
Abstract
The structural stability, spontaneous polarization, piezoelectric response, and electronic structure of AlN and GaN under uniaxial strain along the [0001] direction are systematically investigated using HSE06 range-separated hybrid functionals. Our results exhibit interesting behavior. (i) AlN and GaN share the same structural transition from wurtzite to a graphite-like phase at very large compressive strains, similarly to other wurtzite semiconductors. Our calculations further reveal that this well-known phase transition is driven by the transverse-acoustic soft phonon mode associated with elastic instabilities. (ii) The applied tensile strain can either drastically suppress or strongly enhance the polarization and piezoelectricity, based on the value of the strain. Furthermore, large enhancements of polarization and piezoelectricity close to the phase-transition regions at large compressive strains are predicted, similar to those previously predicted in ferroelectric fields. Our calculations indicate that such colossal enhancements are strongly correlated to phase transitions when large atomic displacements are generated by external strains. (iii) Under the same strain, AlN and GaN have significantly different electronic properties: both wurtzite and graphite-like AlN always display direct band structures, while the the bandgap of wurtzite GaN is always direct and that of graphite-like GaN always indirect. Furthermore, the bandgap of graphite-like AlN is greatly enhanced by large compressive strain, but that of wurtzite GaN is not sensitive to compressive strain. Our results are drastically different from those for equibiaxial strain (Duan et al 2012 Appl. Phys. Lett. 100 022104).
Export citation and abstract BibTeX RIS
1. Introduction
Aluminum nitride (AlN) and gallium nitride (GaN), which usually crystallize in the wurtzite structure at ambient conditions, have been extensively investigated owing to their abundant physics and practical applications [1–3]. The structural properties of wurtzite semiconductors are sensitive to external pressure. Recently, intensive theoretical studies have found that there is an intermediate graphite-like phase between wurtzite and rocksalt at high pressures and the three structures are fivefold, fourfold and sixfold coordinated, respectively [4–8]. This is consistent with the well-known fact that external pressure favors more close packed structures, thereby resulting in the increasing coordination number attributed to the bond-length evolution. Since the graphite-like structure is metastable and is difficult to stabilize by applying external pressure, few relevant experimental results have been reported so far. As a valid alternative, accurate first-principles simulations can complement these studies and provide detailed descriptions of the structural and bonding behavior under extreme conditions. The internal lattice parameter u and lattice ratio c/a are good measures for representing the deviation from ideal wurtzite structure but are not sufficient to describe a phase transition to a different symmetry. It is well known that dynamic and elastic instabilities are often responsible for phase transitions. However, due to the discontinuities in the volume, energy and other relevant properties at critical pressures, there are always other driving forces for first-order phase transitions [9, 10]. It is noteworthy that the wurtzite and graphite-like phases, with the space groups P63mc and P63/mmc, respectively, both show hexagonal symmetry. Therefore, study of the effects of pressure on the dynamic and elastic properties will be more helpful in understanding the underlying mechanisms for these phase transitions. Since the graphite-like phase shows distinct physical properties from the wurtzite and rocksalt phases, such pressure-induced structural variations are attractive technologically, such as in the development of high-quality optoelectronic devices. In this paper, our initial work is to reveal the structural transformations as well as the underlying mechanisms for AlN and GaN subjected to uniaxial strain.
Wurtzite structure possesses the highest symmetry compatible with the coexistence of spontaneous polarization and piezoelectric response [11], which are both quite sensitive to changes of the structural parameters. In the presence of external pressure, the electric polarization is divided into spontaneous and piezoelectric parts. The ground-state polarization and piezoelectricity of binary III–V nitrides have been systematically investigated [12–15]. Our recent results show that hydrostatic pressures greatly enhance the polarization and piezoelectricity of AlN, with the maxima appearing simultaneously at the phase transition from wurtzite to graphite-like [16]. The spontaneous polarization and piezoelectric fields modify the band edges, thereby significantly influencing the optical properties of the heterostructures or supercells [12]. Therefore, it is very important to find a feasible way to control the polarization and piezoelectricity in electronic and optical devices. For piezoelectric devices, ZnO has been predicted as a key enabling material due to its strong spontaneous and piezoelectric polarizations [17]. Furthermore, AlN displays much better piezoelectric performance than ZnO at ambient conditions [18], therefore gaining extensive attention for potential applications. As a result, it will be very useful to find a way to further enhance the piezoelectric response for technological applications. At present, such investigations are mainly focused on ferroelectric materials, which have a wide range of applications in medical imaging, detectors, actuators, telecommunication, and ultrasonic devices [19–21]. It has been revealed that polarization rotation near the phase-transition regions results in giant electromechanical effects for ferroelectric perovskites [22]. Our recent calculations point out that the maximum piezoelectric value of AlN under hydrostatic pressures, appearing at the phase transition to graphite-like, is increased by a factor of ∼97 from the equilibrium value and is much larger than those of most common ferroelectric perovskites [16]. However, the polarization always remains along the c axis and no polarization rotation is observed. Therefore, the driving mechanisms are drastically different from those of ferroelectric materials and more work is needed to further explain such phenomena. Such investigations are helpful in broadening the opportunities for piezoelectric applications of wurtzite semiconductors and in better understanding the driving mechanisms of giant piezoelectric responses for ferroelectric materials. In this paper, one area we are interested in is how uniaxial strain affects the piezoelectric response of AlN and GaN, especially near the phase transition.
