



Abstract
We directly observe radiative charge transfer (RCT) in Ne clusters by dispersed vacuum-ultraviolet photon detection. The doubly ionized Ne2+– initial states of RCT are populated after resonant 1s–3p photoexcitation or 1s photoionization of Nen clusters with
initial states of RCT are populated after resonant 1s–3p photoexcitation or 1s photoionization of Nen clusters with  . These states relax further producing Ne+–Ne+–
. These states relax further producing Ne+–Ne+– final states, and the RCT photon is emitted. Ab initio calculations assign the observed RCT signal to the
final states, and the RCT photon is emitted. Ab initio calculations assign the observed RCT signal to the![${}^{}{{\rm{N}}{\rm{e}}}^{2+}(2{{\rm{p}}}^{-2}{[}^{1}{\rm{D}}])\mbox{--}{{\rm{N}}{\rm{e}}}_{n-1}$](https://content.cld.iop.org/journals/1367-2630/20/1/012001/revision2/njpaaa4afieqn4.gif) initial state, while transitions from other possible initial states are proposed to be quenched by competing relaxation processes. The present results are in agreement with the commonly discussed scenario, where the doubly ionized atom in a noble gas cluster forms a dimer which dissipates its vibrational energy on a picosecond timescale. Our study complements the picture of the RCT process in weakly bound clusters, providing information which is inaccessible by charged particle detection techniques.
initial state, while transitions from other possible initial states are proposed to be quenched by competing relaxation processes. The present results are in agreement with the commonly discussed scenario, where the doubly ionized atom in a noble gas cluster forms a dimer which dissipates its vibrational energy on a picosecond timescale. Our study complements the picture of the RCT process in weakly bound clusters, providing information which is inaccessible by charged particle detection techniques.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
The understanding of charge redistribution processes in weakly bound matter has recently made tremendous progress owing to the discovery and thorough investigation of several non-local ultrafast relaxation mechanisms. Among these mechanisms, interatomic Coulombic decay (ICD) and electron transfer mediated decay (ETMD) play an important role. In ICD, the excess energy released in an electronic transition on one site of, for instance, a van-der-Waals cluster, is transferred to ionize a neighboring site [1–4]. In ETMD, a vacancy is filled by an electron from a neighboring site and the donor atom or a third atom becomes ionized [5–7]. Complementary, slower charge redistribution is also possible if the excess energy does not allow for further local or non-local (auto-)ionization. In the so-called radiative charge transfer (RCT), photon emission accompanies a charge transfer after local ionization, double-ionization, or autoionization, in order to minimize the total energy of the system. A somewhat similar process was first observed and extensively investigated for transient molecules created by collisions of cations and dications with neutral atoms in high-pressure reaction drift tubes [8]. However, the RCT process studied here takes place in systems which are bound before the initial excitation happened.
Recently, a signature of RCT was found in experiments on 2p photoionization of Ar dimers [9]. Furthermore, by measuring the 1s photoelectron in coincidence with two Ne+ ions originating from a Coulomb explosion at the equilibrium distance of Ne2+–Ne states, a comprehensive study performed in [10] proposed the prevalence of RCT with respect to other decay channels after inner-shell ionization of Ne dimers. Later on, signatures of RCT have been observed after direct double photoionization of Ne dimers [11], ionization of Ar dimers by electron impact [12], and inner-shell ionization of heteronuclear Ne–Ar dimers [13]. Theoretically [13–15], RCT photon energies were estimated from calculated potential energy curves and the respective decay rates have been reconstructed. In all experimental studies, electron–ion multicoincidence techniques have been used, which are de facto constrained to the detection of charged particles. Therefore, whenever neutral fragments are produced or photons are emitted, a full reaction information is inaccessible by these detection techniques or can only be (in some cases) reconstructed by applying momentum or energy conservation principles. The RCT photons, thus, were not observed in these studies, and the occurrence of RCT was only deduced from energy conservation reasons. In this letter, we report the first direct observation of RCT photons and support it by comparison of the experimental photon emission spectra from core-excited and core-ionized Ne clusters with the results of ab initio calculations.
