
Abstract
In vitro research for the study of type 2 diabetes (T2D) is frequently limited by the availability of a functional model for islets of Langerhans. To overcome the limitations of obtaining pancreatic islets from different sources, such as animal models or human donors, immortalized cell lines as the insulin-producing INS1E β-cells have appeared as a valid alternative to model insulin-related diseases. However, immortalized cell lines are mainly used in flat surfaces or monolayer distributions, not resembling the spheroid-like architecture of the pancreatic islets. To generate islet-like structures, the use of scaffolds appeared as a valid tool to promote cell aggregations. Traditionally-used hydrogel encapsulation methods do not accomplish all the requisites for pancreatic tissue engineering, as its poor nutrient and oxygen diffusion induces cell death. Here, we use cryogelation technology to develop a more resemblance scaffold with the mechanical and physical properties needed to engineer pancreatic tissue. This study shows that carboxymethyl cellulose (CMC) cryogels prompted cells to generate β-cell clusters in comparison to gelatin-based scaffolds, that did not induce this cell organization. Moreover, the high porosity achieved with CMC cryogels allowed us to create specific range pseudoislets. Pseudoislets formed within CMC-scaffolds showed cell viability for up to 7 d and a better response to glucose over conventional monolayer cultures. Overall, our results demonstrate that CMC-scaffolds can be used to control the organization and function of insulin-producing β-cells, representing a suitable technique to generate β-cell clusters to study pancreatic islet function.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
The worldwide prevalence of type 2 diabetes (T2D) has been increasing over the last decades, attaining the status of a global pandemic [1]. T2D is a chronic metabolic disorder characterized by hyperglycemia. It usually occurs when the peripheral tissues cannot effectively use the insulin that pancreas produces. This situation leads to an increased insulin demand and therefore the insulin-producing β-cells respond by activating compensatory pathways to improve their secretory capacity. Over time, β-cells are no longer able to cope with the metabolic demands and T2D develops [1].
In vitro research for the study of T2D is frequently limited by the availability of a functional model for islets of Langerhans. Pancreatic islets are responsible for maintaining glucose homeostasis by secreting the glucose-lowering hormone insulin and its antagonist, glucagon. Cell lines are a suitable alternative to model T2D in vitro and avoid human donor material or primary mouse pancreatic islets. Both mouse insulinoma MIN6 and rat insulinoma INS1E cell lines are commonly used for in vitro research. Nevertheless, INS1E cells have been reported to present better responsiveness to glucose within the physiological range and relatively high insulin content [2, 3].
Monolayer cell cultures have been shown to function differently than cells in vivo, and results of in vitro tests may not accurately reflect cell response occurring in vivo [2]. Pancreatic islets are round-shaped cell aggregations of around 100 µm in diameter. Their size and shape determine their functionality, crucial to orchestrate the metabolic adjustments [4]. Indeed, β-cell aggregations into pseudoislets have been proven to represent a more suitable model to study β-cell function, demonstrating a better biological response than cultured monolayered cells [5, 6]. However, most of these studies use pseudoislets in suspension, therefore not representing an accurate image of the in vivo environment to study their behavior [7, 8].
To solve this problem, biomaterials and tissue engineering appeared as a valid alternative to generate 3D microenvironments. The use of scaffolds has allowed the generation of a wide variety of 3D environments that have enabled to better mimic the in vivo situation of each tissue (e.g. skeletal muscle [9, 10], intestine [11], or liver [12]). With this purpose, hydrogel encapsulation has appeared as the gold standard. This technique allows modulating the external morphology (i.e. lines [10], pillars [11], or meshes [13]), the stiffness, the pore size, or the biochemical cues to promote cell attachment [14, 15] to better fit with the needs of each engineered tissue. However, this approach entails several drawbacks that can end up in cell death. Exposure to the UV light or other toxic crosslinking reagents [16] or the small pore sizes (usually in the nanometer range) can lead to insufficient nutrient diffusion, clumping problems, hypoxia, and difficulties inducing vascularization [17, 18]. These limitations make this approximation non-suitable for all kinds of tissues. Additionally, these problems become more detrimental when cell aggregations are encapsulated [19, 20] and explicitly challenging with β-cells, specialized cell types adapted to sense rapid changes in glucose [21]. Therefore, a perturbation of the glucose-sensing machinery in these cells can entail a suboptimal insulin release.
Extensive efforts have been made to develop the ideal scaffold to support these cells. Such scaffold must be fabricated with biocompatible polymers, suitable for mammalian cell growth. It must be highly porous to allow adequate oxygen and nutrient diffusion, and it also needs to be mechanically stable, with the appropriate structure to avoid shear-stress-induced cell damage [22]. With the intention to aggregate β-cells in a 3D microenvironment, we engineered gelatin and carboxymethyl cellulose (CMC)-based cryogels, which enabled us to design a supportive material for the proliferation and growth of β-cells.
Cryogels, are sponge-like scaffolds with micrometer pore range formed at sub-zero temperatures [23]. This technique entails several advantages compared to other approaches. It allows high pore diameters [24], fundamental to precisely control cell aggregates diameters. Moreover, it provides the mechanical support suitable to manipulate the structure easily [25]. And finally, it enables cell seeding after polymerization of the scaffold, therefore avoiding exposure to harmful crosslinking reagents or UV light. Additionally, both materials have been reported to present excellent biocompatible properties [25, 26]. Gelatin is a derivate of collagen which displays weak mechanical properties and presents the tripeptide Arg-Gly-Asp (RGD), a cell-binding motif [27]. On the other hand, CMC is a derivate compound from cellulose, which has better mechanical stability and good biocompatibility but without the presence of cell-binding motifs [28].