AlN and GaN have large direct bandgaps [18]; thereby environmentally benign Al (Ga)-based semiconductors always have relatively large bandgaps. Such materials have been widely used in optoelectronic devices such as blue–green light emitting diodes and laser diodes as well as high-power, high-temperature, and high-frequency electronic devices [23]. It is well known that the structural and electronic properties of semiconductors can be altered by either applying external pressure or inducing internal strain, or by both, to obtain the desired physical and electrical properties [5, 24]. Since the interatomic distances and relative positions of atoms have a strong influence on the band structure, it is possible to adjust the bandgap (Eg) by uniaxial strain along the [0001] direction. The effects of external strain on the structural and electronic properties of GaN and ZnX (X = O,S, Se and Te) have been systematically investigated using standard density functional theory (DFT) [15, 25–27]. In those works, uniaxial strain along the [0001] direction and equibiaxial strain in the (0001) plane were believed to have an equivalent influence on the band structures. Recently, our results have shown that although AlN and GaN under in-plane strain share the same structural transition from wurtzite to a graphite-like phase, their electronic properties are significantly different. Furthermore, it is more difficult for AlN than for GaN to obtain the graphite-like semi-metallic phase [28]. Such effects of strain on the properties of common semiconductors are not only intriguing in physics, but also attractive technologically, since strain-induced Eg tuning can be used for photovoltaic applications. In this paper, another area we are interested in is to systematically reveal how uniaxial strain affects the band structures of AlN and GaN, especially near the phase transitions.
Since the covalent bonding in semiconductors is highly directional, strain effects depend on the loading direction. In this paper, we study the uniaxial-strain effects on the structural transitions, polarizations, piezoelectric responses and electronic structures of AlN and GaN using total energy as well as linear response calculations. Our work differs from the previous studies in three main aspects. (1) Previous investigations have just obtained the structural transition from wurtzite to the graphite-like phase from the evolution of lattice constants with strain. However, the underlying mechanism still remains unknown. In our work, one key aim is to reveal the driving forces of such phase transitions based on dynamic and elastic properties. (2) We focus on the effects of uniaxial strain on the polarization and piezoelectric response, especially near the phase-transition regions, in order to point out a feasible way to enhance the electromechanical response. However, such systematic studies have seldom been made in previous work. (3) To overcome the bottleneck of severe underestimation of Eg in standard DFT, Heyd–Scuseria–Ernzerhof (HSE06) hybrid functionals [29, 30] are adopted in this work, which have been proved to be highly reliable [28, 31, 32]. However, the previous studies have mainly been performed using standard DFT, which is unable to accurately describe the variation of Eg with strain. The structure of this paper is organized as follows. In the next section we describe the computational methods used in this work. In section 3, we report the main results of this work. Finally, a brief summary is given in section 4.
2. Computational methods
The total energy and band structure calculations are performed using the projector augmented-wave method [33] as implemented in the VASP code [34]. The electronic wavefunctions are described using a plane wave basis set with an energy cutoff of 650 eV. A Monkhorst–Pack k-point mesh of 6 × 6 × 6 is used throughout the calculations to obtain well-converged results. The standard exchange mixing, which contains 25% Hartree–Fock and 75% Perdew–Burke–Ernzerhof-GGA (PBE-GGA) [35], is employed in the HSE06 hybrid functional. To check the setting of the mixing ratio AEXX (denoted in the VASP code), figure 1 shows the calculated Eg as a function of AEXX for equilibrium AlN and GaN; the ground-state experimental Eg values are listed for comparison. Good agreement is achieved at AEXX = 0.25, especially for AlN, supporting the standard exchange mixing adopted in our work. The incorporation of an appropriate portion of the nonlocal exact exchange into the local or semilocal exchange (HSE06 calculations) can yield reasonable bandgaps compared to standard DFT (which will be systematically discussed in the following). Hartree–Fock cannot describe the van der Waals forces since it only accounts for exchange. We have also checked the influence of van der Waals forces on the results for the wurtzite and graphite-like phases, similarly to previous work [36], and found that the main results do not change after such forces have been incorporated into the HSE06 calculations. Therefore, our results demonstrate that the long-range van der Waals forces originating from nonlocal electron–electron correlation do not have a significant influence on the properties of the GaN and AlN systems.

Figure 1. The calculated Eg for equilibrium AlN and GaN as a function of the exchange mixing ratio AEXX. The open symbols refer to the calculated results and the solid ones refer to the ground-state experimental data.
Download figure:
Standard imageThe phonon-dispersion calculations are performed with the direct method implemented in the PHONOPY package [37, 38]. This method uses the Hellmann–Feynman forces calculated for an optimized supercell through VASP [34]. In theory, the larger the supercell, the more accurate the dispersion curves that are obtained. However, since large supercell HSE06 calculations are currently not computationally feasible, we adopt the 2 × 2 × 3 supercell for all phonon calculations and an accuracy of 0.05 THz for the highest optical zone-center phonon frequency is achieved. The piezoelectric constants eiν and elastic constants cμν (here Roman indices go from 1 to 3, and Greek ones from 1 to 6) are calculated using the density functional perturbation theory (DFPT) [39] of the linear response of strain type perturbations [40]. The electric polarization (P) is calculated using the well-known Berry-phase approach [41]. To find the strain-free lattice constants, the lattice vectors and atomic coordinates are fully relaxed until the Hellmann–Feynman force acting on each atom is reduced to less than 0.02 eV Å−1. In the presence of uniaxial strain ( ), relaxation is performed with the lattice constant c fixed until the following conditions are satisfied within a small tolerance: σ11 = σ22 < 0.02 GPa and σij = 0 for i ≠ j, where σ11 and σ22 are the stresses in the (0001) plane. The tensor σ33 is the externally applied uniaxial stress along the [0001] direction and is adjusted by changing step by step. We have examined the accuracy of our calculations by comparing the ground-state results with experimental and other theoretical data [18, 42]. As summarized in table 1, good agreement between theoretical and experimental methods is achieved.
