


ABSTRACT
It has been shown that rotational lines observed in the Horsehead nebula photodissociation region (PDR) are probably not caused by l-C3H+, as was originally suggested. In the search for viable alternative candidate carriers, quartic force fields are employed here to provide highly accurate rotational constants, as well as fundamental vibrational frequencies, for another candidate carrier: 1 1A' C3H−. The ab initio computed spectroscopic constants provided in this work are, compared to those necessary to define the observed lines, as accurate as the computed spectroscopic constants for many of the known interstellar anions. Additionally, the computed Deff for C3H− is three times closer to the D deduced from the observed Horsehead nebula lines relative to l-C3H+. As a result, 1 1A' C3H− is a more viable candidate for these observed rotational transitions. It has been previously proposed that at least C6H− may be present in the Horsehead nebular PDR formed by way of radiative attachment through its dipole-bound excited state. C3H− could form in a similar way through its dipole-bound state, but its valence excited state increases the number of relaxation pathways possible to reach the ground electronic state. In turn, the rate of formation for C3H− could be greater than the rate of its destruction. C3H− would be the seventh confirmed interstellar anion detected within the past decade and the first CnH− molecular anion with an odd n.
Export citation and abstract BibTeX RIS
1. INTRODUCTION
Recent work by Huang et al. (2013b) has questioned the attribution of lines observed in the Horsehead nebula photodissociation region (PDR) to l-C3H+. Quartic force fields (QFFs) computed from high-level ab initio quantum mechanical energies analyzed using perturbation theory at second order (Papousek & Aliev 1982) are known to produce highly accurate spectroscopic constants. Even though the B0 computed by Huang et al. (2013b; 11 262.68 MHz) is within 0.16% of the B-type rotational constant derived from the observations by Pety et al. (2012; 11 244.9474 MHz), the computed De of 4.248 kHz differs by 44.5% from the observed D value of 7.652 kHz. This "error" is more than an order of magnitude larger than any other error for a computed De of a cation (using similar levels of theory) as compared to known high-resolution experimental data. Furthermore, the sextic distortion constant, H, differs by three orders of magnitude. As a result, it is unlikely that l-C3H+ corresponds to the lines observed by Pety et al. (2012).
This result motivates the question, "What is the carrier of these lines?" If these observed lines are, in fact, related to one another, certain inferences can be made about the molecular carrier. To match the rotational constants derived from the transition energies corresponding to the observed lines, the carrier is either linear or quasi-linear, almost certainly composed of three carbon atoms as well as a single hydrogen atom, and closed-shell since there are no splittings in the lines as required for the rotational spectra of open-shell molecules (M. C. McCarthy 2013, private communication). All of these criteria are, in fact, met by l-C3H+, but this cation's difference between observational and high-accuracy theoretical rotational constants, especially the D constant, discussed above and by Huang et al. (2013b), probably rules it out. As a result, the quasi-linear anion, 1 1A' l-C3H−, remains the most likely candidate carrier of the Horsehead nebula PDR rotational lines of interest, especially since anions have been shown to be more abundant in the interstellar medium (ISM) than originally thought (Cordiner et al. 2013), and there has been reason to suspect the presence of C6H− in the Horsehead nebula PDR (Agúndez et al. 2008).
Even though the most stable singlet isomer of C3H− is the cyclic form, c-C3H−, the barrier to isomerization is high enough ( 45 kcal mol−1) for the quasi-linear Cs isomer to be kinetically stable (Lakin et al. 2001). Various mechanisms for interstellar synthesis of this anion are possible (Millar et al. 2007; Larsson et al. 2012; Senent & Hochlaf 2013) and are probably related to those responsible for the creation of the related C2nH− for n = 2–4 anions previously detected in the ISM (McCarthy et al. 2006; Cernicharo et al. 2007; Brünken et al. 2007b). Furthermore, radical C3H in both the linear and cyclic forms has also been detected in the ISM (Thaddeus et al. 1985; Yamamoto et al. 1987), suggesting the possible interstellar existence of the anion.
Additionally, C3H− is of astronomical interest since it has been computationally shown by Fortenberry (2013) to possess not only a rare dipole-bound singlet excited electronic state (the 2 1A' state), but also an even more rare valence excited state (1 1A'') below the electron binding or electron detachment energy. In fact, the valence electronically excited state is the only such state thus far proposed to exist for an anion of this size which also contains only first-row atoms (Fortenberry & Crawford 2011b, 2011a; Fortenberry 2013). The valence excited state and the bent structure of C3H− are both the result of an unfilled π orbital. The two components of the HOMO π-type orbital split when the additional electron in the anion spin-pairs with the lone electron in the radical's π-type HOMO. A carbene and bent structure are thus created. The valence (1 1A'') state of C3H− is then the product of an excitation from the occupied portion of the split π orbital into the unoccupied portion, an uncommon process not present in the C2nH− anions. Furthermore, anions have been proposed as carriers of some diffuse interstellar bands (DIBs; Sarre 2000; Cordiner & Sarre 2007; Fortenberry et al. 2013a), and the two electronically excited states of C3H− may be of importance to the DIBs and to the chemistry of PDRs, as well.