In this study, INS1E cells were seeded onto 3D gelatin and CMC scaffolds to investigate the substrate architecture's effect on the cell's organization and function. We examined cell viability and formation of cell-clusters after 1, 4, and 7 d of culture, and we compared them with cells seeded in a plate. CMC-based scaffolds promoted the formation of INS1E aggregations into pseudoislets, whereas dispersed organization was observed in gelatin-based cryogels. We also show that INS1E pseudoislets ameliorated their response to glucose stimuli and presented a more closely related mature β-cell phenotype than non-organized cells seeded in gelatin-based cryogels or a traditional well-plate.
Our results demonstrate that scaffold biomaterials can be used to control the organization and enhance the function of insulin-producing β-cells. These advantageous properties make this approach an ideal model for the study of pancreatic islet function, representing a valuable tool for 3D diabetes drug testing and development.
2. Materials and methods
2.1. Cryogel fabrication
CMC (Sigma Aldrich, Germany), or gelatin from porcine skin (Sigma Aldrich, Germany) were diluted into MilliQ water with stirring conditions at 45 °C. Once the prepolymer solution is homogeneous, the crosslinking reagents were prepared; MES buffer from MES hydrate (Sigma Aldrich, Germany) at 0.5 M and pH at 5.5, adipic acid dihydrazide (AAD, Sigma Aldrich, Germany) at 50 mg ml−1, and N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride (EDC, Sigma Aldrich, Germany) at 1 μg μl−1 all dissolved in MilliQ water and vortexed to ensure the homogeneity in all the solution. Prepolymer solution, 1 ml of the prepolymer, 50 mM of MES buffer, 1.83 mM of AAD, and 18.9 μM of EDC were added into a tube vigorously pipetted, avoiding early crosslinking before freezing. For stained cryogels, 10.9 μM fluoresceinamine (Sigma Aldrich, Germany) was added to the final prepolymer solution. Then polydimethylsiloxane (PDMS) molds were filled with the final prepolymer solution. These molds consist of a PDMS pool with 1 mm high and 10 mm of diameter over a squared 24 × 24 mm cover glass. After filling, they were placed into a −20 °C freezer for 24 h. The next day, the crosslinked cryogels were removed carefully from the mold and then submerged into consecutive 5 min cleaning steps; 1 × MilliQ water, 1 × 100 mM NaOH (Panreac, Germany), 1 × 10 mM ethylenediaminetetraacetic acid (EDTA, Sigma Aldrich, Germany), 1 × MilliQ and 3 × phosphate buffered saline (PBS, 0.01 M phosphate buffer, 0.0027 M potassium chloride and 0.137 M sodium chloride, pH 7.4, Sigma-Aldrich, Germany). Once finished the cleaning protocol, the cryogels were sterilized for further cell seeding experiments in an autoclave.
2.2. Biomaterial characterization
2.2.1. Pore analysis
For the pore analysis, the fibers of the cryogel were stained, adding 10.9 μM of fluoresceinamine. Once stained, z-stack images were taken in a confocal microscope, and the different pore diameters were quantified with ImageJ version 1.52b software (National Institutes of Health).
Scanning electron microscopy (SEM) observations were performed with a NOVA NanoSEM 230 microscope at 10 kV. Before imaging, cryogel scaffolds were subjected to consecutive ethanol dehydration steps, washing the cryogels with ethanol 50%, 70%, 80%, 90%, 96% (x2), and 99.5%. Once all the water was substituted for ethanol, ethanol was replaced by CO2, performing a critical point dry step. A final stage of carbon sputtering was done before SEM images were taken.
2.2.2. Swelling
Swelling experiments were performed to calculate the water uptake ratio by a cryogel. Cryogels were fabricated as explained previously, and after sterilizing, cryogels were dried at room temperature and weighted. Next, cryogels were submerged into MilliQ water for 24 h, when they reached equilibrium and weighted again. The swelling ratio was calculated as follows:
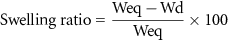
where Weq is the weight in equilibrium and Wd is the dry weight. Three cryogels per condition were measured in this assay.
2.2.3. Stiffness
Compression assays were performed to determine the stiffness of our samples. The compression assays were performed in a Zwick Z0.5 TN instrument (Zwick-Roell) with a 5 N load cell. The experiment was performed with samples at room temperature up to 30% final compression range at 0.1 mN of preloading force and 20%/minute of strain rate. Finally, the Young modulus was calculated from 10% to 20% of compression from the line's slope. In these experiments, three measurements per cryogel and three cryogels per condition were tested.
2.2.4. Permeability assay
Cryogels were placed over a transwell inside a 12 well-plate. 500 ml of 1.5 mM fluorescein (Sigma Aldrich) were added at the transwell's upper compartment, and 1.5 ml of PBS were added in the lower chamber. 100 μl of PBS from the well were taken out in consecutive times. The same amount of fresh PBS was added to the lower compartment to readjust the volume. This procedure was repeated during different times up to an overnight when equilibrium was reached. Finally, the concentration of fluorescein was obtained by absorbance measurements at 494 nm with a Power wave X microplate spectrophotometer.
Permeability was calculated in the linear part of the diffusion curve by the following equation:
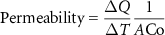
where Q is the milligrams of fluorescein that pass through the cryogel at a specific time, T is the time, A is the area of the cryogel, and Co is the initial concentration of fluorescein. Finally, the permeability of the cryogel was the difference between the total permeability and the permeability of the transwell.
