
Abstract
The adsorption of organic molecules to surfaces is a central issue to achieve fully-functional molecular devices, for which porphyrins are well-studied due to their chemical stability and functional diversity. Herein, we investigate both the physical and the chemical adsorption of the free-base tetraphenylporphyrin 2H-TPP on the Cu(111) surface within the framework of density functional theory and find that the most stable physisorbed configuration is more weakly bound by −0.31 eV than the chemisorbed configuration. We use the electron localization function to investigate the difference in binding mechanisms between strong physisorption and weak chemisorption. We have computed a reaction barrier of 0.12 eV in going from physical binding to chemical bonding to the surface, and a barrier of 50 meV in going between neighboring physical binding sites. Our results support the possibility of realizing free-base porphyrins either physisorbed or chemisorbed on Cu(111) depending on the deposition procedure and experimental conditions.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
The control of large functional molecules on solid surfaces and more particularly coinage metal (Au, Ag, and Cu) surfaces is of great interest for the investigation of new devices with potential applications in nanoscience such as molecular electronics [1–4], photonics [5–9] or spintronics [10–12]. In addition, porphyrins have molecular properties that can be tuned in several ways, for example by functionalization of the core macrocycle [13, 14]. The central core macrocycle has the ability to host various metal atoms, in so-called metalloporphyrins [15], which can be used to add functionalities such as sensing [16, 17] or magnetic properties that could be used as building blocks in storage media [18–20]. In the latter case, the interaction between the metal atom at the center of the macrocycle and the surface atoms can play a major role in the adsorption of the molecule which, in turn, affects the properties of the system [21–24]. In general, the interaction between complex molecules and a substrate is frequently a source of modifications of the molecular properties, such as different molecular conformations, changes in the electronic structure [25–27], variation of the charge or even influencing the magnetic properties [28, 29]. In addition, the type of interaction between the molecule and the surface could lead to differences in, e.g. conductivity and spin lifetimes.
The study of molecular binding on surfaces by means of density functional theory (DFT) is a very active field of research that can reveal the adsorption mechanism and how the molecular conformation and electronic structure vary at different surface sites. Nowadays, physical adsorption on metallic surfaces can be accurately treated by including dispersion corrections in the simulations. However, in the case of porphyrins, it is very challenging to determine the interaction of its different parts with the surface, including assessing if there is strong physisorption or weak chemisorption without proper analysis of the electron density. To make it more straightforward, a way to go is by starting with dividing the large molecules into simpler fragments and assessing their interaction with the metal substrate. Thus, using an inverse design approach to find the most stable physi- and chemisorbed states by part-by-part adsorption of the fragments of the porphyrin ring. This could allow for an easy evaluation of chemically vulnerable electrophilic centers and inversely imply how to understand it in large molecules such as tetraphenylporphyrin (2H-TPP). Although chemisorption of molecules on substrates is associated with (i) immobilization, (ii) molecular conformation (and shape) changes of the molecule, and (iii) significant changes of the molecular density of states (DOS) upon adsorption, all these three effects have been found for porphyrins physisorbed on Cu(111) (see [30, 31]). We investigate these aspects of both physisorption and chemisorption using the free-base 2H-TPP (see figure 1) adsorbed on the Cu(111) surface.
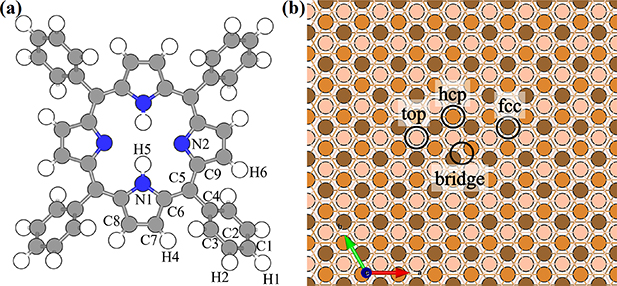
Figure 1. (a) The structure of the free-base tetraphenylporphyrin (2H-TPP) molecule. Gray, blue and white spheres correspond to carbon, nitrogen and hydrogen atoms, respectively. (b) Schematic diagram of Cu(111) slab with different adsorption sites labeled. The light to dark shades of orange balls depict the 1st, 2nd and 3rd layer from the surface.
Download figure:
Standard image High-resolution image2. Computational details
The properties of 2H-TPP on Cu(111) were determined via first-principles DFT calculations using the semi-empirical description of dispersion, DFT-D3 with Becke–Jonson damping, proposed by Grimme [32] with a generalized gradient approximation functional in the parameterization by Perdew–Burke–Ernzerhof (PBE) [33]. Separate calculations were performed with a dispersion-corrected functional optB86b-vdWDF [34]. The projector-augmented wave method [35, 36] has been used for the description of the interaction between the electrons in the core (not explicitly included) and the valence range of energy as implemented in the Vienna ab initio simulation package (VASP) [37–39]. Within VASP, the iterative solutions of the Kohn–Sham equations are performed using a plane-wave basis with an energy cutoff of 460 eV and Gaussian smearing of the initial electronic occupations with a value of 0.2 eV.