2. COMPUTATIONAL DETAILS
The spectroscopic constants and fundamental vibrational frequencies of 1 1A' l-C3H− are computed through the established means of QFFs (Huang & Lee 2008). Starting from a restricted Hartree–Fock (RHF; Scheiner et al. 1987) coupled cluster (Lee & Scuseria 1995; Shavitt & Bartlett 2009; Crawford & Schaefer 2000) singles, doubles, and perturbative triples (CCSD(T); Raghavachari et al. 1989) aug-cc-pV5Z (Dunning 1989; Kendall et al. 1992; Dunning et al. 2001) geometry further corrected for core correlation effects from the Martin–Taylor basis set (Martin & Taylor 1994), a grid of 743 symmetry-unique points is generated. Simple-internal coordinates for the bond lengths and ∠H−C−C are coupled to linear LINX and LINY (Allen et al. 2005) coordinates exactly as those defined in Fortenberry et al. (2012b) for HOCO+. Displacements of 0.005 Å for the bond lengths, 0.005 rad for the bond angle, and 0.005 for the LINX and LINY coordinates and the associated energies computed at each point define the QFF, which is of the form:

where Δi are the displacements and Fij... are force constants (Huang & Lee 2008).
At each point, CCSD(T)/aug-cc-pVXZ (where X = T, Q, 5) energies are computed and extrapolated to the complete basis set (CBS) limit via a three-point formula (Martin & Lee 1996). Additionally, energy corrections are made to the CBS energy for core correlation and for scalar relativistic effects (Douglas & Kroll 1974). The resulting QFF is denoted as the CcCR QFF for the CBS energy, core correlation correction, and scalar relativistic correction, respectively, (Fortenberry et al. 2011). The augmented Dunning basis sets have been shown by Skurski et al. (2000) to be reliable for computations of anionic properties. An initial least-squares-fit of the CcCR energy points leads to a minor transformation of the reference geometry such that the gradients are identically zero. This geometry and the resulting force constants are then employed in the rovibrational computations. All electronic structure computations make use of the MOLPRO 2010.1 quantum chemical package (Werner et al. 2010), and all employ the Born–Oppenheimer approximation making the QFFs identical for the isotopologues.
The QFF is fit from the 805 redundant total energy points with a sum of squared residuals on the order of 3 × 10−17 a.u.2 Cartesian derivatives are then computed from the QFF with the INTDER program (Allen et al. 2005). From these, the SPECTRO program (Gaw et al. 1991) employs second-order vibrational perturbation theory (VPT2) to generate the spectroscopic constants (Papousek & Aliev 1982) and vibrational frequencies (Mills 1972; Watson 1977). After transforming the force constants into the Morse-cosine coordinate system so that the potential possesses proper limiting behavior (Dateo et al. 1994; Fortenberry et al. 2013b), vibrational configuration interaction (VCI) computations with the MULTIMODE program (Carter et al. 1998; Bowman et al. 2003) also produce vibrational frequencies. The VCI computations make use of similar basis set configurations as those utilized by Fortenberry et al. (2012a, 2012b) in similar quasi-linear tetra-atomic systems.
3. DISCUSSION
The force constants computed in this study are listed in Table 1. The CcCR geometrical parameters and spectroscopic constants are given in Table 2 for both 1 1A' l-C3H− and the deuterated isotopologue. The equilibrium dipole moment is computed with respect to the center-of-mass with CCSD(T)/aug-cc-pV5Z to be 2.16 D. The C−C−C Rα vibrationally-averaged bond angle is nearly collinear at 174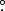 540 while the vibrationally-averaged ∠H−C−C is 109491. These values are in line with those computed by Lakin et al. (2001). As has been discussed by Fortenberry (2013) for C3H−, the C1 carbon atom adjacent to the hydrogen atom shown in Figure 1 is a carbene-type carbon containing a lone pair which leads to a longer C1 −C2 bond length compared to the shorter C2 −C3 bond length. Even though this result differs from the CCSD(T) results from Lakin et al. (2001), their reported CASSCF and HF results give bond lengths similar to ours, leading us to conclude that the CCSD(T) C−C bond lengths are mislabeled in the paper by Lakin et al. (2001). The vibrationally averaged geometrical parameters change slightly upon deuteration. Similar bond angles of the heavy atoms have been computed for the trans-HOCO+, HOCS+, and HSCO+ systems (Fortenberry et al. 2012a, 2012b) with very good agreement present for known experimental data.
540 while the vibrationally-averaged ∠H−C−C is 109491. These values are in line with those computed by Lakin et al. (2001). As has been discussed by Fortenberry (2013) for C3H−, the C1 carbon atom adjacent to the hydrogen atom shown in Figure 1 is a carbene-type carbon containing a lone pair which leads to a longer C1 −C2 bond length compared to the shorter C2 −C3 bond length. Even though this result differs from the CCSD(T) results from Lakin et al. (2001), their reported CASSCF and HF results give bond lengths similar to ours, leading us to conclude that the CCSD(T) C−C bond lengths are mislabeled in the paper by Lakin et al. (2001). The vibrationally averaged geometrical parameters change slightly upon deuteration. Similar bond angles of the heavy atoms have been computed for the trans-HOCO+, HOCS+, and HSCO+ systems (Fortenberry et al. 2012a, 2012b) with very good agreement present for known experimental data.

Figure 1. CcCR equilibrium geometry of 1 1A' l-C3H−.