In Ne clusters, the RCT transitions are predicted to occur in the vacuum-ultraviolet (VUV) spectral range [10], where dispersed photon detection from dilute targets is experimentally challenging. So far, no study of photon emission in the VUV range after 1s ionization or excitation of Ne clusters has been reported. Nonetheless, several investigations have been carried out on solid, liquid, and high-pressure gas phase Ne [16–24]. In few works spectra have been reported, which indeed exhibit a signal in the relevant emission range. However, none of these features has been interpreted in terms of RCT. In other noble gases, mostly in Ar, emissions in the ultraviolet spectral range have been attributed to charge transfer processes in ion–atom collisions [25–29]. Recently [30–32], fluorescence detection has been successfully introduced as a tool to identify and characterize interatomic processes in clusters.
The process relevant for the present study is schematically illustrated in figure 1 on the example of the Ne dimer. After the absorption of an x-ray photon, the dominant relaxation channel of the core-ionized Ne dimer is the one-site Auger decay into Ne2+(2p−2)–Ne states, with the probability of 67% [10]. Obviously, an isolated dication Ne2+(2p−2) has no further electronic decay possibility. Since in a dimer the state with two singly ionized Ne+(2p−1) cations is energetically favorable, the charge is further redistributed by the transfer of an electron from the neutral atom to the dication. For energy conservation, a photon is emitted, and two Ne+ cations are created by the subsequent Coulomb explosion. Alternatively, one-site dicationic Ne2+(2p−2)–Ne states can be populated by resonant core excitation. For the 1s–3p excitation in Ne, there is a 6% probability of direct shake-off processes to produce doubly ionized states. Moreover, many states of the  character, which are directly populated by resonant Auger decay, will further autoionize into the
character, which are directly populated by resonant Auger decay, will further autoionize into the ![${\mathrm{Ne}}^{2+}(2{{\rm{p}}}^{-2}{[}^{1}{\rm{S}},{}^{1}{\rm{D}},{}^{3}{\rm{P}}])$](https://content.cld.iop.org/journals/1367-2630/20/1/012001/revision2/njpaaa4afieqn6.gif) states. In total, the population of the RCT initial states by the 1s–3p excitation in Ne dimers was estimated as 32% [33, 34].
states. In total, the population of the RCT initial states by the 1s–3p excitation in Ne dimers was estimated as 32% [33, 34].

Figure 1. Sketch of the process under investigation, exemplified by Ne dimers. After photoionization in step (a) ![$[{\mathrm{Ne}}_{2}+{\hslash }\omega \to {\mathrm{Ne}}^{+}(1{{\rm{s}}}^{-1})-\mathrm{Ne}+{e}_{{\rm{photo}}}^{-}]$](https://content.cld.iop.org/journals/1367-2630/20/1/012001/revision2/njpaaa4afieqn7.gif) , the core-ionized state relaxes by Auger decay into one-site dicationic states in step (b)
, the core-ionized state relaxes by Auger decay into one-site dicationic states in step (b)  ]. The relaxation process is completed by radiative charge transfer (RCT) in step (c)
]. The relaxation process is completed by radiative charge transfer (RCT) in step (c)  ], and the created repulsive states fragment by Coulomb explosion. Because of the very long radiative lifetime, vibrational motion toward the equilibrium geometry of Ne2+–Ne sets in prior to RCT.
], and the created repulsive states fragment by Coulomb explosion. Because of the very long radiative lifetime, vibrational motion toward the equilibrium geometry of Ne2+–Ne sets in prior to RCT.