2.3. Cell culture
Rat pancreatic β-cell line INS1E cells were cultured in RPMI-1640 with 11.1 mM glucose, supplemented with 10 mM HEPES (Gibco), 2 mM L-glutamine (Gibco), 1 mM sodium-pyruvate (Gibco), 0.05 mM de 2-mercaptoethanol (Thermofisher), 10% fetal bovine serum (FBS) (v/v) (Thermofisher) and 1% penicillin/streptomycin (v/v) (Thermofisher) (complete media). When cells reached confluency, cells were trypsinized with 0.25 trypsin/0.1% EDTA and plated in a new flask at 1:4 density. Cells were maintained in an incubator at 37 °C and 5% CO2.
2.4. Cell seeding
Cryogels were dried for 30 min. After trypsinization of the cells, 200.000 cells mixed with a drop of 20 μl of medium were seeded in each cryogel. After seeding the cells, cryogels were left at room temperature (RT) for 20 min, and a complete RPMI-1640 medium was added and left at the incubator until needed for experimental assay.
2.5. Viability
2.5.1. Live/dead
Viability assays were performed with the live/dead assay kit (Thermofischer) according to manufacturer instructions. The assays were performed at days 1, 4, and 7 of culture after seeding in traditional well plates and gelatin and CMC cryogels. Briefly, the cryogels were washed 5 min with PBS three times to replace culture medium and incubated with the working solution (4 μM EthD-1, 2 μM Calcein AM, and 16.2 μM Hoechst) for 25 min at 37 °C. Then cryogels were washed three times with PBS. Finally, confocal images were taken using a Zeiss LSM 800 confocal microscope. The quantification of live/dead ratio was calculated as follows:
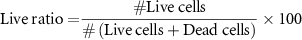
2.5.2. AlamarBlue
AlamarBlue test was performed according to manufacturer specifications. Briefly, the medium was removed from the well plate and substituted for new RPMI-1640 with 11.1 mM of glucose medium with 1:10 dilution of AlamarBlue. After 2 h incubation, 100 μl of each condition was placed in a well of 96 well-plate and read in a Power wave X microplate spectrophotometer at 570 nm wavelength.
2.6. Immunostaining
For confocal analysis, stained cryogels were used. After culturing the cells, cryogels were washed with PBS and fixed with 10% formalin solution (Sigma-Aldrich) for 30 min. Then, cryogels were washed with tris buffered saline (TBS, Canvax Biotech) and permeabilized with 0.1% Triton X-100 (v/v) (Sigma-Aldrich) solution in TBS for 15 min. Cryogels were blocked with 0.5% Triton X-100 (v/v) and 3% donkey serum (v/v) (Sigma-Aldrich) into TBS for 2 h. Cryogels were incubated overnight with primary antibodies against rabbit-anti Ki-67 (1:250, Invitrogen) and mouse anti-insulin (1:500, Origene) in a blocking solution. The following day, cryogels were washed with permeabilization solution and incubated with secondary antibodies for 2 h at room temperature (Alexa-Fluor 647 conjugate anti-mouse 1:200 and Alexa-Fluor 568 conjugate anti-rabbit 1:200, Invitrogen). DAPI (1:1000 Thermofisher) was used to stain nuclei. Finally, cryogels were washed with TBS for 15 min and stored at 4 °C until confocal microscopy acquisition. Images were taken using an LSM 800 from Zeiss.
2.7. Confocal microscopy
Different z-stacks were acquired for pore quantification, and pores were analyzed from images with 20 μm of z-gap between them. For quantification of live/dead and proliferation assays, 20 images per cryogel were taken and then analyzed. All images were acquired using an LSM 800 from Zeiss.
2.8. Gene expression analysis
Total RNA was isolated from the sample cells using the RNeasy MinElute cleanup kit (Qiagen) following the manufacturer's instructions. Of total RNA, 200 ng were used for reverse transcription using high capacity cDNA reverse transcription kit (Applied Biosystems). Quantitative PCR reactions were run using SyberGreen (Invitrogen) in a 7900HT fast real-time PCR system (Applied Biosystems) as described elsewhere [29]. Primer sequences used for gene expression analysis are listed in supplementary table 1 (available online at stacks.iop.org/BF/13/035044/mmedia). Tbp1 was used to normalize the mRNA expression of genes of interest.
2.9. Glucose-stimulated insulin secretion (GSIS)
Cells seeded within the cryogels or in a well plate were preincubated with Krebs-Ringer bicarbonate HEPES buffer solution (115 mM NaCl, 24 mM NaHCO3, 5 mM KCL, 1 mM MgCa2 × 6H2O, 1 mM CaCl2 × 2H2O and 20 mM HEPES, pH 7.4) containing 2.8 mM glucose for 30 min. Then, cells were incubated at low glucose (2.8 mM) for 1 h, followed by incubation at high glucose (16.7 mM) and an additional step at 2.8 mM. After each incubation step, supernatants were collected, and cellular insulin contents were recovered in acid-acetic lysis buffer (glacial acetic acid 5.75%). Insulin concentration was determined by ELISA experiments.
2.10. ELISA (enzyme-linked immunoabsorbent assay)
Briefly, mouse mAB insulin 26.6 nM of capture antibody (Novus) was diluted into coating buffer (0.05 M of sodium carbonate and sodium bicarbonate, pH 9.6) and placed into 96-well plate at 4 °C O/N. Next, samples were placed into the plate, and a calibration curve, previously optimized, was performed. After protein attachment into the primary antibody, the biotinylated secondary anti-insulin antibody was placed at 0.2 nM. Next, streptavidin was added at 4.73 nM (Thermo Scientific). Then, citrate buffer (0.04 M sodium citrate, pH 5.4, 96 ug ml−1 of tetramethylbenzidine and 0.004% of oxygen peroxide) was added to start the reaction. Finally, 4 M sulfuric acid was added to stop the reaction. The colorimetric quantification was made with a Power wave X microplate spectrophotometer at 490 nm of wavelength.