In order to determine the equilibrium lattice parameter for bulk Cu, a (14 × 14 × 14) Monkhorst–Pack [40] k-point mesh was used. The resulting lattice constant of Cu was found to be 3.606 Å (which is in good agreement with the experimental lattice constant of 3.597 Å [31]). The relaxed bulk unit cell was used to construct a Cu(111) slab which was modeled as a 12 × 12 hexagonal cells consisting of four atomic layers with 576 Cu atoms in total and a large vacuum of around ∼20 Å. The geometry optimization of the molecule—surface systems was performed using only the Γ-point in the Brillouin zone sampling (k-point sampling convergence is shown in figure S1 in the Supplementary Material) and utilizing the conjugate gradient algorithm. The relaxation of all atoms was achieved when the force acting on each atom was less than 0.01 eV Å−1. During the relaxation, the cell size and shape were not allowed to change but only the atomic positions with the restriction that the bottom layer of the slab model was kept fixed.
The binding energy was defined as
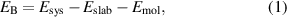
where  ,
,  and
and  correspond to the total energy of the system, surface slab, and molecule respectively.
correspond to the total energy of the system, surface slab, and molecule respectively.
The integration of basins of attractors was done by using the YT algorithm [41] as implemented in the critic2 software [42, 43], while the Virtual NanoLab software [44] was used to set up the simulation cells and supercells. The visualization of the electron localization function (ELF) has been carried out with VESTA [45]. The energy barrier was calculated using the climbing image nudged elastic band (CI-NEB) method [46, 47]. Five intermediate images were constructed to simulate the reaction path between the chemisorbed 2H-TPP to the physisorbed 2H-TPP. Due to the computational effort associated with CI-NEB simulations of such large systems, only DFT-D3 calculations were performed in which the convergence criteria were relaxed to 0.03 eV Å−1 per atom.
3. Results and discussion
We have studied the 2H-TPP adsorption on Cu(111) (see figure 1). From earlier work, we know that these kinds of molecules have a large number of meta-stable adsorption configurations due to their high flexibility and the relative uniformity of the Cu(111) surface. The majority of metal-free porphyrins (that have been characterized from theory and experiment) are bound by either physical binding or chemical bonding [30, 31, 48–53] where the porphyrin can be bound in either a 'saddle' or a more strongly bound 'inverted' conformer [54, 55]. Metalloporphyrins are, on the other hand, usually bound chemically through the central metal atom [22]. In part, physisorption seems to be connected to low-temperature deposition and handling since using high-temperature deposition results in chemisorption also for metal-free porphyrins, see e.g. 2H-porphyrin on the Cu(110) surface and 2H-TPP on the Cu(111) surface [56–58].
To complicate matters, whether there is physisorption or chemisorption is not trivially revealed through heights and distances in general, and for molecular adsorption on metal surfaces in particular. Nonetheless, the two different modes of adsorption have different electron density signatures: repulsion for physical binding; and mixing/hybridization for chemical bonding (for a more thorough description, see [59]). Thus, we have determined which configurations are physisorbed and chemisorbed through ELF analysis (see bottom panel in figure 2) where we identify physical binding by the lack of chemical bonds. ELF analysis has been used with considerable success for the analysis of the nature of binding [31, 59]. This approach requires only the information of the system's electron density, and can, thus, replace more complicated methods, such as charge difference density that can lead to difficulties in interpretation due to inherent non-equilibrium components in its computation. This is done through the determination of the sharing of electrons between the molecule and the substrate, which signifies chemisorbed. If there is no sharing of electrons the molecule is physisorbed.