Download figure:
Standard image High-resolution image{kind=link}
Table 1. The Simple-internal CcCR QFF Quadratic, Cubic, and Quartic Force Constants (in mdyn/Ån radm)a for l-C3H−
| F11 | 10.191 889 | F431 | 0.0711 | F1111 | 318.24 | F4432 | 0.24 | F5531 | 0.12 |
| F21 | 0.841 962 | F432 | −0.4022 | F2111 | 0.44 | F4433 | 0.44 | F5532 | 0.22 |
| F22 | 7.312 189 | F433 | −0.0735 | F2211 | −1.98 | F4441 | 0.45 | F5533 | −0.34 |
| F31 | 0.068 029 | F441 | −0.5015 | F2221 | 5.28 | F4442 | −0.34 | F5541 | −0.01 |
| F32 | −0.006 196 | F442 | 0.1723 | F2222 | 220.01 | F4443 | 0.56 | F5542 | 0.07 |
| F33 | 4.558 746 | F443 | −0.0586 | F3111 | 0.16 | F4444 | −0.81 | F5543 | −0.17 |
| F41 | −0.066 879 | F444 | −0.7769 | F3211 | −0.08 | F5111 | 0.06 | F5544 | 0.51 |
| F42 | 0.515 214 | F511 | −0.0809 | F3221 | 0.80 | F5211 | 0.10 | F5551 | 0.07 |
| F43 | 0.217 498 | F521 | −0.0018 | F3222 | −1.41 | F5221 | −0.39 | F5552 | 0.18 |
| F44 | 0.650 100 | F522 | −0.3714 | F3311 | 0.72 | F5222 | 0.80 | F5553 | 0.12 |
| F51 | 0.070 974 | F531 | −0.0597 | F3321 | −0.80 | F5311 | 0.09 | F5554 | −0.12 |
| F52 | 0.081 130 | F532 | −0.2190 | F3322 | −0.54 | F5321 | 0.21 | F5555 | 1.95 |
| F53 | 0.069 481 | F533 | −0.0030 | F3331 | −0.82 | F5322 | 0.54 | F6611 | 0.14 |
| F54 | 0.064 059 | F541 | −0.0774 | F3332 | 0.46 | F5331 | −0.04 | F6621 | −0.49 |
| F55 | 0.404 485 | F542 | 0.0376 | F3333 | 145.05 | F5332 | −0.07 | F6622 | 0.64 |
| F66 | 0.168 044 | F543 | −0.0694 | F4111 | −0.17 | F5333 | −0.46 | F6631 | 0.03 |
| F111 | −64.7214 | F544 | −0.1425 | F4211 | −0.02 | F5411 | 0.14 | F6632 | 0.06 |
| F211 | 0.5759 | F551 | −0.4284 | F4221 | −0.27 | F5421 | 0.12 | F6633 | −0.14 |
| F221 | −3.2972 | F552 | −0.9210 | F4222 | −0.32 | F5422 | −0.49 | F6641 | −0.07 |
| F222 | −43.3783 | F553 | −0.0900 | F4311 | 0.15 | F5431 | −0.05 | F6642 | −0.04 |
| F311 | 0.0659 | F554 | 0.0071 | F4321 | −0.03 | F5432 | 0.31 | F6643 | −0.10 |
| F321 | −0.3042 | F555 | −0.1839 | F4322 | −0.46 | F5433 | −0.09 | F6644 | 0.08 |
| F322 | −0.0840 | F661 | −0.1710 | F4331 | −0.06 | F5441 | 0.01 | F6651 | −0.04 |
| F331 | 0.1287 | F662 | −0.3467 | F4332 | −0.31 | F5442 | 0.14 | F6652 | 0.02 |
| F332 | 0.2601 | F663 | −0.0476 | F4333 | −1.42 | F5443 | 0.24 | F6653 | −0.03 |
| F333 | −28.7819 | F664 | 0.0133 | F4411 | −0.63 | F5444 | −0.06 | F6654 | −0.10 |
| F411 | −0.2017 | F665 | −0.0708 | F4421 | 1.45 | F5511 | 0.57 | F6655 | 0.23 |
| F421 | 0.3282 | F4422 | −1.71 | F5521 | 0.14 | F6666 | 0.86 | ||
| F422 | −0.6518 | F4431 | −0.17 | F5522 | 1.56 |
Note. a1 mdyn = 10−8 N; n and m are exponents corresponding to the number of units from the type of modes present in the specific force constant.