), relaxation is performed with the lattice constant c fixed until the following conditions are satisfied within a small tolerance: σ11 = σ22 < 0.02 GPa and σij = 0 for i ≠ j, where σ11 and σ22 are the stresses in the (0001) plane. The tensor σ33 is the externally applied uniaxial stress along the [0001] direction and is adjusted by changing step by step. We have examined the accuracy of our calculations by comparing the ground-state results with experimental and other theoretical data [18, 42]. As summarized in table 1, good agreement between theoretical and experimental methods is achieved.
Table 1. Lattice constants (a,c/a and u), Eg and elastic constants (cij) for ground-state wurtzite AlN and GaN. a,Eg and cij are in Å, eV and GPa, respectively. The experimental values are from [18] and other theoretical data are from [42].
| a | c/a | u | Eg | c11 | c12 | c13 | c33 | c44 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| AlN: | Ours | 3.064 | 1.602 | 0.381 | 6.12 | 406 | 138 | 106 | 383 | 122 |
| Exp. | 3.111 | 1.600 | 0.380 | 6.19 | 411 | 149 | 99 | 389 | 125 | |
| Others | 3.103 | 1.602 | 0.382 | 410 | 142 | 110 | 385 | 123 | ||
| GaN: | Ours | 3.232 | 1.630 | 0.376 | 3.02 | 335 | 132 | 99 | 373 | 86 |
| Exp. | 3.190 | 1.627 | 0.377 | 3.17 | 377 | 160 | 114 | 209 | 81 | |
| Others | 3.180 | 1.626 | 0.377 | 369 | 132 | 96 | 406 | 102 |
3. Results and discussion
In the presence of out-of-plane stress, a new lattice parameter c corresponds to a specified uniaxial strain given by = 33 = (c − c0)/c0 and a commensurate equibiaxial in-plane stress-free strain given by 11 = 22 = (a − a0)/a0, where a0 and c0 are the strain-free lattice constants. The top panels in figure 2 summarize the calculated dependence of σ33, which displays the same trends for AlN and GaN. The tensile σ33 all first increase and then decrease with , with a maximum of ∼45 GPa (34 GPa) appearing at = 0.16 (0.14) for AlN (GaN). This indicates that the calculated ideal tensile strengths along the polar c axis are ∼45 GPa for AlN and ∼34 GPa for GaN, which are the maximum stresses required to break the perfect crystals. The structural instabilities at large are emphasized by the dependence of the elastic constants c13 and c33, which become negative when > 0.16 for AlN and > 0.14 for GaN, as shown in the bottom panels of figure 2. On the other hand, for compressive , the σ33 magnitudes drastically increase when −0.08 < < 0 for AlN and −0.12 < < 0 for GaN, which is well known and anticipated, consistent with previous theoretical findings [16]. The most unexpected finding is that the magnitude of σ33 first decreases and then increases with further increase of , with the minimum appearing at =− 0.14 (−0.18) for AlN (GaN). The abnormal behavior of σ33 when −0.14 < <− 0.08 for AlN and −0.18 < <− 0.12 for GaN is attributed to the evolution of the elastic constant c33, which always decreases with compressive and becomes negative in these ranges, as shown in figure 2(b). In the abnormal ranges, although the elastic stability criteria c44 > 0 and c11 > |c12| are true,  is false, indicating that the wurtzite structures become elastically unstable. More interestingly, after the discontinuous changes from negative to positive at =− 0.14 (−0.18) for AlN (GaN), the c13 and c33 increase linearly with further increase of compressive . The new elastic trends lead to great enhancement of the σ33 magnitudes and the reappearance of elastic stabilities for high-pressure structures in these ranges. As a conclusion, phase transitions are usually predicted at the critical .
is false, indicating that the wurtzite structures become elastically unstable. More interestingly, after the discontinuous changes from negative to positive at =− 0.14 (−0.18) for AlN (GaN), the c13 and c33 increase linearly with further increase of compressive . The new elastic trends lead to great enhancement of the σ33 magnitudes and the reappearance of elastic stabilities for high-pressure structures in these ranges. As a conclusion, phase transitions are usually predicted at the critical .
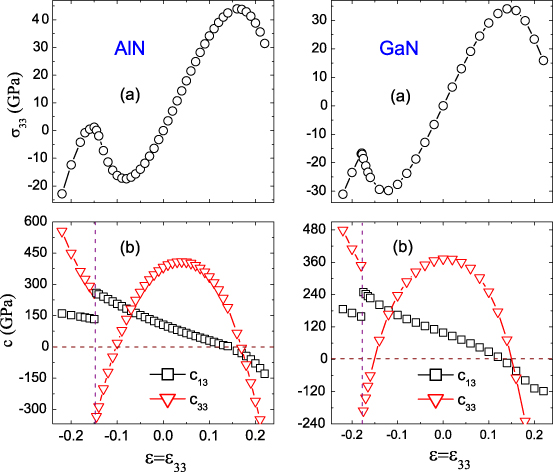
Figure 2. (a) Out-of-plane stress σ33, and (b) elastic constants c13 and c33 as a function of .