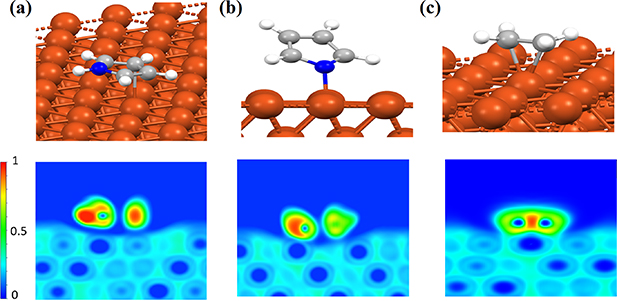
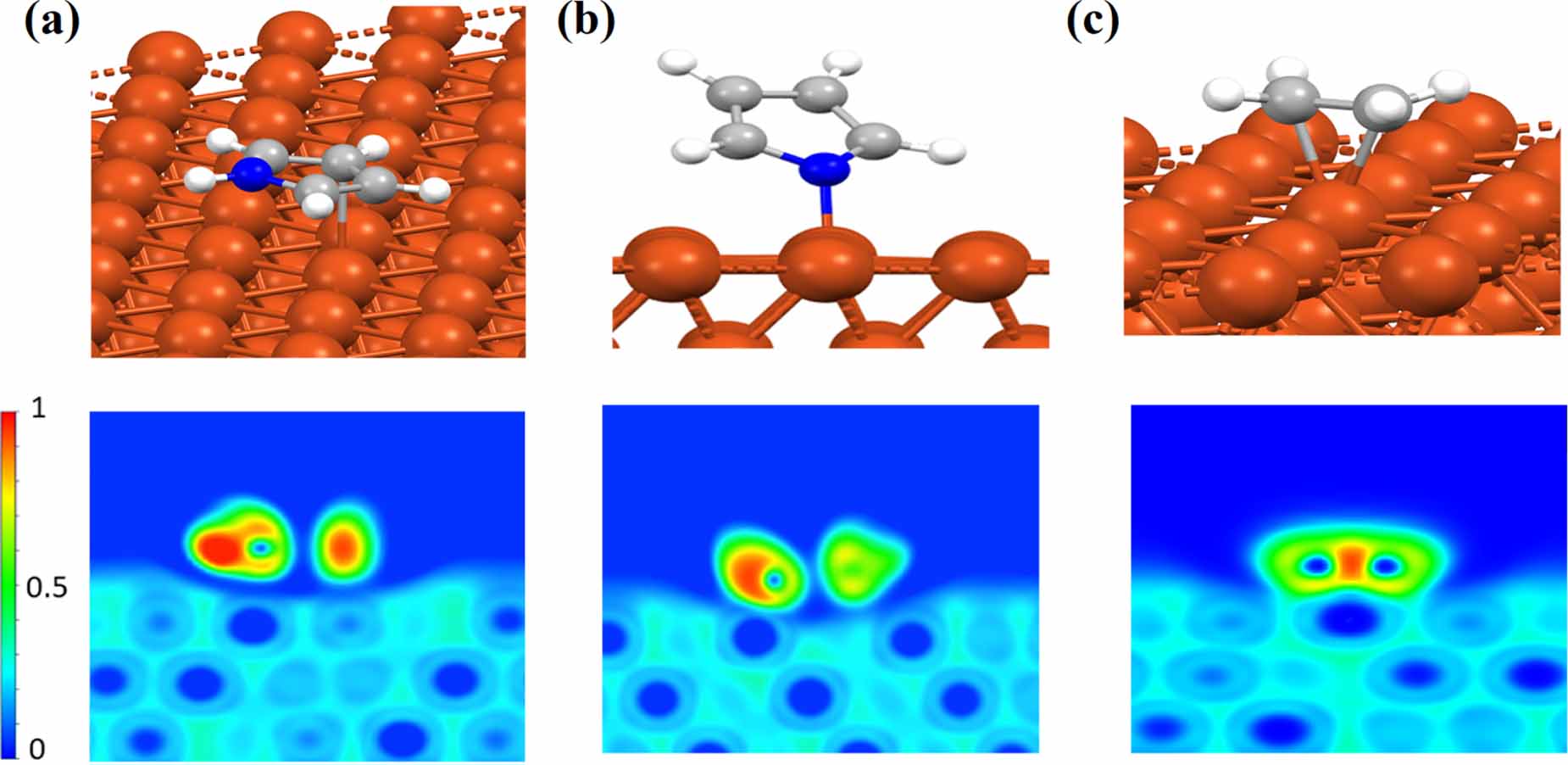
Figure 2. The optimized geometry and 2D-ELF plots of molecular fragments adsorption on Cu(111) for (a) aminic pyrrole, (b) iminic pyrrole and (c) ethene.