Download table as: ASCIITypeset image
Table 2. The Zero-Point (Rα Vibrationally-averaged) and Equilibrium Structures, Rotational Constants, CCSD(T)/aug-cc-pV5Z Dipole Moment, Vibration-Rotation Interaction Constants, and Quartic and Sextic Distortion Constants of 1 1A' l-C3H− and the Deuterated form with the CcCR QFF
| C3H− | Previousa | C3D− | |
|---|---|---|---|
| r0(C1 −H) | 1.119 438 Å | 1.116 446 Å | |
| r0(C1 −C2) | 1.351 595 Å | 1.351 753 Å | |
| r0(C2 −C3) | 1.282 845 Å | 1.282 620 Å | |
| ∠0(H−C1 −C2) | 109491 |
109530 | |
| ∠0(C1 −C2 −C3) | 174540 |
174643 | |
| A0 | 529 134.2 MHz | 295 539.6 MHz | |
| B0 | 11 339.66 MHz | 10 626.03 MHz | |
| C0 | 11 087.35 MHz | 10 238.74 MHz | |
| DJ | 4.954 kHz | 4.544 kHz | |
| DJK | 0.702 MHz | 0.316 MHz | |
| DK | 217.543 MHz | 94.897 MHz | |
| d1 | −0.112 kHz | −0.253 kHz | |
| d2 | −0.023 kHz | −0.052 kHz | |
| HJ | 3.344 mHz | 16.516 mHz | |
| HJK | 3.221 Hz | 2.151 Hz | |
| HKJ | −3.229 kHz | −0.745 kHz | |
| HK | 358.867 kHz | 90.731 kHz | |
| H1 | 0.132 mHz | 0.634 mHz | |
| H2 | 0.203 mHz | 0.612 mHz | |
| H3 | 0.037 mHz | 0.133 mHz | |
| τaaaa | −873.001 MHz | −380.872 MHz | |
| τbbbb | −0.021 MHz | −0.021 MHz | |
| τcccc | −0.019 MHz | −0.017 MHz | |
| τaabb | −2.766 MHz | −1.619 MHz | |
| τaacc | −0.081 MHz | 0.319 MHz | |
| τbbcc | −0.020 MHz | −0.018 MHz | |
| Φaaa | 355 640.661 Hz | 89 988.504 Hz | |
| Φbbb | 0.001 Hz | 0.004 Hz | |
| Φccc | 0.000 Hz | 0.001 Hz | |
| Φaab | 390.158 Hz | 703.204 Hz | |
| Φabb | 4.265 Hz | 3.112 Hz | |
| Φaac | −3 614.354 Hz | −1 445.590 Hz | |
| Φbbc | 0.000 Hz | 0.001 Hz | |
| Φacc | −0.271 Hz | 0.151 Hz | |
| Φbcc | 0.001 Hz | 0.002 Hz | |
| Φabc | 4.570 Hz | 3.618 Hz | |
| αA 1 | 27 922.5 MHz | 11 662.9 MHz | |
| αA 2 | −725.5 MHz | −917.5 MHz | |
| αA 3 | 484.8 MHz | 170.2 MHz | |
| αA 4 | −35 092.1 MHz | −16 226.3 MHz | |
| αA 5 | −3 103.1 MHz | −4 597.5 MHz | |
| αA 6 | 12 333.4 MHz | 9 042.1 MHz | |
| αB 1 | 4.2 MHz | 6.9 MHz | |
| αB 2 | 83.5 MHz | 77.2 MHz | |
| αB 3 | 45.1 MHz | 40.3 MHz | |
| αB 4 | −12.0 MHz | −8.4 MHz | |
| αB 5 | −47.1 MHz | −48.4 MHz | |
| αB 6 | −48.6 MHz | −45.9 MHz | |
| αC 1 | 14.8 MHz | 18.3 MHz | |
| αC 2 | 78.6 MHz | 70.1 MHz | |
| αC 3 | 38.4 MHz | 39.3 MHz | |
| αC 4 | 16.0 MHz | 12.9 MHz | |
| αC 5 | −16.1 MHz | −15.2 MHz | |
| αC 6 | −78.5 MHz | −69.8 MHz | |
| re(C1 −H)b | 1.106 939 Å | 1.110 Å | ⋅⋅⋅ |
| re(C1 −C2) | 1.349 832 Å | 1.289 Å | ⋅⋅⋅ |
| re(C2 −C3) | 1.281 900 Å | 1.363 Å | ⋅⋅⋅ |
| ∠e(H−C1 −C2) | 109529 |
1092 |
⋅⋅⋅ |
| ∠e(C2 −C3 −C4) | 174571 |
1712 |
⋅⋅⋅ |
| Ae | 530 044.3 MHz | 524.5 GHz | 295 106.5 MHz |
| Be | 11 352.05 MHz | 11.2 GHz | 10 636.73 MHz |
| Ce | 11 114.02 GHz | 10.9 MHz | 10 266.68 MHz |
| μc | 2.16 D | ⋅⋅⋅ | ⋅⋅⋅ |
| μx | 1.63 D | ⋅⋅⋅ | ⋅⋅⋅ |
| μy | 1.41 D | ⋅⋅⋅ | ⋅⋅⋅ |
Notes. aCCSD(T)/aug-cc-pVQZ QFF results from Lakin et al. (2001). bThe equilibrium geometries are identical among isotopologues from the use of the Born–Oppenheimer approximation. cThe C3H− coordinates (in Å with the center-of-mass at the origin) used to generate Born–Oppenheimer dipole moment components are: H, 1.733414, −0.910473, 0.000000; C1, 1.276456, 0.098036, 0.000000; C2, −0.069613, −0.016965, 0.000000; C3, −1.352424, −0.004605, 0.000000.
The most notable values in Table 2 are the rotational constants and the quartic centrifugal distortion (D-type) constants. For 1 1A' l-C3H−, the B0 rotational constant is 11 339.66 MHz while C0 is 11 087.35 MHz. The equilibrium constants are slightly larger, but both sets are in reasonable agreement with those computed by Lakin et al. (2001). The D-type constants have not been vibrationally-averaged, and DJ, most prominently, is 4.954 kHz.
Direct comparison between these explicitly computed values and those deduced from the Horsehead nebula PDR spectrum observed by Pety et al. (2012) is not possible since the isomer of C3H− of interest here is not perfectly linear. Pety et al. (2012) assume a linear structure in order to fit the effective rotational constant, Beff, and the effective centrifugal distortion constant, Deff and use the second-order fitting equation,

to compute the affiliated rotational constants. C3H− is non-linear and requires the following related equation from McCarthy et al. (1997):

with the assumption that K = 0 forcing c = 8. As such, we can set Equation (2) equal to Equation (3). The (J + 1) term in Equation (3) is equal to 2 Beff, and the (J + 1)3 term in Equation (3) is equal to 4 Deff. Using the CcCR computed A0, B0, C0, and DJ values, where DJ is the only equilibrium constant, Beff is computed to be 11 213.51 MHz, and Deff is 8.795 kHz. Hence, direct comparison between the CcCR C3H− derived effective rotational constants and those obtained from the lines observed by Pety et al. (2012) is possible.