2.11. Statistical analysis
Data are expressed as the mean ± SEM of at least three independent experiments with three replicates each. Statistical significance was determined by a two-tailed Student t-test and one-way ANOVA with post hoc Tukey test as appropriate using GraphPad Prism version 8.3.0. Results were considered significant at p < 0.05.
3. Results and discussion
3.1. Cryogel scaffold characterization
The difficulty of obtaining pancreatic islets from human patients or rodents conceives a big deal to study T2D in vitro. The limited availability of primary pancreatic islets has prompted investigators to use cell lines to study β-cell function to model this disease. However, two-dimensional monolayer cell cultures fail to recapitulate the main key characteristics of primary β-cells. The lack of a 3D structure has been proven to be one of the main problems associated with decreased functionality [30]. Despite this limitation, many in vitro approaches focused on generating 3D functional pancreatic tissue using hanging-drop methods [31] or cell encapsulation into hydrogels [13, 32]. The origin of the pancreatic cells is from animals, cadaveric donors, or immortalized cell lines. However, having cell aggregations of 100 µm (the average size of a pancreatic islet) drives encapsulation problems, such as lack of oxygen and nutrient diffusion [20, 21].
Thereby, it is complicated to engineer a fully functional pancreatic tissue. To solve the problems exposed in β-cell obtention and cell encapsulation, we combined clustering cell ability with cryogelation to generate the scaffold. We developed a proper scaffold for in vivo mimicking of β-cells while improving its diffusion with this approximation. These scaffolds were generated at sub-zero temperatures. Thus, while the material fibers crosslink between them, water–ice crystals are formed. When thawed, these ice crystals lead to empty pores (figure 1(a)). This scaffold has a sponge-like highly interconnected structure with a controllable pore size. This feature makes this scaffold a good material to handle due to its elastic properties (supplementary video 1). Moreover, the good mechanical stability and elasticity allows manipulating and moving the scaffold from one place to another without breaking it or suffering any damage (figure 1(b)).
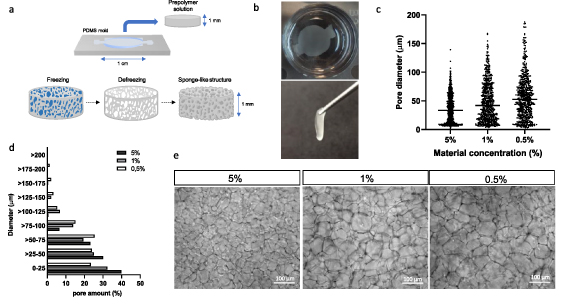
Figure 1. General overview of the study. (a) Cryogel fabrication process scheme. Cylinder-like scaffolds were generated, placing the prepolymer solution in a PDMS mold. After, the prepolymer solution within the mold is placed at −20 °C. Thus, when material crosslinks, water–ice crystals are formed. When thawed, this ice leads to the porosity of the scaffold. (b) Images of the cryogel scaffold when deep in water (left) and when tweezered (right), proving the mechanical resistance of the scaffold. (c) Pore distribution using different concentrations of the material. (d) Percentage of pores in different diameter ranges according to the concentration of the material. Of note, the higher the concentration, the higher the percentage of pores between 0 and 25 μm of length. (e) General magnifying glass images of the scaffold structure among different material concentrations. Scale bar = 100 μm.
Download figure:
Standard image High-resolution imageOne of the remarkable properties of cryogelation is the possibility to modulate the pore size. For this, we studied different material (gelatin and CMC) concentrations and quantified the diameter of each pore. Regardless of the material type, in the case of 5% (w/v), porosity ranged from 10 up to 100 μm of diameter while in 1%, pores ranged from 10 to 150 μm and at 0.5%, pores ranged up to 200 μm (figure 1(c)). Percentage of pores in different diameter ranges according to the concentration of the material are shown in figure 1(d). Of note, the higher the concentration, the higher the percentage of pores between 0 and 25 μm of length. By observing the fiber mesh, we can note this ascendant porosity range (figure 1(e)). Our goal was to generate β-cell aggregations that match in size with primary pancreatic islets, which are very heterogeneous in size. Therefore, the pore distribution of our scaffold should also present a wide range distribution. Additionally, in vivo small pancreatic islets are more common than big ones [33, 34]. Knowing all this, we concluded that with 1% cryogels, we achieved the porosity that suited all the needs exposed.
3.2. CMC cryogel has good physical properties, similar to the native pancreas
We tested two different materials to develop this new approximation, each with various beneficial properties to aggregate β-cells. CMC is a biocompatible biomaterial with good mechanical stability and non-degradable by mammalian cells [35]. Importantly, in vitro and in vivo evaluations of those cellulose-based materials have demonstrated excellent biocompatibility [25, 26]. The other material studied is gelatin, a biocompatible biomaterial, biodegradable, and with RGD cell adhesive points, but with low mechanical stability [27]. In this case, gelatin was selected because it was previously studied that it can enhance pseudoislet formation [30, 36].
To prove that the pore distribution fits our needs and does not vary between materials, we studied the pore distribution of the 1% CMC cryogels and 1% gelatin cryogels (figure 1(a)). We could observe that by changing the material, the pore distribution did not change. The pores, as expected, range from 10 up to 170 μm. Despite a high amount of small pores found in the cryogel, big pores were also observed. As previously reported, this pore distribution fits with the range that we want to generate cell aggregations.