Download figure:
Standard image High-resolution imageIn order to see whether both physisorption and chemisorption can be realized for the 2H-TPP molecule on Cu(111), we start our investigation by analyzing the molecular fragments of the 2H-TPP porphyrin ring's interactions with the surface. The porphyrin ring consists of two different kinds of pyrrole conjugations; one is an amine (NH), and one is an imine (N), which are shown in figures 2(a) and (b). The molecules were placed at different binding sites and in different orientations on the Cu slab such that all possible binding sites can be analyzed systematically in a straight-forward way through geometry optimization (using standard structural relaxation techniques) followed by ELF analysis. Here, we present only the most stable configurations (see figure 2). The stable configurations found for the aminic pyrrole molecular fragment (C4H4NH) in the 2D-ELF plot (figure 2(a), lower panel) shows that the aminic N physisorbs to the surface. For the two C atoms opposite N–H we have found a weak chemical interaction with one Cu atom (the 2D-ELF cuts through the center of the CC bond), while the geometric center of the molecule lays on a top position of the Cu(111). Moreover, we found through Bader charge population analysis a diminishingly small amount of charge transfer  (e is the elementary charge) from Cu(111) to C4H4NH. However, the highly reactive iminic molecule in figure 2(b), binds strongly to the Cu atoms at the top position via the nitrogen atom with a significant amount of charge gain of
(e is the elementary charge) from Cu(111) to C4H4NH. However, the highly reactive iminic molecule in figure 2(b), binds strongly to the Cu atoms at the top position via the nitrogen atom with a significant amount of charge gain of  for the C4H4N molecule. Moreover, we also test an ethene as a C=C double bond fragment which chemisorbs more strongly to the top site of Cu(111), as seen in the 2D-ELF plot in the lower panel in figure 2(c), with a significant amount of charge gain
for the C4H4N molecule. Moreover, we also test an ethene as a C=C double bond fragment which chemisorbs more strongly to the top site of Cu(111), as seen in the 2D-ELF plot in the lower panel in figure 2(c), with a significant amount of charge gain  for the C2H4 molecule.
for the C2H4 molecule.
With the interaction elucidation for the molecular fragment interactions with Cu(111) it is evident that the aminic nitrogens (NH) atoms do not form chemical bonds with the underlying Cu atoms contrary to the readily reactive iminic nitrogens (N). With these results in mind, in order to realize a chemisorbed state for the 2H-TPP molecule, the iminic parts of the porphyrin ring and conjugated CC bond in the aminic rings should preferably be centered on top of Cu atoms. Such a configuration was indeed the strongest chemical bonded one found for the 2H-TPP molecule. The most stable geometry of 2H-TPP chemisorbed on the Cu(111) surface is shown in figure 3(a) where both opposite iminic (N) are placed above Cu atoms. Our results for the chemisorbed configuration are in reasonable agreement with the estimated heights from experiments (see below) [51, 57, 60]. The binding energy of the chemisorbed 2H-TPP on Cu(111) is −4.80 eV (−3.77 eV with optB86b-vdWDF), which is a more stable configuration by 0.31 eV in comparison to the physisorbed configuration [59].
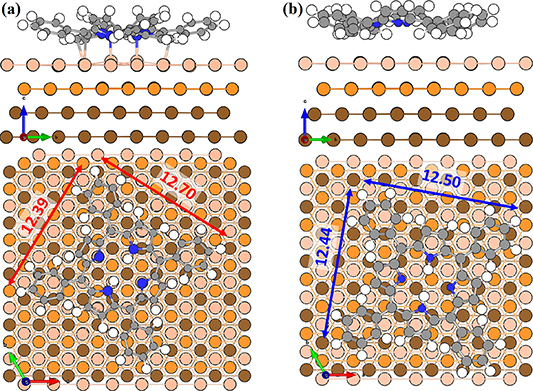
Figure 3. The DFT relaxed geometries of 2H-TPP on the Cu(111) surface (a) chemisorbed 2H-TPP, and (b) physisorbed 2H-TPP in side and top views, respectively. The light to dark shade of orange is used to represent 1st, 2nd, and 3rd layer of Cu(111). The gray, blue and white represent C, N and H respectively.
Download figure:
Standard image High-resolution imageIn the case of the physisorbed 2H-TPP configuration, we found it to be more challenging to converge. We have found the physisorbed configuration by starting by moving the molecule to put the more reactive iminic N atoms centered on three possible starting sites i.e. (i) two opposite hcp-hollow, (ii) two opposite fcc-hollow sites and (iii) one hcp-hollow in combination with one fcc-hollow site (see figure 1(b) for labeling of the surface sites). We have found that (iii) is the only configuration that converges into a physisorbed local minima (see figure 3(b)). However, this agrees well to the most stable physisorption configuration of Br4TPP on Cu(111) [30]. While the other two converge to different chemisorbed configurations.
The binding energy of the most stable physisorbed configuration was calculated to be −4.49 eV (−3.42 eV with optB86b-vdWDF). Configurationally it is similar to the physisorbed conformations previously reported for Br4TPP in [30, 31] with binding energies in a similar range. It is important to note that the successful computation of the physisorbed and the chemisorbed configurations depends on the proper inclusion of dispersion, either in a functional form (vdW-functional) or in a semi-empirical form (DFT + D3). Although the physical binding contribution for each atom may be small, the total vdW contribution to the binding is essential and contributes on the eV scale to the binding energy.