The second-order fit of the lines observed by Pety et al. (2012) indicates that the carrier must have a B-type constant that is very close to 11 244.9474 MHz and a D-type quartic distortion constant that is around 7.652 kHz. The Beff computed with the A0, B0, and C0 rotational constants by the above approach is very close, off by 31.44 MHz or 0.28%. This is roughly the same difference between the observed B and that of l-C3H+ (Huang et al. 2013b). However, the 8.795 kHz Deff for 1 1A' l-C3H− is much closer to the 7.652 kHz D derived from the lines observed by Pety et al. (2012) in the Horsehead nebula than the linear cation (Huang et al. 2013b). Even so, this Deff of 8.795 kHz differs from the observation by 1.14 kHz or 14.93%.
Table 3 provides some insight into the accuracies that can be expected for calculated rotational constants of similar molecules. Related quasilinear molecules studied previously have all been cations. Hence, within Table 3, the cation B and D-type constants listed are more correctly understood to be Beff and Deff as is the case for C3H− (i.e., Equation (3) is used). Calculation of the vibrationally-averaged Beff values incorporate B0 and C0 while the equilibrium Beff values incorporate Be and Ce. Calculation of Deff for each of the bent, quasilinear systems utilizes A0, B0, and C0 and the equilibrium DJ value since vibrational averaging is not available for the D-type constants. The lone exception to this definition of Deff is the C3H− Deff computed with Ae, Be, and Ce given in the second line of Table 3, which actually lowers the C3H− Deff value to 8.366 kHz, a difference of 0.714 kHz or 9.34% from that determined by Pety et al. (2012). Finally, since all of the anions observed in the ISM have been linear, directly comparable B0, Be, and De constants have been computed explicitly and are listed in Table 3. Note that all of the experimental B and D values contained in Table 3 correspond to vibrationally averaged constants.
Table 3. Errors in the Computation of B (in MHz) and D (in kHz) for Linear Molecules and Beff (in MHz) and Deff (in kHz) for Quasilinear Molecules
| Theoretical | B/Beff | D/Deff | |||||
|---|---|---|---|---|---|---|---|
| Molecule | B0 or Be | Experiment | Theory | % Error | Experiment | Theory | % Error |
| C3H−a | Equilibrium | 11244.9474 | 11233.04 | 0.11% | 7.652 | 8.366 | 9.3% |
| Vib.-avg. | 11244.9474 | 11213.51 | 0.28% | 7.652 | 8.795 | 14.9% | |
| HSCO+b | Vib.-avg. | 5636.866 | 5637.60 | 0.01% | 3.1 | 3.116 | 0.5% |
| HOCO+c | Vib.-avg. | 10691.58265 | 10705.44 | 0.13% | 4.580576 | 4.511 | 1.5% |
| NNOH+d | Vib.-avg. | 11192.9214 | 11198.57 | 0.05% | 7.764972 | 7.604 | 2.1% |
| HOCS+e | Vib.-avg. | 5726.66011 | 5730.22 | 0.06% | 1.064 | 1.107 | 4.0% |
| C2H−f | Equilibrium | 41639.20 | 41781.0 | 0.34% | 0.09697 | 0.0946 | 2.4% |
| Vib.-avg. | 41639.20 | 41614.0 | 0.06% | ||||
| C4H−g | Equilibrium | 4654.9449 | 4625.6546 | 0.63% | 0.5875 | 0.55 | 6.4% |
| Vib.-avg. | 4654.9449 | 4653.9 | 0.02% | ||||
| C6H−h | Vib.-avg. | 1376.86298 | 1376.9 | 0.00% | 0.03235 | 0.0270 | 16.5% |
| C8H−i | Vib.-avg. | 583.30404 | 583.2 | 0.02% | 0.0042 | 0.0033 | 16.7% |
| CN−j | Equilibrium | 56132.7562 | 56152 | 0.03% | 186.427 | 185 | 0.8% |
| Zero-point | 56132.7562 | 56126.5 | 0.01% | ||||
| C3N−k | Equilibrium | 4851.62183 | 4850 | 0.03% | 0.68592 | 0.628 | 8.4% |
| C5N−l | Equilibrium | 1388.860 | 1387.8 | 0.08% | 0.033 | 0.0300 | 9.1% |
| Vib.-avg. | 1388.860 | 1386.2 | 0.19% | ||||
Notes. aThis work with the observed lines described by Pety et al. (2012). bCcCR QFF data Fortenberry et al. (2012a) and experimental data from Ohshima & Endo (1996). cCcCR QFF data from Fortenberry et al. (2012b) and experimental data from Bogey et al. (1988b). dCcCR QFF data from Huang et al. (2013a), experimental Beff from McCarthy & Thaddeus (2010), and experimental Deff computed from the constants given in Bogey et al. (1988a). eCcCR QFF data from Fortenberry et al. (2012a) and experimental data from McCarthy & Thaddeus (2007). fCcCR QFF data from Huang & Lee (2009) and experimental data from Brünken et al. (2007a). gB0 from the CCSD(T)/cc-pVTZ Be corrected for vibrational averaging with CCSD(T)/cc-pVDZ; CCSD(T)/cc-pVDZ De; and experimental data are from Gupta et al. (2007). The RCCSD(T)/aug-cc-pVQZ Be is from Senent & Hochlaf (2010). hCCSD(T)/cc-pVTZ Be corrected for vibrational averaging with CCSD(T)/cc-pVDZ, CCSD(T)/cc-pVDZ De, and experimental data from McCarthy et al. (2006). iCCSD(T)/cc-pVTZ Be corrected for vibrational averaging with SCF/DZP, SCF/DZP De, and experimental data from Gupta et al. (2007). jCCSD(T)/aug-cc-pCV5Z Be, CCSD(T)/aug-cc-pCVQZ De, and experimental data from Gottlieb et al. (2007) with CCSD(T)/MTcc B0 from Lee & Dateo (1999). kCCSD(T)/aug-cc-pCV5Z Be and De from Kołos et al. (2008) (ΔB0 is reported to be 0.606 MHz giving a % error of about 0.02%) and experimental data from Thaddeus et al. (2008). lCCSD(T)/aug-cc-pCV5Z Be and B0 with CCSD(T)/aug-cc-pVQZ De from Botschwina & Oswald (2008) with experimental data from Cernicharo et al. (2008).