Download figure:
Standard imageSince dynamic instabilities are often responsible for structural transformation, five phonon-dispersion curves at different values are plotted in figure 3 for AlN and GaN. The previous related works were mainly performed using standard DFT, where the DFT-PBE (LDA) always overestimates (underestimates) the optimized volume of the unit cell at fixed . This leads to very weak (strong) nearest-neighbor force constants and a significant underestimation (overestimation) of phonon frequencies, especially for the optical modes [43]. The HSE06 adopted in this work has been proven to be more accurate than the DFT-PBE and LDA for the phonon frequency and dispersion [43]. The reciprocal lattice point (0, 0, 0.06) near the zone center is denoted hereafter as Y, owing to its interesting behavior, as shown in panel (b). There are twelve phonon branches for the wurtzite unit cell: one longitudinal-acoustic (LA), two transverse-acoustic (TA), three longitudinal-optical (LO), and six transverse-optical (TO) ones, as shown in panel (d). Since the Al (Ga) atom is heavier than the N atom, the internal vibrations of Al (Ga) atoms are mainly correlated to the low-frequency modes. On the other hand, due to the difference between Al and Ga atoms, the modes of AlN shift to higher frequencies than those of GaN at different . The six TO modes are separated from the other branches by the well-known phonon bandgap for the wurtzite phase, which is most noticeable in the equilibrium structure, as shown in figure 3(d). At ambient conditions, the gap of GaN is larger than that of AlN. Our calculated results further reveal that the tensile always reduces the gaps for AlN and GaN, whereas the gaps are first reduced when −0.14 < < 0 for AlN and −0.18 < < 0 for GaN and then enlarged unexpectedly as the compressive increases further. This abnormal phonon behavior is consistent with the novel elastic trends and emphasizes the occurrence of phase transition at the critical .
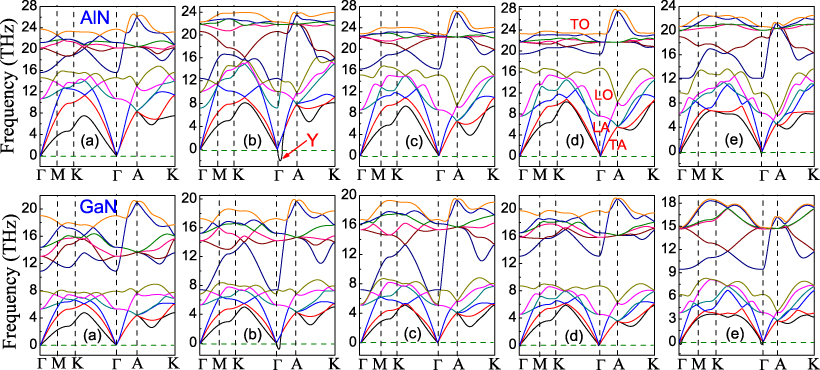
Figure 3. Phonon-dispersion curves for AlN and GaN. AlN: (a) =− 0.18, (b) =− 0.14, (c) =− 0.11, (d) = 0.0 and (e) = 0.17; GaN: (a) =− 0.20, (b) =− 0.18, (c) =− 0.17, (d) = 0.0 and (e) = 0.16.
Download figure:
Standard imageIt is shown that the phonon modes become unstable when > 0.16 for AlN and > 0.14 for GaN (see figure 3(e)). In other words, the wurtzite phase is stabilized by tensile when < 0.16 (0.14) for AlN (GaN), consistent with the results from the elastic properties. Our investigations further reveal that the wurtzite structure remains stable for a broad compressive range until the TA frequencies at Y become imaginary near =− 0.14 for AlN and =− 0.18 for GaN, as shown in figure 3(b). This is drastically different from the results for the elastic properties, where elastic instabilities are observed when −0.14 < <− 0.08 for AlN and −0.18 < <− 0.12 for GaN; phonon instabilities are still not obtained at =− 0.11 (−0.17) for AlN (GaN) (see figure 3(c)). The most interesting finding is that when <− 0.14 for AlN and <− 0.18 for GaN, the modes become stable once again, as shown in figure 3(a). The anomalous behavior of the phonon modes confirms the occurrence of a phase transition at the critical . Schematic representations of the eigenvectors for the TA(Y) soft phonon modes at the critical are shown in figure 4. The eigenvectors of the Al (Ga) cation and the N anion are parallel and along the [110] direction. Since the particular atomic displacements are responsible for the soft modes, which result in the structural instabilities, the atomic movements along this particular direction are closely correlated with the new-found phase transitions.
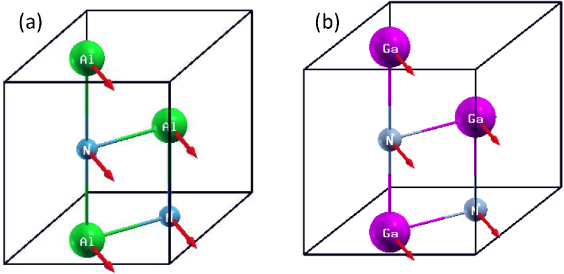
Figure 4. Atomic displacements along the [110] direction, perpendicular to the Al–N (Ga–N) plane, at the critical TA(Y) mode softening strains: (a) =− 0.14 for AlN and (b) =− 0.18 for GaN.