As can be seen in figures 3(a) and (b) the phenyl legs are tilted with regard to the surface, while the free molecule is relatively flat. One can assume that there is a loss in physical binding with the surface of the legs that is compensated by a stronger interaction through the molecular core and decreased steric repulsion. This saddle shape is more pronounced for the chemisorbed configuration. In general, the conformation of molecules on surfaces depends on the balance between binding surface interactions and steric effects. In table 1, we report the largest differences in bond lengths when physisorbed and chemisorbed compared with the gas phase (The bond lengths are listed in full in table S1 in the supplementary material). Overall, we found diminishingly small bond length differences for the converged structures. However, a difference of 0.31 eV (0.36 eV with optB86b-vdWDF) in binding energies was noted.
Table 1. Comparison of bond lengths (in Å) of 2H-TPP in the gas phase, and when either physisorbed or chemisorbed on Cu(111) calculated with DFT-D3 (optB86b-vdWDF), with numbering of atoms according to figure 1(a). All bond lengths are shortened in the bonded states except for one shown in bold.
| Bond | Free molecule | Physisorbed | Chemisorbed |
|---|---|---|---|
| C6–N1 | 1.377 (1.377) | 1.381 (1.381) | 1.391 (1.390) |
| C9–N2 | 1.372 (1.371) | 1.375 (1.374) | 1.393 (1.393) |
| C5–C6 | 1.406 (1.406) | 1.415 (1.416) | 1.415 (1.419) |
| C7–C8 | 1.375 (1.375) | 1.390 (1.391) | 1.406 (1.406) |
| C5–C9 | 1.413 (1.412) | 1.418 (1.419) | 1.433 (1.423) |
| C9–C10 | 1.458 (1.458) | 1.456 (1.455) | 1.430 (1.436) |
| C10–C11 | 1.361 (1.360) | 1.362 (1.361) | 1.377 (1.371) |
Qualitatively, the intramolecular structure of 2H-TPP changes only slightly upon adsorption. However, several subtle changes take place. Upon adsorption, the C9–C10 bond is slightly shortened, while the C6–N1, C9–N2, C5–C6, C7–C8, C5–C9, C10–C11 bonds are slightly elongated (numbering of atoms according to figure 1(a)). All other bond lengths differ by only 0–0.007 Å. In the two most stable 2H-TPP adsorption configurations on Cu(111) (figure 3), the symmetry of the molecule is largely preserved. However, there also exists a physisorbed configuration that is more rectangular, cf. Br4TPP on Cu(111) in [30, 31], with higher energy.
The distances between the 2H-TPP molecule and the surface are presented in table 2. Comparing the distances between the two configurations shows quantitatively the difference in height between physisorption and chemisorption. For the average height of the carbon and nitrogen atoms from the top layer Cu (column one in table 2) we found the physisorbed configuration 0.403 Å higher than the chemisorbed, which is not drastic. In fact, the top-to-bottom height of the saddle shaped 2H-TPP on Cu(111) is much bigger, 1.614 Å for the physisorbed and 1.784 Å for the chemisorbed configuration, as measured discounting the hydrogen atoms (cf figures 3(a) and (b)). In the shortest distances, though, the difference between C–Cu and N–Cu bond lengths and physical binding distances are revealed. The chemisorbed configuration has C–Cu bonds of 2.283 Å, while the shortest distance in the physisorbed case is 2.655 Å. The N–Cu chemical bonds for the iminic nitrogens are 2.169 Å, while in the physisorbed case, the shortest distances between N and Cu are 3.464 Å. The Cu atoms bonded to C are also displaced, mostly in the vertical direction. We note that these Cu atoms have moved towards the molecule enough to be seen visually in figure 3(a).
Table 2. Heights and the shortest distances to Cu for 2H-TPP physisorbed or chemisorbed on Cu(111) in Å.  (C/N) is the average height of the C and N atoms to the Cu surface,
(C/N) is the average height of the C and N atoms to the Cu surface,  (C) and
(C) and  (N) are the minimum heights of a C atom and N atom respectively to the Cu surface;
(N) are the minimum heights of a C atom and N atom respectively to the Cu surface;  (C) and
(C) and  (N) are the shortest distances between a Cu atom and a C and N atom respectively. The distances we get for optB86b-vdWDF are very similar to DFT-D3 (they are listed in table S2 in the Supplementary Material).
(N) are the shortest distances between a Cu atom and a C and N atom respectively. The distances we get for optB86b-vdWDF are very similar to DFT-D3 (they are listed in table S2 in the Supplementary Material).