Download table as: ASCIITypeset image
From Table 3, the quasi-linear cations listed below C3H− show strong correlation between the computed Beff from the use of B0 and C0 and the Beff derived from the various experiments. Additionally, the Deff values computed the same way with the equilibrium DJ also show good, albeit not as strong, correlation between theory and experiment. Unfortunately, C3H− has errors that are larger than this. However, this probably results from a combination of basis set incompleteness and higher-order correlation effects. Even though aug-cc-pVXZ basis sets used at the CCSD(T) level of theory have been shown to be effective in the computation of anionic properties (Skurski et al. 2000; Fortenberry & Crawford 2011b), higher-order properties such as the D-type constants are more susceptible to even the smallest errors. This is clear for the cations as well, where the Deff values are not as accurate as the Beff values.
The known interstellar anions and the related C2H− system, which has not yet been detected in the ISM, are linear and have B and D computed directly, either as B0 or Be and De. Note that the theoretical rotational constants are not as accurate for the anions as they are for the cations. Most notably, the Be/B0 and De values computed with a CCSD(T)/aug-cc-pCV5Z cubic force field for C5N− by Botschwina & Oswald (2008) are directly used in the identification of this anion in the ISM (Cernicharo et al. 2008). As listed in Table 3, agreement between computed B values and that necessary to match the observed rotational lines actually worsens when B0 is used instead of Be, more than doubling the percent error. This is the same behavior as what is currently found for C3H−. Additionally, the De percent error for C5N−, as compared to observation, is 9.1%, almost exactly what it is for C3H− when using the equilibrium rotational constants. The force field employed by Botschwina & Oswald (2008) also includes core correlation like the CcCR QFF. Hence, the present rotational constants are in the same accuracy range for C3H− as those used to detect C5N− in the ISM. Furthermore, the calculated De values compared to experiment for C6H− and C8H− actually have a larger percent error than Deff for C3N−, C5N−, or even C3H−.
Comparison of the sextic distortion constant, Heff, is not as straightforward. There is a dearth of data on how the computation of this value for anions compares to experiment. HJ, which is an equilibrium value, is not exactly Heff, but they are probably related. Even though H obtained by Pety et al. (2012) is 560 mHz and HJ for C3H− is 3.344 mHz, this is an order of magnitude closer agreement than this same H compared to the He for l-C3H+, 0.375 mHz (Huang et al. 2013b). Additionally, the same basis set and correlation errors for anions that affect the calculation of D will be present for H. As a result, we can only say here that as far as H is concerned for comparison to the lines observed in the Horsehead nebula by Pety et al. (2012), 1 1A' C3H− is a better candidate than l-C3H+.
Even though lower levels of theory have been used to reproduce rotational constants of the detected, linear interstellar anions, C3H− is the only anion examined here that is not linear. It is known that basis set effects can be pronounced in the computation of bond angles in anions (Lee & Schaefer 1985; Huang & Lee 2009) where the average change in a bond angle computed with a standard basis set and one augmented to include diffuse functions is around 10. For example, the equilibrium ∠C−C−C in C3H− from a simple CCSD(T)/cc-pVTZ QFF is 17332, while this same angle is 17420 with a CCSD(T)/aug-cc-pVTZ QFF. In fact, the resulting 088 difference by simply adding diffuse functions to the standard basis set is actually larger than the equilibrium ∠C−C−C difference between the CCSD(T)/aug-cc-pVTZ QFF and that from the CcCR QFF, 038. As a result, Beff for C3H− is slower to converge with respect to the basis set chosen relative to the linear anions. This is made clear in that the CCSD(T)/cc-pVTZ QFF vibrationally averaged Beff is 11 056.74 MHz whereas the corresponding CcCR Beff is 11 213.51 MHz, an increase of 156.77 MHz. The linear anions are able to use lower level levels of theory in order to approach the experimental rotational constants, but higher levels of theory are required for the non-linear anion. The fact that Beff computed with the equilibrium rotational constants is closer to the B derived from the observations by Pety et al. (2012) than Beff computed with the vibrationally averaged rotational constants is coincidental. However, the important point is that the C3H− vibrationally averaged Beff approaches the corresponding observed value as more accurate QFFs are employed, and the remaining error is typical.