Download figure:
Standard image High-resolution image2. Methods
The present experiment was performed at the PLEIADES beamline of Synchrotron Soleil, France, in its multibunch operation mode. Clusters were produced by supersonic expansion of Ne gas at a stagnation pressure of 6 bar into an expansion chamber through a pinhole-nozzle of 32 diameter, cooled with a liquid helium flow cryostat to 36.3 K. Using the formalism of [35], these conditions result in a mean cluster size of
diameter, cooled with a liquid helium flow cryostat to 36.3 K. Using the formalism of [35], these conditions result in a mean cluster size of  . After passing a 1.5 mm diameter skimmer (Beam Dynamics, Inc.), the cluster jet was crossed with the linearly polarized synchrotron radiation. In the inner-shell excitation range, the exit slit of the plane grating beamline monochromator was set to 100 μm, resulting in an exciting-photons bandwidth of about 750 meV. The excitation energy was calibrated using the atomic Ne 1s–3p resonance position of 867.12 eV [36]. In addition, reference photon emission spectra were recorded in the inner-valence excitation regime. In this exciting-photon energy range of about 47 eV, a photon bandwidth of about 20 meV was achieved by using a 400
. After passing a 1.5 mm diameter skimmer (Beam Dynamics, Inc.), the cluster jet was crossed with the linearly polarized synchrotron radiation. In the inner-shell excitation range, the exit slit of the plane grating beamline monochromator was set to 100 μm, resulting in an exciting-photons bandwidth of about 750 meV. The excitation energy was calibrated using the atomic Ne 1s–3p resonance position of 867.12 eV [36]. In addition, reference photon emission spectra were recorded in the inner-valence excitation regime. In this exciting-photon energy range of about 47 eV, a photon bandwidth of about 20 meV was achieved by using a 400  exit slit. The reference spectra were recorded for somewhat smaller cluster size of
exit slit. The reference spectra were recorded for somewhat smaller cluster size of  . Nevertheless, it was ensured that the reference spectrum is converged and does not change with increase of the mean cluster size.
. Nevertheless, it was ensured that the reference spectrum is converged and does not change with increase of the mean cluster size.
For spectrally resolved detection of emitted photons, a well-established set-up for photon-induced fluorescence spectrometry was used [37]. Briefly, the fluorescence photons, entering a 1 m normal-incidence spectrometer (McPherson) through a 1 mm entrance slit perpendicular to the plane spanned by synchrotron radiation and cluster jet, were dispersed by a gold-coated reflection grating with 1200 lines mm−1 and detected with a position sensitive open-face microchannel plate detector equipped with a wedge-and-strip anode. Positions and widths of known atomic lines after valence emission from an atomic gas jet were used to calibrate the fluorescence wavelengths and to estimate the detection resolution. With the present set-up configuration, a wavelengths resolution of about 1.5 nm was achieved.
3. Results
Figure 2 depicts the photon emission spectra recorded for the 1s–3p resonantly excited and 1s ionized Ne clusters. The 1s–3p resonance in clusters was identified by scanning the exciting-photon energy across the edge and recording the total VUV-fluorescence yield. The measured energy position of 868.3 eV of that resonance agrees with previous results on large clusters and thin solid films [38, 39]. These spectra do not exhibit any significant features outside the displayed range within the whole range of interest from 50–120 nm. The spectrum does not change for either 1s excitation or ionization. Only the statistic is drastically enhanced for the former, although both were recorded with identical total acquisition times. This is due to the remarkably larger cross section of the excitation compared to ionization.
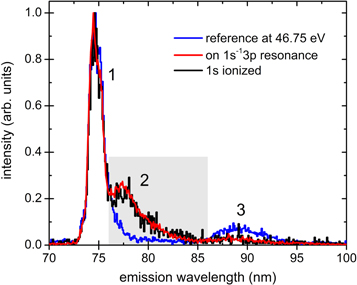
Figure 2. VUV-photon emission spectra from Ne clusters for three exciting-photon energies. Blue curve: at the resonant valence excitation at 46.75 eV; red curve: on the 1s–3p cluster resonance at 868.3 eV; and black curve: just above the 1s ionization threshold at 871.3 eV. Within the range of interest 50–120 nm, these spectra do not exhibit any further significant emission features outside the displayed interval. The signal within the light gray shaded range from 76–86 nm is attributed to RCT. The difference between the spectrum for the core-excitation and the reference spectrum, highlighted by the gray shaded area, is displayed in figure 4.