Download figure:
Standard imageTo reveal the predicted structure, we systematically study the evolution of the in-plane strain 11, lattice ratio c/a, internal lattice parameter u, and Born effective charge  with respect to , as shown in figure 5. When >− 0.14 for AlN and >− 0.18 for GaN, the calculated 11, u, and
with respect to , as shown in figure 5. When >− 0.14 for AlN and >− 0.18 for GaN, the calculated 11, u, and  display a nonlinear response with respect to ; a linear trend is observed for c/a. When <− 0.14 (−0.18) for AlN (GaN), c/a < 1.20 and u = 0.50. This implies the structural transition from wurtzite to a graphite-like phase, similar to those previously predicted for other forms of pressures [15, 16, 26–28]. Therefore, the new structure predicted by elastic and phonon instabilities at large compressive is the graphite-like phase, whose structure is shown in figure 6, together with the wurtzite structure for comparison. Furthermore, the predicted transition values are in good agreement with those from the evolution of c/a and u. As a result, the TA soft phonon modes associated with the elastic instabilities are the driving forces for such well-known phase transitions. In the graphite-like phase, each cation (anion) has bonds with two adjacent anions (cations) with the same bonding length along the c axis, in addition to bonds with three anions (cations) in the basal plane.
display a nonlinear response with respect to ; a linear trend is observed for c/a. When <− 0.14 (−0.18) for AlN (GaN), c/a < 1.20 and u = 0.50. This implies the structural transition from wurtzite to a graphite-like phase, similar to those previously predicted for other forms of pressures [15, 16, 26–28]. Therefore, the new structure predicted by elastic and phonon instabilities at large compressive is the graphite-like phase, whose structure is shown in figure 6, together with the wurtzite structure for comparison. Furthermore, the predicted transition values are in good agreement with those from the evolution of c/a and u. As a result, the TA soft phonon modes associated with the elastic instabilities are the driving forces for such well-known phase transitions. In the graphite-like phase, each cation (anion) has bonds with two adjacent anions (cations) with the same bonding length along the c axis, in addition to bonds with three anions (cations) in the basal plane.

Figure 5. (a) In-plane strain 11, (b) lattice ratio c/a, (c) internal lattice parameter u (in units of the lattice c), and (d) Born effective charge  as a function of . The transition from wurtzite to a graphite-like phase occurs at ≅− 0.14 (−0.18) for AlN (GaN) (dashed line) and the charge satisfies
as a function of . The transition from wurtzite to a graphite-like phase occurs at ≅− 0.14 (−0.18) for AlN (GaN) (dashed line) and the charge satisfies  .
.
Download figure:
Standard image
Figure 6. (a) The equilibrium wurtzite unit cell and (b) the graphite-like phase unit cell under large compressive uniaxial strain. The c axis is along the [0001] direction in each case. Adapted with permission from [27]. Copyright 2010 American Institute of Physics.
Download figure:
Standard imageThe electric polarization (P) originates from two contributions: the lack of centrosymmetry and the deviation from the ideal wurtzite structure for which  and u = 0.375. The former is characterized by u, which describes the shortest bonding length between two adjacent atoms along the c axis; the latter has a strong influence on
and u = 0.375. The former is characterized by u, which describes the shortest bonding length between two adjacent atoms along the c axis; the latter has a strong influence on  . The u and
. The u and  , whose dependence is shown in figures 5(c) and (d), are responsible for P. Large u corresponds to high symmetry along the polar direction, which suppresses P, whereas large
, whose dependence is shown in figures 5(c) and (d), are responsible for P. Large u corresponds to high symmetry along the polar direction, which suppresses P, whereas large  enhances P. As increases from compressive to tensile, u and
enhances P. As increases from compressive to tensile, u and  both first decrease and then increase for wurtzite AlN and GaN. On the other hand, the graphite-like phase shows a higher symmetry than the wurtzite phase, with the coordination number increasing from four to five. It is well known that the wurtzite phase displays the highest symmetry compatible with the coexistence of spontaneous P and piezoelectric response. The graphite-like structure does not possess polar and piezoelectric properties. In other words, the foregoing structural transformation is a polar–nonpolar phase transition, which is similar to that from the ferroelectric to the paraelectric phase in a ferroelectric field [19–21].
both first decrease and then increase for wurtzite AlN and GaN. On the other hand, the graphite-like phase shows a higher symmetry than the wurtzite phase, with the coordination number increasing from four to five. It is well known that the wurtzite phase displays the highest symmetry compatible with the coexistence of spontaneous P and piezoelectric response. The graphite-like structure does not possess polar and piezoelectric properties. In other words, the foregoing structural transformation is a polar–nonpolar phase transition, which is similar to that from the ferroelectric to the paraelectric phase in a ferroelectric field [19–21].