The harmonic and anharmonic vibrational frequencies for both 1 1A' l-C3H− and l-C3D− are given in Table 4. Positive anharmonicities are present in both isotopologues for the ν5 C1 −C2 −C3 bending and the ν6 torsional modes. VPT2 and VCI produce fundamental vibrational frequencies from the CcCR QFF that are quite consistent. The largest deviation between the methods, 1.0 cm−1, is found for the ν4 H−C1 −C2 bending mode. Comparison of the C3H− CcCR QFF vibrational frequencies, whether computed using VPT2 or VCI, to those computed by Lakin et al. (2001) is roughly consistent for ν1-ν4. The ν5 anharmonic frequencies differ by more than 50 cm−1, though the ω5 harmonic frequencies are very similar (i.e., the difference in the ν5 fundamental frequency is mostly due to differences in the anharmonic correction). The torsional mode is nearly identical between the two studies, though in this case the harmonic frequencies differ by more than 50 cm−1. It is hoped that the present QFF computations of the fundamental vibrational frequencies provided here will assist in the characterization of this anion in current and future studies of the ISM or simulated laboratory experiments at infrared wavelengths in addition to studies in the sub-millimeter spectral region.
Table 4. The C3H− and C3D− CcCR QFF Harmonic, VCI, and VPT2 Fundamental Vibrational Frequencies in cm−1
| C3H− | Previousa C3H− | C3D− | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Mode | Description | Harmonic | VCI | VPT2 | Harmonic | Anharm. | Harmonic | VCI | VPT2 |
| ν1(a') | C1 −H stretch | 2881.9 | 2714.4 | 2713.9 | 2863 | 2723 | 2122.9 | 2036.4 | 2035.5 |
| ν2(a') | C2 −C3 stretch | 1843.9 | 1804.3 | 1804.4 | 1831 | 1828 | 1832.9 | 1796.5 | 1796.5 |
| ν3(a') | C1 −C2 stretch | 1117.1 | 1108.0 | 1107.9 | 1091 | 1120 | 1112.0 | 1100.9 | 1101.0 |
| ν4(a') | H−C1 −C2 bend | 1037.8 | 1012.1 | 1011.1 | 1002 | 1022 | 817.0 | 803.8 | 802.7 |
| ν5(a') | C1 −C2 −C3 bend | 406.7 | 419.4 | 418.9 | 393 | 368 | 379.1 | 382.4 | 381.9 |
| ν6(a'') | torsion | 281.0 | 296.8 | 296.1 | 349 | 297 | 278.9 | 286.7 | 286.1 |
Note. aCCSD(T)/aug-cc-pVQZ QFF results from Lakin et al. (2001).
Download table as: ASCIITypeset image
4. ASTROCHEMICAL CONSIDERATIONS
The lines observed by Pety et al. (2012) are present in the Horsehead nebula PDR but not in the dense core. Typically, a PDR is defined in terms of shells starting from the exterior shell dominated by an influx of far-ultraviolet (FUV) photons. In this region, the photons are most often absorbed by polycyclic aromatic hydrocarbons (PAHs) and dust particles. However, electrons are also produced in these regions from various mechanisms involving the aforementioned larger molecular particles as well as from interactions with atoms or small molecules. As the FUV flux is reduced from shielding resulting from the PAHs and dust, the H2 shell is formed. Moving further in to the region, CO begins to form, and, finally, O2 formation is present in the dense core when the photon shielding is high enough (Tielens 2005; Wolfire 2011). In fact, PDRs are believed to be a major cache of the interstellar molecular abundance due to the stability of the dense cores.
It could be assumed that such a large flux of high-energy photons in the outer shells would remove any excess electron from an anion or even from many neutral radicals. However, this same process results in a veritable sea of elections that could attach to neutrals and actually lead to the creation of anions even in the Horsehead nebula PDR (Millar et al. 2007). Additionally, many anions are known to be surprisingly stable (Hammer et al. 2003; Simons 2008, 2011; Fortenberry & Crawford 2011b, 2011a; Fortenberry 2013), and electron attachment rates are also believed to be quite high in these regions (Millar et al. 2007). Several anions have also been shown to possess dipole-bound excited states, or threshold resonances, which may play a significant role in the creation and recreation of interstellar anions (Güthe et al. 2001; Carelli et al. 2013). The mechanism of radiative attachment (RA), outlined by Carelli et al. (2013) as radiative stabilization, describes attachment of an electron to a neutral species, A, through creation of the excited electronic state of the resultant anion:

Relaxation can take place such that the electronic ground state of the anion would be present (Carelli et al. 2013; Millar et al. 2007; Herbst & Osamura 2008).
The dipole-bound (and only) excited state of a small anion may function as the necessary excited state for RA. Dipole-bound states are known to exist for each of C4H−, C6H−, and C8H− (Pino et al. 2002). In order for such a state to be present, the dipole moment of the corresponding neutral, a radical for these systems, must be on the order of 2 D or larger (Simons 2008, 2011). For the 2Π ground states of C6H and C8H, the dipole moments are large enough to support a singlet dipole-bound excited state. C4H, 2Σ+ in its ground state (Fortenberry et al. 2010), has a relatively small dipole moment at 0.8 D (Graf et al. 2001). Hence, in order for C4H− to form, the radical must either excite out of the weakly dipolar  state into the large-dipole A 2Π state before undergoing RA, or it must form through another manner besides RA. As discussed by Gupta et al. (2007) and McCarthy & Thaddeus (2008), the need for radical excitation followed by RA could explain the very low [C4H−/C4H] ratio observed toward various interstellar objects (Agúndez et al. 2008; Cordiner et al. 2013). Even though these two states of C4H are "nearly degenerate" (Taylor et al. 1998), some additional energy is required to populate the A 2Π state, which, in turn, lowers the probability of electron attachment. Furthermore, C2H is also 2Σ+ in its ground state, but the excitation energy into the large-dipole A 2Π state is more than double its counterpart in C4H (Fortenberry et al. 2010), which may shed light on the even lower [C2H−/C2H] ratio proposed by Agúndez et al. (2008). C6H− and C8H− could be present in the Horsehead nebula PDR, as has been suggested from observations and modeling by Agúndez et al. (2008), but these longer anions may only be accessible from their dipole-bound excited states. However, C3H− has more than just a dipole-bound excited state.