Download figure:
Standard image High-resolution imageThe spectra exhibit three distinct features: a relatively sharp emission at 74.7 nm (labeled as 1 in figure 2), a broad peak at 77.5 nm (labeled as 2) and a very faint feature at about 88.0 nm (labeled as 3). Feature 1 is the well-known universal emission after various excitations of Ne clusters and solids [16–18, 22, 24, 31]. It is the result of fluorescing atoms (the  transition) after desorption from the surface [24] and a variety of excitonic states in solid Ne [21, 22], which are not resolved in the present experiment. The presence of this general feature in the emission spectrum after inner-shell excitation is not surprising, because a considerable amount of Auger electrons and photoelectrons scatters in the cluster, behaving like a broadband electron impact excitation source [40]. A weak signal in the wavelength range encompassing features 2 and 3 has been observed in solid/liquid neon [21] and attributed to emissions from valence-excited, unrelaxed
transition) after desorption from the surface [24] and a variety of excitonic states in solid Ne [21, 22], which are not resolved in the present experiment. The presence of this general feature in the emission spectrum after inner-shell excitation is not surprising, because a considerable amount of Auger electrons and photoelectrons scatters in the cluster, behaving like a broadband electron impact excitation source [40]. A weak signal in the wavelength range encompassing features 2 and 3 has been observed in solid/liquid neon [21] and attributed to emissions from valence-excited, unrelaxed  molecules.
molecules.
In order to identify a possible contribution of RCT to the measured spectra, a reference spectrum was recorded at 46.75 eV, which corresponds to the resonant inner-valence bulk excitation [41]. Here, all valence-excited states of Ne clusters will be populated either directly or by electron impact subsequent to autoionization of the 2s–3p excited state. Importantly, this exciting-photon energy is below the double ionization potential (60.9 eV, calculated for Ne dimers in [42]), and, thus, states that undergo RCT cannot be populated at this energy. One can see from figure 2, that features 1 and 3 are present in the reference spectrum, while feature 2 is strongly suppressed. It appears exclusively after inner-shell excitation/ionization in the emission wavelength range from 76–86 nm (gray shaded range in figure 2). Note also that the intensity ratio of different features may vary between the spectra because all spectra were normalized to their maximum for comparison.
To explain the present observations, we performed ab initio calculations of the potential energy curves of the RCT initial ![${\mathrm{Ne}}^{2+}(2{{\rm{p}}}^{-2}\,{[}^{1}{\rm{S}},{}^{1}{\rm{D}},{}^{3}{\rm{P}}])$](https://content.cld.iop.org/journals/1367-2630/20/1/012001/revision2/njpaaa4afieqn16.gif) –Ne and final
–Ne and final  states in the Ne dimer, as well as the corresponding radiative decay widths. Calculations were performed using configuration interaction singles and doubles method as implemented in the GAMES-US program suite [43]. The correlation consistent aug-cc-pCVTZ basis set was used for Ne [44]. The radiative lifetimes computed at the equilibrium distance of the ground neutral state of the Ne dimer (
states in the Ne dimer, as well as the corresponding radiative decay widths. Calculations were performed using configuration interaction singles and doubles method as implemented in the GAMES-US program suite [43]. The correlation consistent aug-cc-pCVTZ basis set was used for Ne [44]. The radiative lifetimes computed at the equilibrium distance of the ground neutral state of the Ne dimer ( ) lie between 80 ns and 80
) lie between 80 ns and 80  for different RCT initial states. This disparity can be explained by the different overlaps between the hole orbitals of Ne2+ and occupied orbitals of neighboring Ne. For this reason, the RCT of the
for different RCT initial states. This disparity can be explained by the different overlaps between the hole orbitals of Ne2+ and occupied orbitals of neighboring Ne. For this reason, the RCT of the ![${\mathrm{Ne}}^{2+}(2{{\rm{p}}}^{-2}{[}^{1}{\rm{D}}])$](https://content.cld.iop.org/journals/1367-2630/20/1/012001/revision2/njpaaa4afieqn20.gif) –Ne
–Ne  state populating the
state populating the  state is very efficient due to the strong overlap between the 2pz orbitals of the two Ne atoms. The efficiency of RCT increases almost exponentially as the two Ne atoms approach each other. At the internuclear distance of 2 Å, which roughly corresponds to the inner turning points of the one-site
state is very efficient due to the strong overlap between the 2pz orbitals of the two Ne atoms. The efficiency of RCT increases almost exponentially as the two Ne atoms approach each other. At the internuclear distance of 2 Å, which roughly corresponds to the inner turning points of the one-site  –Ne dicationic potential energy curves, the radiative lifetimes become 0.1 ns–2 μs. Therefore, RCT occurs mainly at shorter internuclear distances close to these inner turning points.