Additionally, the porosity was analyzed through SEM images (figure 2(b)) and confocal images (figure 2(c)). In SEM images acquired after dehydration and critical point dry, it can be observed that pore distribution is heterogeneous, and pores from many different sizes were formed. Following the same tendency, in confocal images, where fibers are stained in green, many different pore sizes can be observed, both in CMC and gelatin cryogels (figure 2(c)). Also, in confocal images, it can be appreciated that there are no significant visual differences in pore size between CMC and gelatin cryogels.
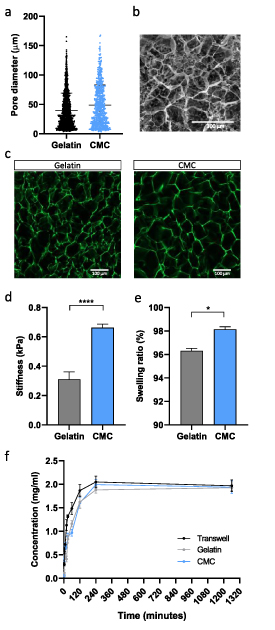
Figure 2. Mechanical characterization of the scaffold. (a) Comparison of the pore distribution between gelatin and CMC cryogels with the same material concentration (1% (w/v)). (b) SEM images of the cryogel porosity. Scale bar = 300 μm. (c) Aminofluorescein stained fibers of gelatin (left) and CMC (right) obtained with confocal microscopy. Scale bar = 100 μm. (d) Stiffness results from different cryogels. (e) Swelling ratio. (f) Diffusion profile of fluorescein through the scaffold. Results are expressed as the mean ± SEM. *p < 0.05, ****p < 0.0001.
Download figure:
Standard image High-resolution imageKnowing that the porosity is in the desired range to form pseudoislets with a similar size to the in vivo, the next step was to check our scaffold mechanical properties. As we wanted to mimic the extracellular matrix and the pancreatic islet environment, the stiffness is an essential property. The ECM mainly has the objective to support cells and plays an important role in cell viability and functionality by doting the cells of specific biochemical and physical signals [37]. Moreover, knowing that cells modulate their behavior in different substrate stiffness [39, 40, 41], maintaining a similar stiffness as the pancreas should help the cells to function and differentiate better. Compression assays were performed to study bulk stiffness by studying scaffolds Young modulus. In elastic solids with a determined section and length, the main stiffness variable is the Young modulus, so studying this intensive material property we can get trustable stiffness values. These CMC cryogel are stiffer than gelatin (0.67 ± 0.08 kPa vs 0.30 ± 0.1 kPa) (figure 2(d)) and the stiffness achieved correlates well with the proper stiffness defined for pancreatic tissue. As the pancreas is a soft tissue, its stiffness ranges from 0.1 to 10 kPa [40]. The pancreas seems to respond properly to this interval's lower stiffness, as cells can increase insulin mRNA expression and glucose sensitivity [41]. In other approaches, the stiffness of native healthy pancreas was set as approximately 1 kPa when measured by magnetic resonance elastography [42, 43], a value that fits with the scaffold stiffness achieved. Also, soft scaffolds favor cell coalescence and preserve the cluster-like organization, while in stiff substrates, the extracellular-cell interactions cause cell scattering and loss of islet-like structure [44].
Another feature that we wanted to improve is the diffusion of nutrients through the scaffold. Swelling is the water uptake capability of a hydrogel, an indirect measurement of pore interconnectivity [45]. The high pore diameter distribution and the high pore interconnectivity, typical from cryogels, enhances this swelling property [46]. Also, the better are these properties, the faster diffusion among all the scaffold. After only 24 h, our cryogel reached an equilibrium, with a swelling ratio of 98.14 ± 0.32% for CMC and 96.30 ± 0.38% for gelatin cryogel (figure 2(e)). Although this property is higher in CMC cryogels than in gelatin, as expected, both ratios are higher than 95%. This high percentage indicates that the scaffold's structure is highly interconnected, as water could colonize all the scaffold structure after drying.
Moreover, one of the strong points of this approach is the high diffusion of oxygen and nutrients through the scaffold because high permeability of the scaffold. A fluorescein permeation experiment was performed to test scaffold features. We could observe that the control sample, where the transwell was placed without cryogel, reaches equilibrium faster than the cryogels. However, these conditions reached equilibrium equally after 240 min (figure 2(f)). Calculating the scaffold's permeability, we obtained values of 5.72 mm s−1 in CMC cryogels and 0.61 mm s−1 in gelatin cryogels. This difference in permeability indicates that CMC cryogels are more permeable than gelatin cryogels [46]. This rapid equilibrium reached shows that the cryogel has barely any interaction as a diffusion barrier. After 3 min, fluorescein can be found in the lower part of the transwell. This high permeability ensures us to generate a microporous cryogel able to sustain cells in all the scaffold's depths with no hypoxia or lack of nutrient problems.
We can conclude that our scaffold satisfies all the mechanical and physical needs of the β-cells. Overall, these results show a well-defined and reproducible method to afford non-degradable and microporous cell supportive scaffold.
3.3. CMC-based scaffold enhances INS1E pseudoislet formation
To generate a functional 3D structure able to support β-cells, we seeded INS1E cells onto the scaffolds. After cell seeding, INS1E morphology was evaluated in gelatin and CMC-based scaffolds at days 1, 4, and 7. Interestingly, at day 1 after seeding, instead of the typical monolayer architecture, cells cultured in 3D CMC cryogel scaffolds formed round-shaped clusters (figure 3(b)), morphologically resembling pancreatic islets (figure 3(a)). In contrast, a dispersed organization was observed in gelatin-based cryogels (figure 3(b)). The difference in cell organization observed in CMC and gelatin scaffolds can be explained by the presence or absence of the cell adhesion motifs in these structures. Gelatin is known to contain RGD (arginine-glycine-aspartic acid) motifs, cell adhesion sites found in several ECM proteins [47]. Hence, gelatin has a profound effect on the ability of cells to adhere to this material. On the other hand, CMC cryogels do not present cell-binding motifs, so it displays shallow adhesion properties for anchorage-dependent growth of INS1E cells, promoting cells to interact between them and to cluster together, forming islet-like structures.