Figure 7(a) shows P as a function of . For equilibrium wurtzite AlN and GaN, the spontaneous P values are 0.098 C m−2 and 0.027 C m−2, respectively, which are consistent with the other corresponding theoretical values of 0.081 and 0.027 C m−2 [13, 3]. For the ideal wurtzite structure, the spontaneous P is nonvanishing due to the nonsuperposition of positive and negative electric charge centers along the polar c axis. When <− 0.14 (−0.18) for AlN (GaN), P = 0, emphasizing the structural transition from the wurtzite to the graphite-like phase. Our calculated results further reveal that P displays the same trend as u and  for the wurtzite phase, whose values first decrease and then increase with . This implies that
for the wurtzite phase, whose values first decrease and then increase with . This implies that  plays the dominant role in the behavior of P. The minimum P values appear at ≅ 0.06 for AlN and ≅ 0.02 for GaN, whereas the maximum values appear at the phase transition.
plays the dominant role in the behavior of P. The minimum P values appear at ≅ 0.06 for AlN and ≅ 0.02 for GaN, whereas the maximum values appear at the phase transition.
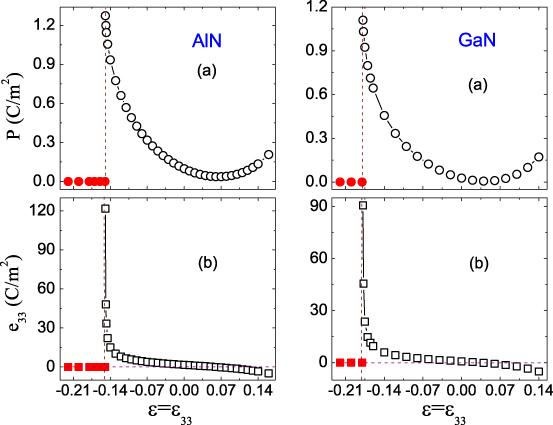
Figure 7. (a) Electric polarization P (C m−2) and (b) piezoelectric coefficient e33 (C m−2) as a function of . The open symbols refer to the wurtzite phase and the solid ones refer to the graphite-like phase.
Download figure:
Standard imageFigure 7(b) shows the piezoelectric coefficient e33 as a function of . For equilibrium wurtzite AlN and GaN, the e33 values are 1.65 C m−2 and 0.76 C m−2, respectively, in good agreement with the other corresponding results of 1.46 and 0.73 C m−2 [3, 13]. When <− 0.14 (−0.18) for AlN (GaN), e33 = 0, emphasizing the predicted phase transition. However, as increases, the e33 value of the wurtzite phase first strongly decreases until it reaches zero at ≅ 0.06 (0.02) for AlN (GaN), and then its absolute value gradually increases. The maximum value of ∼122 C m−2 (91 C m−2) appears at the phase transition, which is increased by a factor of ∼74(118) from the ground-state value for AlN (GaN), and is much larger than those of most common ferroelectric perovskites. Therefore, two important conclusions are obtained. (1) The externally applied tensile can either suppress or enhance the piezoelectric response, based on the value of . (2) The compressive greatly enhances the piezoelectricity, especially near the phase-transition region.
The strongest piezoelectric responses near the phase-transition region were originally predicted in ferroelectric materials [19, 21, 22], which as well as wurtzite semiconductors possess spontaneous P and piezoelectricity. The driving mechanism has been revealed to be polarization rotation [19, 22]. However, since the P of the wurtzite phase only remains along the c axis (the [0001] direction), it is impossible to observe polarization rotation, i.e., only the magnitude of P changes with . This is because there is only one polar direction for the wurtzite phase. It is well known that the e33 describes the derivative of P with respect to . The maximum e33, appearing at the phase transition, corresponds to the maximum slope of the P versus curve, as shown in figure 7(a). On the other hand, the zero slope, related to the minimum P, leads to e33 = 0 at ≅ 0.06 (0.02) for AlN (GaN). At large tensile , the e33 becomes negative, which just reflects the change in the sign of the slope; what we care about most is its absolute value. Very similar trends are predicted for the strain-induced colossal enhancement in piezoelectricity when approaching the phase-transition region; therefore, our investigation suggests that polarization rotation is not a necessary condition for such great enhancement in piezoelectricity; instead, it is the change in the P magnitude with that leads to such an amazing phenomenon.
The predicted novel piezoelectric response can also be summarized from the elastic behavior. As shown in figure 2(b), the c33 of the wurtzite phase first increases until it reaches the maximum at ≅ 0.06 (0.02) for AlN (GaN), and then it monotonically decreases with further increase of . This suggests that these wurtzite materials first become hard and then soft along the polar c axis in these ranges. Based on the conclusion for ferroelectric materials, the softer the crystal lattice, the larger the electromechanical response [44], it is easy to find that as increases, the piezoelectric response is first suppressed and then strongly enhanced for the wurtzite phase, with the weakest behavior appearing at ≅ 0.06 (0.02) for AlN (GaN). The predicted trends are in good agreement with the directly calculated results, as shown in figure 7(b), thereby indicating that this conclusion is applicable to wurtzite semiconductors. Such investigations are technologically very helpful, in view of the conversion between electric energy and mechanical energy, in enhancing the performance of piezoelectric devices.