state into the large-dipole A 2Π state before undergoing RA, or it must form through another manner besides RA. As discussed by Gupta et al. (2007) and McCarthy & Thaddeus (2008), the need for radical excitation followed by RA could explain the very low [C4H−/C4H] ratio observed toward various interstellar objects (Agúndez et al. 2008; Cordiner et al. 2013). Even though these two states of C4H are "nearly degenerate" (Taylor et al. 1998), some additional energy is required to populate the A 2Π state, which, in turn, lowers the probability of electron attachment. Furthermore, C2H is also 2Σ+ in its ground state, but the excitation energy into the large-dipole A 2Π state is more than double its counterpart in C4H (Fortenberry et al. 2010), which may shed light on the even lower [C2H−/C2H] ratio proposed by Agúndez et al. (2008). C6H− and C8H− could be present in the Horsehead nebula PDR, as has been suggested from observations and modeling by Agúndez et al. (2008), but these longer anions may only be accessible from their dipole-bound excited states. However, C3H− has more than just a dipole-bound excited state.
A few rare anions possess valence excited electronic states between the dipole-bound state and the ground electronic state (Fortenberry & Crawford 2011a, 2011b). As mentioned in the Introduction, 1 1A' C3H− is, thus far, the only anion composed solely of first-row atoms (and hydrogen) to possess a valence singlet excited state (Fortenberry 2013). The presence of two excited electronic states with the same spin multiplicity should increase the production of C3H− since multiple relaxation pathways exist. Beginning from the dipole-bound state, the excited anion can relax within the RA mechanism to the ground electronic state either directly or via the valence excited state first. Enough C3H− may then exist in a steady state to counterbalance the destructive photons present in this region.
If the Horsehead nebula PDR abundances of l-C3H+ from Pety et al. (2012) can be inferred to actually be 1 1A' C3H−, the [C3H−/C3H] ratio could be as high as 0.30 in the Horsehead nebula PDR. This is not as high as the upper limit proposed for [C6H−/C6H] at 8.9, but it is an order of magnitude larger than [C4H−/C4H] (Agúndez et al. 2008) as can be expected since the ground electronic state of C3H is strongly dipolar and that of C4H is not. The amount of C3H− should decrease as the observations move toward the dense molecular core due to the higher reactivity of this anion. The reaction cross-section of anions is much larger than in neutrals (Eichelberger et al. 2007), and C3H− could go through various destructive processes (Millar et al. 2007; Larsson et al. 2012) as the molecular density increases. Alternatively, this anion could exist within the observed sightline but on the outer edge of the PDR where the photon flux is small enough for a measurable population to be stable. In this region a longer path length of such material is also present away from the high AV dense core. Either way, the existence of 1 1A' C3H− in the Horsehead nebula PDR is feasible.
5. CONCLUSIONS
Since the link between l-C3H+ and the lines observed in the Horsehead nebula PDR by Pety et al. (2012) has recently been strongly questioned by Huang et al. (2013b), another viable candidate is necessary. The rotational lines seem to require a closed-shell quasi-linear structure composed of three carbon atoms along with a hydrogen atom. 1 1A' C3H− appears to be the most likely candidate. Here, the CcCR QFF has determined a Beff for this anion to be in error by 0.28% from that required to fit the observed lines. The use of the equilibrium rotational constants fortuitously lowers the error to 0.11%. However, the error reduction and error magnitudes themselves are in line with the computed C5N− rotational constants used in its interstellar detection. Additionally, the discrepancy between the Ae, Be, and Ce computed C3H− Deff and the Deff deduced from the observed interstellar rotational lines is similar to the De errors for C4H−, C3N−, and C5N− and less than that of C6H−, which are all reported for CCSD(T) computations, i.e., similar levels of theory. Hence, the consistency of the errors for C3H− with other anions previously detected in the ISM coupled with its matching the required spectral criteria and the rationale for its existence involving its valence and dipole-bound excited states, make this anion the strongest candidate carrier for the Horsehead nebula PDR lines and, potentially, the seventh and most recent anion detected in the ISM. It would also be the first detected interstellar odd-numbered carbon monohydrogen chain anion.
R.C.F. is currently supported on a NASA Postdoctoral Program Fellowship administered by Oak Ridge Associated Universities. NASA/SETI Institute Cooperative Agreement NNX12AG96A has funded the work undertaken by X.H. Support from NASA's Laboratory Astrophysics "Carbon in the Galaxy" Consortium Grant (NNH10ZDA001N) is gratefully acknowledged. The U.S. National Science Foundation (NSF) Multi-User Chemistry Research Instrumentation and Facility (CRIF:MU) award CHE-0741927 provided the computational hardware, and award NSF-1058420 has supported T.D.C. The CheMVP program was used to create Figure 1. The authors also acknowledge many others for their contributions to our astronomical understanding of this subject. These include, most notably: Dr. Michael C. McCarthy of the Harvard-Smithsonian Center for Astrophysics, Dr. Naseem Rangwala of the University of Colorado, Dr. Lou Allamandola of the NASA Ames Research Center, and Dr. Christiaan Boersma of the NASA Ames Research Center and San Jose State University.