–Ne dicationic potential energy curves, the radiative lifetimes become 0.1 ns–2 μs. Therefore, RCT occurs mainly at shorter internuclear distances close to these inner turning points.
In order to compute the photon emission spectra, we diagonalized the complex vibrational Hamiltonian  for each two-site dicationic initial RCT state. Here, TR is the nuclear kinetic energy, V(R) is the potential energy of the decaying state, and
for each two-site dicationic initial RCT state. Here, TR is the nuclear kinetic energy, V(R) is the potential energy of the decaying state, and  is the respective radiative decay width. In this way, we obtained the energies, widths, and wave functions of the individual vibrational states. We further assumed that the photoionization and Auger decay transitions are instantaneous and the vibrational wave packet is vertically transferred from the initial ground state onto the RCT initial states. Under this assumption, amplitudes for the population of the individual vibrational levels are given by the corresponding Frank–Condon overlaps with the v = 0 vibronic state of the Ne2 ground state. The energy difference between adjacent vibrational states are much larger than their decay widths. Under these conditions, the resulting spectra are given by the incoherent sum of the individual spectra due to the decay of each vibrational level [45]. The latter spectra were obtained by projecting the corresponding vibrational densities, multiplied with the R-dependent decay widths
is the respective radiative decay width. In this way, we obtained the energies, widths, and wave functions of the individual vibrational states. We further assumed that the photoionization and Auger decay transitions are instantaneous and the vibrational wave packet is vertically transferred from the initial ground state onto the RCT initial states. Under this assumption, amplitudes for the population of the individual vibrational levels are given by the corresponding Frank–Condon overlaps with the v = 0 vibronic state of the Ne2 ground state. The energy difference between adjacent vibrational states are much larger than their decay widths. Under these conditions, the resulting spectra are given by the incoherent sum of the individual spectra due to the decay of each vibrational level [45]. The latter spectra were obtained by projecting the corresponding vibrational densities, multiplied with the R-dependent decay widths  , onto the repulsive potential energy curves of the final states. This procedure is schematically visualized in figure 3.
, onto the repulsive potential energy curves of the final states. This procedure is schematically visualized in figure 3.

Figure 3. Sketch of the theoretical model used to compute the RCT spectra. In a free dimer, vibrational levels of the  –Ne state are populated according to the corresponding excitation Franck–Condon factors. The respective densities are visualized by the gray shaded functions. Thereafter, each density is multiplied by the R-dependent decay widths
–Ne state are populated according to the corresponding excitation Franck–Condon factors. The respective densities are visualized by the gray shaded functions. Thereafter, each density is multiplied by the R-dependent decay widths  and mapped onto the RCT final state
and mapped onto the RCT final state  . The observed photon spectrum (gray spectrum in right panel) is obtained by incoherent summation of contributions from all vibrational states. If the dimer is embedded in a cluster, it first relaxes vibrationally during the RCT lifetime. Therefore, only the vibrational ground state (visualized by the magenta shadowed functions) does contribute to the photon emission spectrum (magenta spectrum in right panel).
. The observed photon spectrum (gray spectrum in right panel) is obtained by incoherent summation of contributions from all vibrational states. If the dimer is embedded in a cluster, it first relaxes vibrationally during the RCT lifetime. Therefore, only the vibrational ground state (visualized by the magenta shadowed functions) does contribute to the photon emission spectrum (magenta spectrum in right panel).