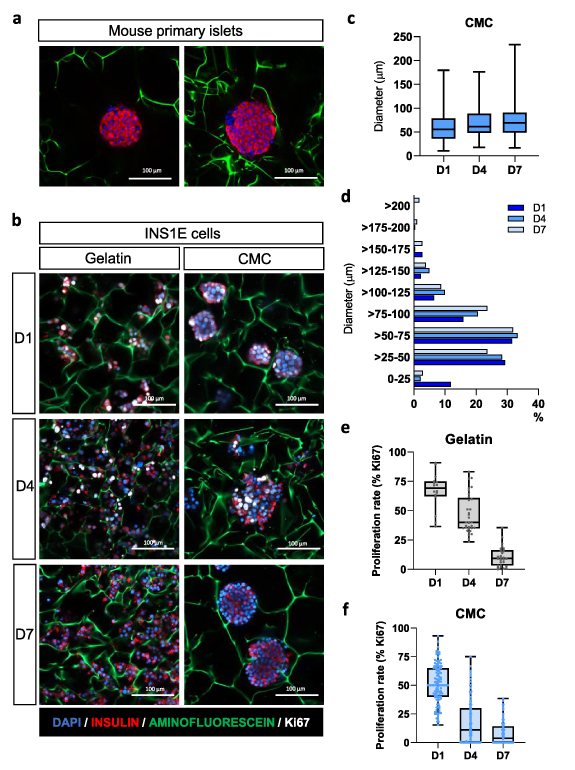
Figure 3. Pseudoislets generated in the scaffold have a high resemblance with primary pancreatic islets. (a) Immunostaining of mouse primary pancreatic islets within a 1% CMC-scaffold stained for insulin (red), Ki67 (white) and nuclei (DAPI). Aminofluorescein was used to stain the fibers of the cryogel (green). Scale bar = 100 μm. (b) Representative images of INS1E cells inside the scaffold at days 1, 4, and 7 stained as in a. Note that INS1E cells within CMC scaffolds aggregate forming pseudoislets already 1 d after seeding. In contrast, cells within the gelatin are spread out. Scale bar = 100 μm. (c) Diameter of the pseudoislets on days 1, 4, and 7. (d) Diameter distribution of the pseudoislets formed inside the CMC cryogel along the week. (e) and (f) Proliferation rate (calculated as the percentage of Ki67-positive β-cells concerning the total number of β-cells) of INS1E cells inside the (e) gelatin cryogel and (f) CMC cryogel. Results are expressed as dot-box plots indicating the first quartile, the median, the third quartile, and the minimum and maximum values.
Download figure:
Standard image High-resolution imageConfocal image analysis of INS1E clusters revealed that at day 1 pseudoislets were about 60.6 µm in diameter, and they increased in size during the first 7 d, reaching an average diameter of 75.5 µm after 1 week of culture. At this point, we obtained a heterogeneous pseudoislet population in size, ranging from 16.8 to 216.7 µm (figure 3(c)). Primary rodent pancreatic islets present a considerable heterogeneity in size and shape, varying from small cell clusters to larger islets [48, 49]. Several studies have revealed that islet heterogeneity influences the insulin secretory response of β-cells, so heterogeneity should be an essential consideration when understanding T2D pathogenesis, both at a single-cell and islet level [51, 52, 53]. At day 1, up to 12% of the clusters ranged from 0 to 25 µm, whereas at days 4 and 7, aggregations smaller than 25 µm represented less than 3% (figure 3(d)). Pseudoislets bigger than 200 µm were only observed at day 7. Of note, percentages of clusters higher than 25 µm diameter correlate with those of the scaffold porous sizes (figure 1(d)), indicating that cells keep proliferating until they reach the porous diameter.
Indeed, cells within the gelatin-based cryogel presented high proliferation rates at day 1 after seeding (67.0 ± 3.9%), determined by immunostaining of Ki67, but this ratio decreased to 46.8 ± 2.9% at day 4 and 10.8 ± 1.6% at day 7 (figure 3(e)). This trend was also observed in cells cultured within the CMC-based scaffold, presenting 51.3 ± 1.6% at day 1, 17.1 ± 1.5% at day 4, and 8.0 ± 1.0% at day 7 (figure 3(f)). In rodent islets, the proliferative capacity of β-cells is confined to the early stages of life, linked to an immature functional phenotype [52, 53]. Thus, reduced proliferative capacity is one of the characteristics of mature β-cells, and maturation of β-cells defines their functional identity. Therefore, a strategy to obtain a heterogeneous population of islet cell clusters with low proliferation capacity offers excellent potential to engineer a model for the study of β-cell function and viability.