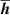 (C/N) (C/N) |
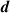 (C) (C) |
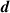 (N) (N) |
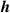 (C) (C) |
 (N) (N) | |||
|---|---|---|---|---|---|---|---|
| Aminic | Iminic | Aminic | Iminic | ||||
| Physisorbed | 3.236 | 2.655 | 3.396 | 3.464 | 2.527 | 3.163 | 3.218 |
| Chemisorbed | 2.833 | 2.283 | 3.207 | 2.169 | 2.164 | 2.659 | 2.167 |
To examine the existence of chemical bonding we, once again, scrutinize the ELF at the interface for the physisorbed and the chemisorbed configuration in figure 4. For both the physisorbed and the chemisorbed configuration, it is clear that the aminic nitrogens (NH) atoms do not form chemical bonds with the underlying Cu atoms. This is consistent with the aminic pyrrole molecule adsorbed on Cu(111) in figure 2. However, for the iminic nitrogens (N), there is a marked difference. In the physisorbed configuration (figure 4(e)) it interacts with the substrate through vdW forces, while in the chemisorbed configuration (figure 4(b)) N forms a chemical bond with a Cu atom. Again, this is consistent with the result of the iminic pyrrole molecular fragment in figure 2. Similarly, as for the physisorbed aminic pyrrole, the C7–C8 carbons in figure 4(f), show ELF signature of physical binding (a blue ribbon between the carbon atoms and the substrate) and chemical bonding in figure 4(c). The chemisorbed case has a 2D-ELF similar to that of ethene chemically bound on Cu(111), see figure 2. This is supported by the difference between the ELF-profiles for the C7–C8 fragments interaction with Cu as given in figure 5, where for the chemisorbed configuration, there is no minimum in the ELF between the C and Cu atom, while for the physisorbed configuration, there is a minimum. A minimum in the ELF-profile, at values between n = 0.05 to n = 0.5, between the interacting atoms, is significant of repulsion in the two constituents and touching atoms, as seen for hydrogen bonds [61] and strong dispersion (cf graphene on a Cu(111) substrate in [59]).
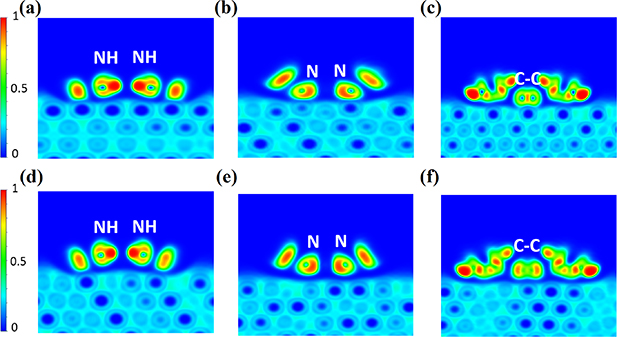
Figure 4. ELF plots along the planes passing through the chemisorbed/physisorbed 2H-TTP@Cu(111) (a)/(d) aminic pyrrole rings, (b)/(e) iminic pyrrole rings and (c)/(f) C7–C8 of aminic pyrrole ring (for numbering of the carbon atoms see figure 1).
Download figure:
Standard image High-resolution image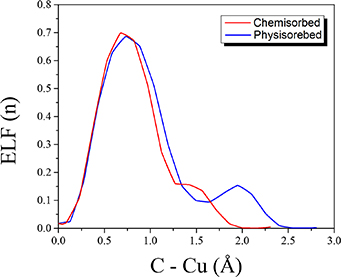
Figure 5. ELF-profiles plots of the chemisorbed and physisorbed configuration of 2H-TPP on Cu(111) between a pyrrole carbon and copper atom from the center of the C-atom core at 0 Å to the Cu core at 2.30 Å (chemisorbed configuration) and 2.80 Å (physisorbed configuration).
Download figure:
Standard image High-resolution imageOnly the chemisorbed configuration has sharing of electrons (chemical bonds) and electronic states that are hybridized, with mixing between the molecule and the Cu-surface. There is, thus, a difference in the binding mechanism between the two configurations. However, we note that both configurations have binding energies of over ∼3 eV. The situation with molecules being attached strongly to a surface by either physical binding or chemical bonding is rather unusual. In fact, the strong physical binding found for 2H-TPP that does not involve bond dipoles (Keesom binding) has only been described for a limited set of cases. Physisorbed molecules are usually bound more loosely.
The ELF analysis shows that there are physical binding contributions from the aminic nitrogens (NH) and the phenyl legs for both configurations. The only chemical bonds formation contributions come from the iminic nitrogens and the pyrrole C7–C8 carbons, that also show significant charge redistributions. Evidently, the labeling of the configurations as physisorbed and chemisorbed has been done based on the ELF analysis. For a quantitative measure of the electron transfer between the surface and the molecule, we have performed a Bader charge population analysis [60]. The analysis shows a stark difference in net charge transfer between the 2H-TPP molecule and the Cu(111) surface for the chemisorbed and physisorbed cases. In the case of chemisorbed 2H-TPP the total charge transfer is calculated  , whereas it is diminishingly small for the case of physisorbed
, whereas it is diminishingly small for the case of physisorbed  .