Download figure:
Standard image High-resolution imageThe experiment was performed not with dimers, but rather with large clusters. It has been suggested that singly or doubly ionized atoms in a noble gas cluster form within picoseconds [46] an embedded dimer with a random neighbor, and this dimer efficiently dissipates its vibrational energy into the cluster lattice. To compute the RCT photon spectrum in the presence of intracluster vibrational relaxation, we relied on the fact that relaxation times in rare gas clusters are tens to hundreds picoseconds [47, 48]. These times are about an order of magnitude shorter than the computed RCT lifetimes. Therefore, in a cluster vibrational relaxation takes place prior to photon emission. Consequently, only the lowest vibrational state of the dicationic potential energy curve contributes to the RCT spectrum (see also figure 3).
The calculated RCT photon spectra for free dimers and intracluster vibrationally relaxed dimers are shown in figure 4. The three contributions from RCT initial states ![${\mathrm{Ne}}^{2+}(2{{\rm{p}}}^{-2}\,{[}^{1}{\rm{S}},{}^{1}{\rm{D}},{}^{3}{\rm{P}}])$](https://content.cld.iop.org/journals/1367-2630/20/1/012001/revision2/njpaaa4afieqn30.gif) –Ne are well separable. The states
–Ne are well separable. The states ![${\mathrm{Ne}}^{2+}(2{{\rm{p}}}^{-2}\,{[}^{1}{\rm{D}}])$](https://content.cld.iop.org/journals/1367-2630/20/1/012001/revision2/njpaaa4afieqn31.gif) –Ne and
–Ne and ![${\mathrm{Ne}}^{2+}(2{{\rm{p}}}^{-2}\,{[}^{1}{\rm{S}}])$](https://content.cld.iop.org/journals/1367-2630/20/1/012001/revision2/njpaaa4afieqn32.gif) –Ne are populated directly with the statistical ratio of 7:1 by the Auger decay of the
–Ne are populated directly with the statistical ratio of 7:1 by the Auger decay of the ![${\mathrm{Ne}}^{+}\,(1{{\rm{s}}}^{-1}\,{[}^{2}{\rm{S}}])$](https://content.cld.iop.org/journals/1367-2630/20/1/012001/revision2/njpaaa4afieqn33.gif) –Ne states. The
–Ne states. The ![${\mathrm{Ne}}^{2+}(2{{\rm{p}}}^{-2}\,{[}^{3}{\rm{P}}])$](https://content.cld.iop.org/journals/1367-2630/20/1/012001/revision2/njpaaa4afieqn34.gif) –Ne state is assumed to be populated by the radiative decay of the
–Ne state is assumed to be populated by the radiative decay of the ![${\mathrm{Ne}}^{2+}(2{{\rm{s}}}^{-1}2{{\rm{p}}}^{-1}\,{[}^{3}{\rm{P}}])$](https://content.cld.iop.org/journals/1367-2630/20/1/012001/revision2/njpaaa4afieqn35.gif) –Ne Auger final state. In order to extract the pure RCT contribution from the experimental spectra, we subtracted the reference spectrum from the spectrum taken at the 1s–3p inner-shell excitation in the region of interest (light gray shaded range in figure 2). This difference spectrum is depicted in figure 4 for comparison, and it excludes possible contributions from excited dimers [21]. The experimental signal agrees very well with the theoretical RCT spectrum from the vibrationally relaxed
–Ne Auger final state. In order to extract the pure RCT contribution from the experimental spectra, we subtracted the reference spectrum from the spectrum taken at the 1s–3p inner-shell excitation in the region of interest (light gray shaded range in figure 2). This difference spectrum is depicted in figure 4 for comparison, and it excludes possible contributions from excited dimers [21]. The experimental signal agrees very well with the theoretical RCT spectrum from the vibrationally relaxed ![${\mathrm{Ne}}^{2+}(2{{\rm{p}}}^{-2}{[}^{1}{\rm{D}}])$](https://content.cld.iop.org/journals/1367-2630/20/1/012001/revision2/njpaaa4afieqn36.gif) –Ne initial state at 77.8 nm. This wavelength corresponds to the transition at internuclear distances of around 2.25 Å, (see figure 3), and it confirms the scenario of vibrational relaxation. At the peak edges, there are intensity artefacts due to the signal normalization before the subtraction. We thus assign feature 2, observed in the experimental spectrum of figure 2, to RCT.