The electronic properties can also be modified by the uniaxial . Four band structures at different values are plotted in figure 8 for AlN and GaN. The reciprocal lattice point (0,1/2,1/3) is denoted hereafter as X, due to its interesting behavior in the conduction bands, as shown in panel (c). For wurtzite AlN and GaN (see figures 8(b)–(d)), the Eg is always direct, with the valence-band maximum (VBM) and conduction-band minimum (CBM) at Γ. This is drastically different from wurtzite AlN under equibiaxial , where the Eg is either direct or indirect based on the value of [28]. As a conclusion, uniaxial and equibiaxial show different influences on the band structure for AlN; this is attributed to the different elastic responses to uniaxial and biaxial . However, in other theoretical work [26, 45], uniform uniaxial along the [0001] direction was believed to be equivalent to equibiaxial in the (0001) plane. Our results further reveal that the band structures of graphite-like AlN and GaN are significantly different (see figure 8(a)). The Eg of AlN is direct, with the VBM and CBM at Γ, whereas that of GaN is indirect, with the VBM at H and the CBM at Γ. This is also different from graphite-like AlN under equibiaxial , where an indirect Eg is observed at large tensile [28]. Therefore, one key difference between AlN and GaN under uniaxial is that both wurtzite and graphite-like AlN always display direct band structures, while the Eg of wurtzite GaN is always direct and that of graphite-like GaN is always indirect.
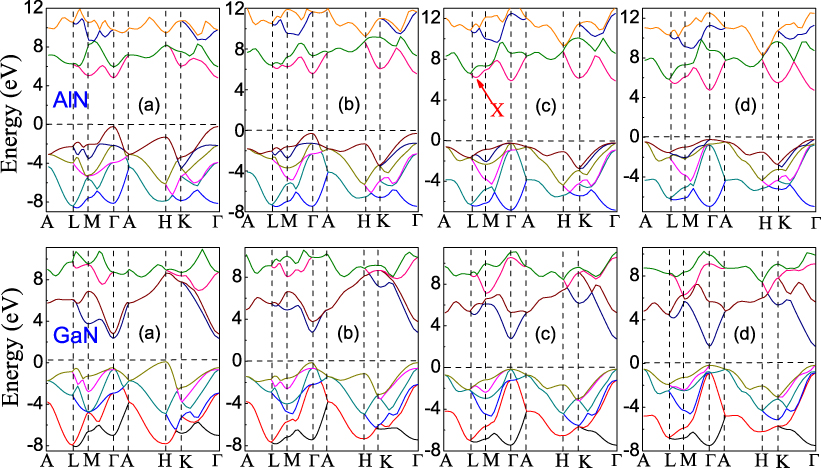
Figure 8. Band structures for AlN and GaN. AlN: (a) =− 0.18, (b) =− 0.08, (c) = 0.00 and (d) = 0.17; GaN: (a) =− 0.20, (b) =− 0.12, (c) = 0.00 and (d) = 0.16. The crystal structures are graphite-like in (a), and wurtzite in (b), (c) and (d). The Fermi level is set to zero in each figure.
Download figure:
Standard imageTo better reveal the underlying mechanisms for the abnormal band structures, figure 9 shows the minima of the conduction states and the maxima ofthe valence states at different points, where the CBM or VBM most probably appear, as a function of . As shown in figure 9(a), the CBM of AlN (GaN), whose states are mainly attributed to the N s and Al (Ga) s orbitals, is always located at Γ. This suggests a dominant role of N s and Al (Ga) s orbital hybridization in the Γ state. This is consistent with previously reported work [46], where the electronic structure of ground-state GaN was investigated by soft x-ray spectroscopy and first-principles methods. Meanwhile, for compressive , the Al and Ga s orbitals also show important contributions to the M and X states. This leads to the finding that the Γ,M and X states share similar trends, especially the Γ and M states of graphite-like AlN. It is noteworthy that N py and Al (Ga) s hybridization is observed in the X state, which results in the X energy being much larger than the Γ energy in this range. This is attributed to the higher energy level of the N p orbital compared with the N s one. In contrast, for tensile , the X state is mainly occupied by the N s orbital and the M state by the N pz one. Consequently, the Γ and X states also share similar trends and the M point is higher than the Γ one in energy.
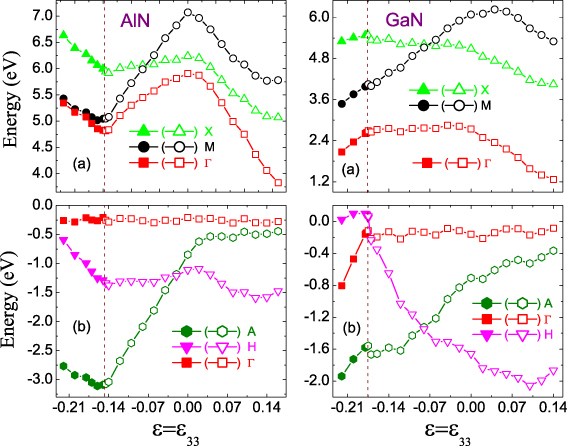
Figure 9. The uniaxial dependence of (a) the minimum of the conduction states and (b) the maximum of the valence states at different points. The open symbols refer to the wurtzite phase and the solid ones refer to the graphite-like phase. The Fermi level is set to zero in each case.