3.4. CMC-based scaffold maintains cell viability and promotes β-cell identity
Since 3D pseudoislets may have less access to nutrients, it was of interest to establish cell viability along one week of culture. Cell viability was assessed at culture days 1, 4, and 7 by a live/dead assay (figure 4(a)). We found that after 7 d of culture, encapsulated cells retained their viability compared to non-encapsulated cells, and both gelatin and CMC scaffolds presented a similar percentage of viability (figures 4(a) and (b)). Changes in viability or cell proliferation can be easily detected with the AlamarBlue test. Encapsulated cells at day 7 showed decreased metabolic activity (figure 4(c)). As no differences were observed in cell viability (figure 4(a)), this decreased metabolic activity correlates with a reduced proliferation ratio, confirming our previous results (figure 3). Overall, these results demonstrated that the highly porous cryogels were suited for engineering cell-supportive tissue scaffolds, facilitating the diffusion of oxygen and nutrients, and enabling cell viability for up to 7 d.
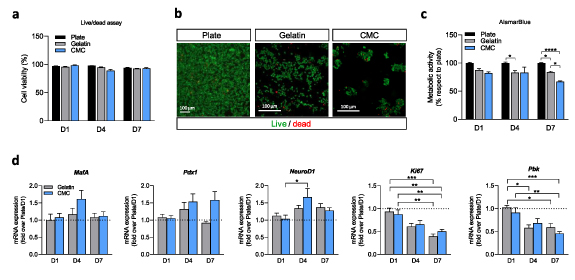
Figure 4. Cell viability and β-cell identity are preserved when pseudoislets are generated inside the scaffold. (a) Analysis of live/dead INS1E cells at day 1, 4, and 7 seeded in a traditional plate and within the scaffolds. (b) Representative fluorescent images were taken by confocal microscopy of cells seeded in a monolayer and gelatin and CMC-based scaffolds. Live cells are marked with calcein AM in green, and dead cells are marked with EthD-1 in red. Note that cells within the CMC cryogel appear as clusters. Scale bar = 100 μm. (c) Alamar blue test of the cells in monolayer at the plate, monolayer on the gelatin cryogel and in the form of pseudoislets in the CMC cryogels, at days 1, 4, and 7. Data are shown relative to cells seeded in a plate. (d) Gene expression analysis of the β-cell identity markers, MafA, Pdx1, and NeuroD1, and proliferation markers Ki67 and Pbk. Gene expression was normalized against Tbp1. Results are expressed as mean ± SEM from three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Download figure:
Standard image High-resolution imageThe ability of CMC scaffolds to efficiently aggregate single cells into engineered pseudoislets, with round-shaped structures similar to native islets, prompted us to examine the gene expression profile of these pseudoislets over time compared to gelatin-based monolayer INS1E cells and INS1E cells cultured without a 3D scaffold. We first focused on the genes encoding MafA, Pdx1, and NeuroD1, three β-cell specific transcription factors (TFs) involved in β-cell functionality. Although many TFs have been involved in the maintenance of the β-cell identity, these specific transcriptional regulators have been demonstrated to play a crucial role in maintaining the function of the insulin-producing cells. Indeed, it has been demonstrated that these TF activates the insulin gene expression in a coordinated and synergistic manner in response to increased glucose levels. Furthermore, the fine-tune regulation of these TF ensures β-cell identity [54, 55, 58].
Interestingly, cells supported within the cellulose-based scaffold presented a gradually increased expression level of the β-cell specific marker Pdx1 concerning cells cultured in a well plate (figure 4(d)), even though results are not statistically significant. The results obtained are consistent with previous works demonstrating how reaggregating cells to form 3D spheroids significantly enhances the gene expression profile of β-cells [56]. Similarly, recapitulating endocrine cell clustering in culture has been demonstrated to foster the maturation of human stem-cell-derived β-cells [57].
This better-differentiated phenotype of β-cells when cultured within a 3D extracellular matrix, is consistent with the decreased proliferation markers, Ki67 and Pbk (figure 4(d)), corroborating the balance between an increased β-cell identity and a reduced ability to proliferate of these cells [52].
3.5. Cell aggregation improves GSIS and can be used as a suitable cellular model for the study of the β-cell function
Several findings indicate that islet architecture has a pivotal role in determining β-cell functionality as cell-cell interactions are fundamental for the correct cellular function [58, 59]. Indeed, it has been described that the secretory response of structurally coupled β-cells is higher than that of insulin-producing β-cells not arranged within the islet architecture [60, 61]. INS1E cells traditionally seeded in a monolayer do not present reproducible responses to dynamic glucose stimulations [6].
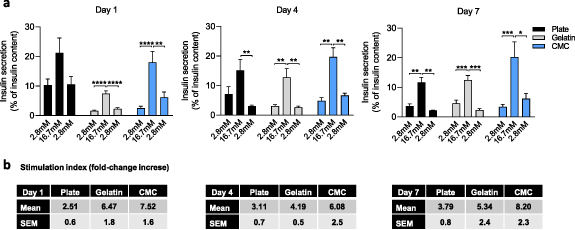
To determine whether pseudoislet formation within the cryogel correlates with increased β-cell function, we tested the dynamic response of pseudoislets to glucose. To check islet functionality, a GSIS assay, which defines the ability of β-cells to secrete the suitable amount of insulin in response to proportional extracellular glucose stimuli, was performed in all conditions. As shown in figure 5(a), cell clustering improved the insulin secretion stimulation index under high glucose stimulation concerning the basal insulin secreted in low glucose conditions. This result demonstrates the benefit of cell aggregation in islet functionality. For primary islets of Langerhans, a threshold stimulation index of at least five defines a functional response, and often immortalized β-cell lines do not reach this threshold level or display a reproducible behavior. Indeed, at day 1, INS1E cells seeded in a 48 well-plate presented a 2.51 ± 0.6 fold increase of insulin secretion when cells were challenged with 16.7 mM compared to cells incubated with 2.8 mM glucose. Cells seeded in gelatin cryogels showed a 6.47 ± 1.8 fold increase. Interestingly, we reached a fold increase of 7.52 ± 1.6 of insulin secretion when CMC-based pseudoislets were challenged with 16.7 mM glucose. This trend was repeated along the week, indicating that the stimulation index for insulin response to glucose is significantly higher in pseudoislets than dispersed and non-organized cells (figure 5(b)).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. Pseudoislet formation enhances β-cell responsiveness to glucose. (a) Glucose-stimulated insulin secretion (GSIS) assay at day 1, 4, and 7 in plate monolayers, gelatin cryogels, and CMC cryogels. For the GSIS assays, cells were incubated for 1 h at 2.8 mM glucose, followed by 16.7 mM glucose, and returning to basal condition (2.8 mM glucose). Results are expressed as the percentage of insulin secreted related to the corresponding sample ± SEM's total insulin content from three independent experiments, each one including at least three different replicates per condition. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (b) Stimulation index, calculated as the fold-change increased between the insulin secreted at basal levels (first incubation with 2.8 mM glucose) and after challenging the cells with 16.7 mM glucose. Results are expressed as the mean ± SEM.