–Ne initial state at 77.8 nm. This wavelength corresponds to the transition at internuclear distances of around 2.25 Å, (see figure 3), and it confirms the scenario of vibrational relaxation. At the peak edges, there are intensity artefacts due to the signal normalization before the subtraction. We thus assign feature 2, observed in the experimental spectrum of figure 2, to RCT.

{kind=link}
{kind=link}
{kind=link}
Figure 4. Simulated RCT photon spectra for free dimers (dashed black curve) and vibrationally relaxed dimers embedded in a cluster (solid black curve) compared to the experimental signal (red solid curve), obtained by subtraction of the reference spectrum from the spectrum recorded at the 1s–3p resonance within the gray shaded range of figure 2. The measured signal is normalized to the spectrum computed for dimers embedded in a cluster at their maxima.
Download figure:
Standard image High-resolution image{kind=link}
Finally, theory predicts two additional contributions to the spectrum owing to RCT from the ![${\mathrm{Ne}}^{2+}(2{{\rm{p}}}^{-2}\,{[}^{1}{\rm{S}}])$](https://content.cld.iop.org/journals/1367-2630/20/1/012001/revision2/njpaaa4afieqn37.gif) –Ne and
–Ne and ![${\mathrm{Ne}}^{2+}(2{{\rm{p}}}^{-2}\,{[}^{3}{\rm{P}}])$](https://content.cld.iop.org/journals/1367-2630/20/1/012001/revision2/njpaaa4afieqn38.gif) –Ne initial states (see figure 4). In these calculations, however, the signal from
–Ne initial states (see figure 4). In these calculations, however, the signal from ![${\mathrm{Ne}}^{2+}(2{{\rm{p}}}^{-2}\,{[}^{3}{\rm{P}}])$](https://content.cld.iop.org/journals/1367-2630/20/1/012001/revision2/njpaaa4afieqn39.gif) –Ne state emerges only due to the assumption of a local radiative decay of the
–Ne state emerges only due to the assumption of a local radiative decay of the ![${\mathrm{Ne}}^{2+}(2{{\rm{s}}}^{-1}2{{\rm{p}}}^{-1}\,{[}^{3}{\rm{P}}])$](https://content.cld.iop.org/journals/1367-2630/20/1/012001/revision2/njpaaa4afieqn40.gif) –Ne state. In reality, as confirmed experimentally in [10], this radiative transition is quenched by non-adiabatic charge transfer into
–Ne state. In reality, as confirmed experimentally in [10], this radiative transition is quenched by non-adiabatic charge transfer into  states of the dimer. The present analysis suggests that, if the dimer is embedded in a large cluster, ICD or ETMD with non-nearest neighbors are additional competing pathways, and this initial RCT state is not populated at all. On the contrary, the
states of the dimer. The present analysis suggests that, if the dimer is embedded in a large cluster, ICD or ETMD with non-nearest neighbors are additional competing pathways, and this initial RCT state is not populated at all. On the contrary, the ![${\mathrm{Ne}}^{2+}(2{{\rm{p}}}^{-2}{[}^{1}{\rm{S}}])$](https://content.cld.iop.org/journals/1367-2630/20/1/012001/revision2/njpaaa4afieqn42.gif) –Ne state is directly populated by the Auger decay of the core hole. However, in a large cluster the
–Ne state is directly populated by the Auger decay of the core hole. However, in a large cluster the ![${\mathrm{Ne}}^{2+}(2{{\rm{p}}}^{-2}\,{[}^{1}{\rm{S}}])$](https://content.cld.iop.org/journals/1367-2630/20/1/012001/revision2/njpaaa4afieqn43.gif) –
– surface lies among the manifold of surfaces of Ne+–Ne+–Ne*–Ne
surface lies among the manifold of surfaces of Ne+–Ne+–Ne*–Ne character. Therefore, we propose a complete depopulation of this RCT initial state within the lifetime of the radiative decay by non-adiabatic couplings between these surfaces. Such mechanism can alternatively be described as an ETMD process, where electron transfer is accompanied by an excitation into a band of bound electronic states. Thus created excited dicationic states will fragment by a Coulomb explosion, followed by an atomic emission in the wavelengths range of feature 1. Importantly, the
character. Therefore, we propose a complete depopulation of this RCT initial state within the lifetime of the radiative decay by non-adiabatic couplings between these surfaces. Such mechanism can alternatively be described as an ETMD process, where electron transfer is accompanied by an excitation into a band of bound electronic states. Thus created excited dicationic states will fragment by a Coulomb explosion, followed by an atomic emission in the wavelengths range of feature 1. Importantly, the ![${\mathrm{Ne}}^{2+}(2{{\rm{p}}}^{-2}\,{[}^{1}{\rm{D}}])$](https://content.cld.iop.org/journals/1367-2630/20/1/012001/revision2/njpaaa4afieqn46.gif) –
– surface lies well below this manifold and can thus decay only by RCT.
