


Abstract
Photocatalytic CO2 reduction reaction (CO2RR) is believed to be a promising remedy to simultaneously lessen CO2 emission and obtain high value-added products, but suffers from the thwarted activity of photocatalyst and poor selectivity of product. Over the past decade, aided by the significant advances in nanotechnology, the atom manufacturing of photocatalyst, including vacancies, dopants, single-atom catalysts, strains, have emerged as efficient approaches to precisely mediate the reaction intermediates and processes, which push forward in the rapid development of highly efficient and selective photocatalytic CO2RR. In this review, we summarize the recent developments in highly efficient and/or selective photocatalysts toward CO2RR with the special focus on various atom manufacturing. The mechanisms of these atom manufacturing from active sites creation, light absorbability, and electronic structure modulation are comprehensively and scientifically discussed. In addition, we attempt to establish the structure–activity relationship between active sites and photocatalytic CO2RR capability by integrating theoretical simulations and experimental results, which will be helpful for insights into mechanism pathways of CO2RR over defective photocatalysts. Finally, the remaining challenges and prospects in this field to improve the photocatalytic CO2RR performances are proposed, which can shed some light on designing more potential photocatalysts through atomic regulations toward CO2 conversion.
Export citation and abstract BibTeX RIS
Recommended by Dr Tao Wang
1. Introduction
The ever-increasing depletion of fossil fuels along with excessive CO2 emissions has aroused serious energy crisis, climate change and environmental pollution [1–8]. To address these problems, capture and conversion of CO2 into highly reusable value-added chemical fuels is becoming one of the most attractive and promising strategy to simultaneously ameliorate the energy shortage and environmental issues over the long haul [9–13]. Among many approaches being explored so far, solar driven CO2 reduction reaction (CO2RR) to produce low-carbon fuels has been regarded as one of the ultimate and ideal technologies because of its zero-carbon emission process and direct utilization of abundance solar energy [14–17].
The pioneering report on solar driven CO2RR can be tracked back to 1978, when Halmann demonstrated photoelectrochemical CO2RR over p-GaP photocathode [18]. Inoue et al realized the photocatalytic CO2RR to form various C1 products, such as CH4, HCOOH and CH3OH by using different photocatalyst in the following year [19]. Since then, developing the cleanest, sustainable and low-cost 'solar fuel' technology, photocatalytic CO2RR, to produce high value-added products, has attracted significant attention [20–23]. Until now, numerous photocatalytic materials used for photocatalytic hydrogen evolution reaction (HER), have been attempted to do photocatalytic CO2RR, such as TiO2, CdS, WO3, polymeric carbon nitride (PCN), layered double hydroxides (LDHs), to name a few [24–43]. However, for the same photocatalyst, the CO2RR performance generally is at least a few times lower than its HER activity [44–47]. Therefore, continuous efforts to develop highly efficient photocatalysts to achieve high CO2 conversion rate and product selectivity are necessary and urgent.
As a 'solar fuel' technology, the high quantum efficiency of photocatalytic CO2RR is always desired. Several general strategies, such as heterojunction construction, morphology control, doping, material compositing, associated with the light harvesting and photo-generated charge separation are encouraged [48–53]. However, many explored photocatalysts investigated to date were limited to poor product selectivity due to the competitive HER in aqueous media, because of the thermodynamic stability of CO2 and the multi-electron reaction process. Regulating the adsorption and activation of CO2 on the surface of photocatalysts has been considered as key issues for highly efficient photocatalytic CO2RR [10, 54–64]. In this regard, the required energies for adsorption and activation of CO2 are determined by the electronic structure of adsorbed atoms, in which the coordinate state of atom, atom–atom bond, atom–atom distance and atom–atom species can profoundly affect the process [65]. For example, the vacancies can effectively lower the coordination numbers of adjacent metal atoms, building the unsaturated surroundings for enhanced CO2 adsorption and reactivity. In particular, the specific sites consisted of unsaturated surface sites proximal to vacancies with Lewis acidic nature and the adjacent Lewis basic surface hydroxide site can develop the surface frustrated Lewis pairs, which shows stronger acidity and basicity for promoting the dissociation of CO2 during the photocatalytic process [66]. Thus, atom manufacturing of a photocatalyst can create the active site required for the adsorption and activation of CO2 to improve the activity and product selectivity of photocatalytic CO2RR.