.
In figure 6, we compare the DOS for 2H-TPP in the gas phase with the molecular projected DOS (PDOS) of the adsorbed molecules. It should be noted here that a Gaussian smearing function (sigma value of 0.025 eV) was used in order to reproduce the molecular states. The DOS was calculated using 1000 points to have a good resolution.
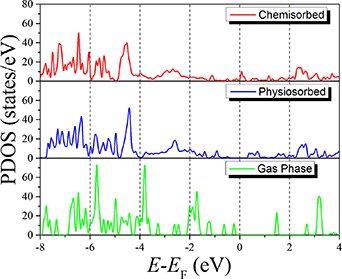
Figure 6. The molecular projected density of states (PDOS) for free 2H-TPP (gas phase) compared to 2H-TPP@Cu(111) in chemisorbed and physiosorbed configuration using (DFT + D3). The energy scale is relative to the Fermi energy.
Download figure:
Standard image High-resolution imageIn accordance with what was found for Br4TPP@Cu(111), we find significant changes in the DOS between the molecule in the gas phase and the physisorbed phase. The pronounced molecular states, appearing as sharp peaks in the DOS of the molecule in the gas phase [31], are seen to interact with the metallic states of the Cu slab causing the peaks to be more smeared out. In addition, the highest occupied molecular orbital (HOMO) is seen to be shifted from around −0.2 eV to −1.0 eV and the lowest unoccupied molecular orbital (LUMO) from 1.5 eV to 0.5 eV. Thus, forming a HOMO-LUMO gap of around 1.5 eV.
The DOS of the chemisorbed molecule is very similar to that of the physisorbed molecule. However, the spectrum is shifted to lower energies (around 0.5 eV). As a result, the smeared-out LUMO states in the physisorbed DOS are approaching the Fermi level. This is the consequence of the Fermi level shift for the chemisorbed molecule because of the charge transfer from the Cu slab, as was found in the Bader analysis.
We further investigate the differences in the molecular DOS, we have simulated scanning tunneling microscopy (STM) images. The simulated STM images were calculated using the Tersoff-Hamann approximation [62, 63] at a constant current for different sample bias voltages, as shown in figure 7. The p4vasp software package [64] was used to generate the virtual STM images, and the iso-density values were adjusted for each case to produce the best possible images. The simulated STM images should be proportional to the local DOS and the results certainly reflect the molecular DOS for the two cases with only minor observable changes in the STM images. This is attributed to the fact that the same 2H-TPP orbitals are concerned in this energy region for both configurations.

Figure 7. Simulated STM images at constant current for the chemisorbed and physisorbed configurations. The range of values shown at the top specifies the energy range with respect to EF of the bands that are used for the evaluation of the partial charge densities.
Download figure:
Standard image High-resolution imageSince 2H-TPP (and Br4TPP, and similar metal-free porphyrins) can physisorb on Cu(111) in low-temperature conditions, but chemisorb when deposited at higher temperatures, we have calculated the energy barrier between the two configurations using the CI-NEB method going between the most stable chemisorbed and physiosorbed configuration (see figure 8) using the PBE-D3 method. In this simulation, the bottom layer of the slab model was kept fixed and we defined the shortest reaction path by both translation and rotation of the 2H-TPP molecule as shown in figure 8. Overall, the molecular center shifts through a very short path distance of ∼1.54 Å (see figure 8(a)). For such a short diffusion we have simulated the reaction path through five intermediate images and the resulting energy barrier height of 0.12 eV was obtained for going from physisorption to chemisorption, as shown in figure 8(b). This barrier should be seen in the context of the relatively small energy difference between the two configurations of 0.31 eV (0.36 eV with optB86b-vdWDF). More reactive molecules might chemisorb to metals barrier-less or with almost negligible barriers, but cases with barriers have also been reported in the literature [65].

Figure 8. (a) Schematic representing the diffusion of 2H-TPP between chemisorbed (in red) and physiosorbed (in blue) states on the Cu(111) surface. (b) The barrier energy that 2H-TPP needs to overcome to switch between the chemisorbed (in red) configuration and physisorbed (in blue) configuration. The short reaction path is show in detail in the enlarged inset. The  E is the relative barrier energy compared to the physisorbed configuration of 2H-TPP.
E is the relative barrier energy compared to the physisorbed configuration of 2H-TPP.