surface lies well below this manifold and can thus decay only by RCT.
4. Conclusion
RCT is a well-known fundamental relaxation mechanism, but it was never observed directly. In the present work, we detect for the first time RCT photons after core ionization of Ne clusters and assign the observed signal to the transition from the ![${\mathrm{Ne}}^{2+}(2{{\rm{p}}}^{-2}\,{[}^{1}{\rm{D}}])$](https://content.cld.iop.org/journals/1367-2630/20/1/012001/revision2/njpaaa4afieqn48.gif) –Ne state of vibrationally relaxed dimers within the cluster. This becomes possible because the formation and vibrational relaxation of the embedded dimer takes place faster than the RCT decay, i.e. at least on a picosecond timescale. In general, a theoretical model of vibrationally relaxed dimers should be sufficient for interpretation of relatively slow relaxation processes in large clusters, as demonstrated here. We also propose quenching of RCT from
–Ne state of vibrationally relaxed dimers within the cluster. This becomes possible because the formation and vibrational relaxation of the embedded dimer takes place faster than the RCT decay, i.e. at least on a picosecond timescale. In general, a theoretical model of vibrationally relaxed dimers should be sufficient for interpretation of relatively slow relaxation processes in large clusters, as demonstrated here. We also propose quenching of RCT from ![${\mathrm{Ne}}^{2+}(2{{\rm{p}}}^{-2}\,{[}^{1}{\rm{S}}])$](https://content.cld.iop.org/journals/1367-2630/20/1/012001/revision2/njpaaa4afieqn49.gif) –Ne and
–Ne and ![${\mathrm{Ne}}^{2+}(2{{\rm{p}}}^{-2}\,{[}^{3}{\rm{P}}])$](https://content.cld.iop.org/journals/1367-2630/20/1/012001/revision2/njpaaa4afieqn50.gif) –Ne states by competing decay pathways present in large clusters and confirm a possibility of the population of RCT initial states by resonant core excitation. Our results demonstrate the utility of dispersed photon detection as a complementary method to conventional electron–ion coincidence detection to reveal information on particular charge redistribution processes in weakly bound matter. The current experiment paves the way for future investigations of such processes in dense media, where charged particles, owing to their very short mean free path, may not be able to escape.
–Ne states by competing decay pathways present in large clusters and confirm a possibility of the population of RCT initial states by resonant core excitation. Our results demonstrate the utility of dispersed photon detection as a complementary method to conventional electron–ion coincidence detection to reveal information on particular charge redistribution processes in weakly bound matter. The current experiment paves the way for future investigations of such processes in dense media, where charged particles, owing to their very short mean free path, may not be able to escape.
Acknowledgments
This work was supported by the State of Hesse Initiative for the Development of Scientific and Economic Excellence (LOEWE) within the focus-project Electron Dynamics of Chiral Systems (ELCH) and by the Deutsche Forschungsgemeinschaft (Forschergruppe FOR 1789). We acknowledge Synchrotron Soleil for beamtime allocation (proposal 20140585) and the PLEIADES beamline team for excellent support during the experiments.