overpotential at 10 mA cm−2 is 0.92 V. Compared with the Pt/C catalyst, Co3O4-CeO2@N–C has good ORR/OER bifunctional electrocatalytic activity and stability. The ZABs assembled with Co3O4-CeO2@N–C as the oxygen electrode reached a maximum power density of 110 mW cm−2 at a current density of about 180 mA cm−2, and at the same time, at a constant discharge current density of 10 mA cm−2, it also exhibited a higher discharge specific capacity of 780 mA h g−1 and excellent cycle stability. The superior performance of Co3O4-CeO2@N–C for ORR could be ascribed as: (1) extraordinary redox couple (Ce3+/Ce4+) of CeO2, which leads good redox performance and high oxygen storage capacity; (2) N-doped carbon nanorod can provide graphitic-N, which greatly improve the ORR catalytic performance especially the limiting current density; (3) the introduction of Co stabilizes the specific surface area of CeO2, and increases the redox performance and oxygen storage performance via a synergistic effect. This work provides a promising strategy for the development of high-efficiency electrocatalysts through CeO2 modification and new insights for the development and application of green advanced energy devices.
Acknowledgments
We acknowledge the financial support from the North Minzu University Major Project (ZDZX201802), the National Natural Science Foundation of China (21865002).
Data availability statement
All data that support the findings of this study are included within the article (and any supplementary files).
Declaration of competing interest
The authors declare no competing financial interests.