Download figure:
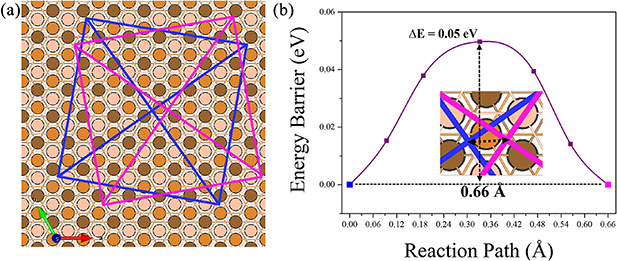
Standard image High-resolution imageTo be able to achieve either physisorption or chemisorption selectively can be important to accomplish certain desired interface properties for molecules on surfaces [66, 67]. For example, chemical bonds provide electrical wiring that could result in Ohmic contact between the molecule and substrate, while physical binding usually results in a Schottky barrier through tunneling of electrons. For 2H-TPP on Cu(111) the two modes of adsorption render very similar DOS, but only the chemisorbed configuration has hybridized states with sharing of electrons between Cu and the molecule and it is expected that these shared states would have an impact on the electron transport and its mechanism. On the other hand, the repulsion between the electron densities of the substrate and a physisorbed molecule offers isolation that could result in longer spin-lifetimes for magnetic molecules. In the case of 2H-TPP on Cu(111), both chemisorption and physisorption are strong enough to result in molecular immobilization on the surface, with a calculated reaction barrier of 0.12 eV between the two local equilibrium states. In addition, the barrier between neighboring physisorbed states was computed to be 50 meV (as seen in figure 9), which is larger than expected. This indicates that the physisorbed state can exist as a metastable configuration rather than being unrestrictedly mobile or transient, especially at low temperature.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 9. (a) Schematic representing the diffusion of 2H-TPP physisorbed at one site (in blue) to a neighboring physiosorbed site (in magenta) on the Cu(111) surface. (b) The short reaction path is show in detail in the enlarged inset. The  E is the relative barrier energy between two neighboring physisorbed configurations of 2H-TPP.
E is the relative barrier energy between two neighboring physisorbed configurations of 2H-TPP.
Download figure:
Standard image High-resolution image{kind=link}
The presence of a barrier could enable control of the binding mechanism upon deposition, which provides a device design strategy at the nanoscale for both single-molecule devices and molecular thin-films. It opens up for the tailoring of the interface properties, for example, electron transport properties or lifetimes of excited states invoked in the molecule.
4. Conclusions
We have studied the two lowest energy configurations of 2H-TPP on Cu(111) and have found that the most stable one is chemisorbed, while the other one is physisorbed, 0.31 eV higher in energy. Their binding energies to the copper substrate are −4.80 and −4.49 eV.
Their mode of binding has been determined using the ELF, which offers a straightforward way to differentiate between strong physical binding and weak chemical bonding. The chemisorbed configuration has an average height of 0.403 Å lower than the physisorbed one. A Bader analysis has revealed that there is negligible charge transfer from the surface to the molecule in physisorbed case but −0.5 e are transferred from the surface to the molecule for chemisorption. The 2H-TPP PDOS, when adsorbed on copper, are significantly different from the free molecular DOS for both configurations, which is remarkable since large changes are traditionally associated with chemisorption but not with physisorption. We, thus, make note that large changes compared to the free molecule's DOS can also be due to strong physical binding. The similarity of the PDOS in the region around the Fermi level of both the physisorbed and chemisorbed molecule is also remarkable and is attributed to the large component of physisorption for all atoms in both configurations, except the iminic nitrogens and the pyrrole carbons that in the chemisorbed case is chemically bonded to copper atoms. We have compared the first principles inclusion of dispersion in the semi-empirical inclusion of dispersion through DFT-D3 with the optB86b-vdWDF functional. In comparison, the geometrical changes were noted to be insignificantly small, whereas in case of the optB86b-vdWDF functional, the binding energies were calculated to be lower than the binding energies calculated with the DFT-D3. We have computed the barrier going from the physisorbed to the chemisorbed configuration to 0.12 eV, which offers an explanation to why metal-free porphyrins can be found to be either physisorbed or chemisorbed to copper substrates, depending on the deposition conditions.
Acknowledgments
We want to thank the Knut and Alice Wallenberg Foundation, Kempe Foundations for their financial support. We are thankful for the allocation of time and resources at the Swedish National Infrastructure for Computing (SNIC) and National Academic Infrastructure for Super-computing in Sweden (NAISS) at HPC2N, PDC and NSC partially funded by the Swedish Research Council through Grant Agreement No. 2018-05973.
Data availability statement
All data that support the findings of this study are included within the article (and any supplementary files).
Supplementary data Convergence test, Atomic distances (0.2 MB PDF)