Download figure:
Standard image High-resolution image{kind=link}
Like other tissues, β-cell functionality is greatly influenced by cell-cell and cell-matrix interactions, controlling basal and stimulated insulin secretion [62, 63]. Remarkably, GSIS defects in monolayer INS1E cell cultures arise from an increased insulin release under basal conditions (2.8 mM glucose) in addition to a decreased insulin secretory response under stimulatory glucose conditions. These results are consistent with other studies showing that aggregating β-cells enhances the secretory responsiveness to nutrients compared with cells configured as monolayers. And it also suggests that β-cells interactions might be sufficient to sustain a normal glucose response.
Therefore, our study validates that a correct structural arrangement is essential for appropriate insulin response, demonstrating a robust GSIS by pseudoislets formed within a cryogel.
4. Conclusions
An increasing need to engineer advanced 3D scaffolds for tissue engineering has emerged to provide cellular structural support and mimic the complicated physical and biochemical properties of the native extracellular matrix. With this improvement, more resemblance tissues can be engineered for many applications as drug screening or disease modeling. Until now, many tissues have been generated in the laboratory. Particular attention must be paid when engineering islet-like structures as an adequate round-shaped islet architecture is necessary to maintain and improve β-cell functionality. Moreover, the formation of islet-like structures or pseudoislets, with the consequent β-cell communications, is required for an appropriate insulin secretory response [64, 65].
Traditionally, pancreatic islets have been encapsulated inside hydrogels, generating a scaffold with interesting properties [13, 32, 66, 67], but lacking oxygen and nutrient diffusion throughout all the scaffold. This decreased diffusion rate usually results in an impaired GSIS profile. In this sense, cryogels appeared as interesting alternatives [24], as they present high pore sizes and therefore better permeation ability. Additionally, cryogels present a strong potential to aggregate single cells into a pseudoislet architecture [24, 56, 68]. Other studies have already exhibited the feasibility of aggregating pancreatic β-cells into microporous scaffolds, however most of them do not reproduce the islet functionality [69, 70, 72, 73] or obviate the role of the 3D microenvironment, not mimicking the physiological environment of the pancreatic islets [5, 56, 58, 71]. And even though previous studies have used other materials such as electrospung gelatin scaffolds to generate ∼100 µm pseudoislets surrounded with a matrix and with promising insulin secretion [30], to the best of our knowledge this is the first study using CMC scaffolds to generate viable and functional pseudoislets.
This study reports a new CMC cryogel scaffold that favors pseudoislet generation and functionality. Here, we proved that the cryogelation allows to generate a sponge-like scaffold with controllable structural properties. We demonstrated that we can create and modulate a wide range of porosity that fits with primary pancreatic islets' size and shape. Our scaffold's diffusion and permeability overcome some of the most problematic conditions, such as the lack of nutrient and oxygen diffusion through all the scaffold. Moreover, the mechanical properties of the cryogels match with the previously reported stiffness of the native pancreas ranging around 1 kPa. All these properties of cryogels favor the viability of the β-cells, promote their β-cell identity and increase their glucose responsiveness.
In summary, in this study, we have generated a new approximation to engineer pancreatic tissue, combining the cryogelation technique with cell aggregation in microporous scaffolds. Because cell clustering improves β-cell identity and functionality, our results demonstrated the feasibility of using these microporous gel materials as 3D scaffolds culturing islet-like cell aggregates as an in vitro model to study T2D and other related diseases.
Acknowledgments
The authors thank Dr Rosa Gasa (IDIBAPS) for providing INS1E cells and Dr Joan Marc Servitja for providing mouse pancreatic islets to perform immunostaining assays.
This Project received financial support from the European Research Council Program under Grants ERC-StG-DAMOC (714317), the European Commission under FET-open program BLOC project (GA- 863037), the Spanish Ministry of Economy and Competitiveness, through the 'Severo Ochoa' Program for Centres of Excellence in R&D (SEV-2016-2019) and 'Retos de investigación: Proyectos I+D+i' (TEC2017-83716-C2-2-R), the CERCA Programme/Generalitat de Catalunya (2014- SGR-1460) and Fundación Bancaria 'la Caixa'- Obra Social 'la Caixa' (Project IBEC-La Caixa Healthy Ageing).
Authors contribution
F V M and J R C contributed to the study design, performance of experiments, data analysis, and writing and review of the manuscript. J R A contributed to the study design, data analysis, and report and review of the manuscript. J R A is the guarantor of this work. It had full access to all the data in the study and takes responsibility for the data integrity and data analysis accuracy.
Data availability statement
All data that support the findings of this study are included within the article (and any supplementary files).