Download figure:
Standard imageFor the valence bands, the A and H points are very close to the Γ in energy. As shown in figure 9(b), the VBM of AlN and GaN are not sensitive to and remain at Γ for broad ranges of . This suggests that the variations of Eg with respect to depend on the evolution of the CBM in these ranges. N p and Al (Ga) p orbital hybridization is identified near the VBM, but the N p orbital shows a greater contribution. Therefore, our calculated results for ground-state GaN are in good agreement with previous experimental and theoretical results [46]. Furthermore, when AlN and GaN are strain-free, the VBM states are mainly occupied by the N px,py and pz orbitals, which overlap in a 'degenerate' manner. More interestingly, for different , the VBM state originates from different electron orbitals: for compressive , it is from the N pz one; for tensile , it is from the N px and py ones. The H states are occupied by the N px,py and pz orbitals, where the N px and py show the same contributions. As the compressive increases, the N px and py orbitals play more and more important roles and in total contribute the same as the N pz one after the phase transition. Furthermore, the N px and py states are pushed up by compressive and their energy values in GaN are more easily affected by . This introduces the direct–indirect bandgap transition in GaN at the phase transition with the CBM shifting to H; a similar result is not observed in AlN. For tensile , the N pz orbital plays the dominant role in the H state and is pushed down by , thereby leading to the H value being much smaller than the Γ value. The A state is attributed to N px,py and Al (Ga) px hybridization, where the N px and py orbitals are dominant. As a result, the A point is very close in energy to the Γ point for tensile .
Figure 10 shows the Eg as a function of . To examine the accuracy of our calculations, the corresponding LDA results obtained from the HSE structures are also listed for comparison. Although the HSE hybrid functional opens up the LDA Eg significantly, the trends are in good agreement. The maximum Eg values, appearing at = 0, are 6.14 eV and 3.02 eV for AlN and GaN, respectively; these values are in good agreement with the corresponding experimental values of 6.19 and 3.17 eV. For wurtzite AlN, the tensile results in a linear decrease in Eg as Eg =− 14.78 + 6.25 eV; the compressive leads to a nonlinear decrease as Eg =− 30.002 + 3.32 + 6.17 eV. On the other hand, the Eg of wurtzite GaN decreases linearly as Eg =− 11.57 + 3.08 eV with tensile , whereas for a broad range of compressive the Eg almost remains unchanged, due to the insensitivity of the CBM and VBM to in this range. However, as the in-plane increases, the Eg of wurtzite GaN drastically decreases [28]. This also implies that uniaxial along the [0001] direction and equibiaxial in the (0001) plane play obviously different roles in the band structures. More interestingly, no phase transitions from semiconductor to semi-metallic are observed for the wurtzite phase. Before the Eg reaches zero, the crystals have been broken up along the polar axis by the tensile . This is significantly different from the behavior previously predicted for in-plane [28], where such a transition induced by tensile is found. For graphite-like AlN, the Eg increases rapidly with , which is attributed to the abnormal behavior of the CBM induced by . This is also different from the results for in-plane [28], thereby further emphasizing the different effects of uniaxial and equibiaxial on the band structure. In contrast, compressive leads to a decrease in the Eg of graphite-like GaN and the important transition to the semi-metallic phase is finally observed.
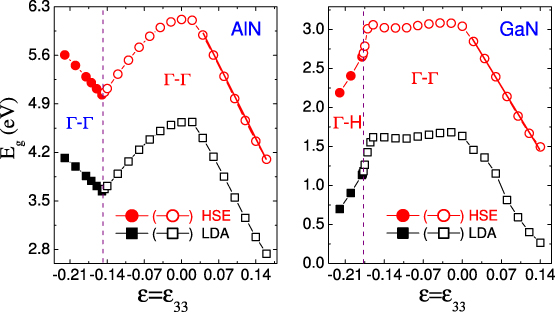
Figure 10. Calculated Eg as a function of . The gap is calculated using two different methods. (i) HSE: the electronic and atomic structures are relaxed using the HSE hybrid functional; and (ii) LDA: a static LDA electronic structure calculation for the HSE optimized structure. The open symbols refer to the wurtzite phase and the solid ones refer to the graphite-like phase.
Download figure:
Standard image4. Summary
In summary, we have systematically studied the effects of uniaxial strain on the properties of AlN and GaN using HSE06 range-separated hybrid functionals. The phase transition from wurtzite to graphite-like, driven by the TA soft phonon mode associated with the elastic instabilities, is predicted, accompanied by a great enhancement of polarization and piezoelectricity occurring near the phase-transition region. The predicted huge electromechanical response originates from the anomalous behavior of P near the phase-transition region. This amazing phenomenon was originally predicted in ferroelectric fields, where the giant piezoelectric response is attributed to polarization rotation. Our investigations show that polarization rotation is not a necessary condition. Furthermore, AlN and GaN have significantly different band structures under the same . Before the semi-metallic phase is observed, the tensile σ33 has reached the ideal tensile strength. More specially, our calculated results further reveal that uniaxial along the [0001] direction and equibiaxial in the (0001) plane play obviously different roles in the band structure, different from the previously predicted behavior. This work sheds light on the origins of the structural phase transition and huge piezoelectric response in wurtzite semiconductors, and it should also stimulate more investigations of bandgap tuning for producing a wide spectrum, thereby enabling a number of important technological applications with better performance in piezoelectric and electronic fields.
Acknowledgments
We thank Dr Leiming Chen for useful discussions. The work is supported by the National Natural Science Foundation of China under Grant Nos 11004242, 10947119 and 11047128, the Fundamental Research Funds for the Central Universities under Grant Nos 2010LKWL01, 2010LKWL02 and 2012LWB54, the Youth Science Funds under Grant Nos 2009A040 and 2009A048, and startup funds from China University of Mining and Technology.