Herein, we principally summarize the recent advancements in atom manufacturing of various semiconductor photocatalysts by means of displaying the regulation patterns to promote photocatalytic CO2RR, accompanied by the corresponding underlying mechanisms. Starting from introducing common types of atom manufacturing, including vacancies, doping, single atom and strain engineering, we briefly discuss their effects on activity and stability of heterogeneous photocatalysts, fundamentals of catalysts in photocatalytic CO2RR, and subsequently describe the effects of different atom manufacturing types on adsorption and activation of CO2. Finally, the remaining challenges and prospects of atom manufacturing of photocatalysts for CO2RR in terms of theoretical simulation, controllable synthesis, advanced characterization techniques, and practical applications are highlighted.
2. Fundamentals of heterogeneous photocatalytic CO2RR
In addition to the basic thermodynamic requirements for semiconductor photocatalysts, photocatalytic CO2RR needs four elementary steps [67, 68]: (1) CO2 adsorption on surface of photocatalyst; (2) CO2 activation by the photo-induced energetic electrons over illuminated photocatalyst; (3) dissociation of the C–O bond and multiple proton-coupled electrons transfers at the active site and (4) desorption of reduced products from the active sites, as shown in figure 1. Among all steps, the activation of CO2 (CO2 •−) is a single electron transfer process from conduction band (CB) of the excited photocatalyst to CO2 molecule, which induces a bending geometry of the CO2 molecule. Because the bending geometry increases the repulsion between the added electron and the free electron pairs on the oxygen atoms, the lowest unoccupied molecular orbital (LUMO) of CO2 is raised, leading to a thermodynamically unfavorable process with electrochemical potential of 1.85 V vs normal hydrogen electrode (NHE). Due to the very negative potential for the activation of CO2, almost no semiconductor has the enough energy to transfer photo-induced electron from CB to free CO2.
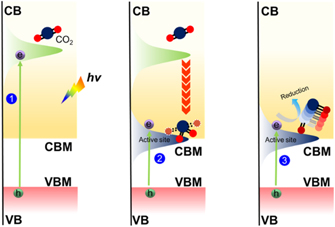
Figure 1. Elementary steps of photocatalytic CO2RR. ① The activation of CO2 (CO2 •−) by a single photo-generated electron. ② The bending and antisymmetric stretching vibrations of CO2 •−. ③ The dissociation of CO2 •−.
Download figure:
Standard image High-resolution imageThrough the chemisorbed CO2 molecules onto a catalyst surface, the linear CO2 structure can transform into a bent structure, resulting in a partially charged species of CO2 δ•−. Chu et al theoretically investigated the photocatalytic CO2RR on metal oxide surface by using ab initio quantum dynamics simulation, which demonstrated that the activation of free CO2 needs a photon energy higher than 5.5 eV, while the activation of chemisorbed CO2 only requires a photon energy larger than 2.2 eV [69]. Due to the electronic structure difference of these being absorbed atoms, the CO2 molecules at these adsorption sites present different geometries, thereby different energies of LUMO. If the LUMO of the absorbed CO2 molecule is close to the CB of a photocatalyst, the photocatalytic CO2RR is easily taken place on these being absorbed atoms. Creating atoms that can absorb and stabilize the CO2 molecule with low activation energy is the critical step to break through the bottleneck of CO2RR over a photocatalyst.
3. Atom manufacturing
Atom manufacturing can be categorized into vacancy, heteroatom doping, single-atom decoration, strain defects (figure 2), etc. The vacancy and heteroatom doping are two typic defects at the atom level originating from native defect and nonnative defect of materials [70]. This kind of atom manufacturing can be easily performed in the process of materials synthesis or post-modification, which has been widely studied for a long time in photocatalyst modification. It is noted that, due to different valance state and atom radius of heteroatom from the native atom, the heteroatom doping is accompanied by the formation of vacancies in many cases. Both vacancies and heteroatom substitution can effectively lower the coordination numbers of adjacent atoms, creating the unsaturated surroundings for CO2 absorption and activation. In addition, the atomic vacancies or heteroatoms substitution generally cause the electronic structure change of photocatalysts, which probably improves the light absorption ability and alters the charge separation behaviors. Single-atom decoration is one of newly emerged atom manufacturing in recent decades, in which single atoms can be stabilized in a special coordination structure. The single-atom decoration does not present periodic or symmetrical structure, while the chemical coordination structure is highly consistent. Compared to vacancies and heteroatoms doping, single-atom decoration does not affect the electronic structure of photocatalyst in general, but offers additional and uniform active site owing to the homogeneous chemical coordination structure [71]. Through altering coordination structure of the decorated single atom, the absorption and activation energy of CO2 can be modulated. In comparison with the change of local structure caused by above atom manufacturing, strain engineering can homogeneously modify the electronic structure of crystal planes, grain boundaries or interface by changing the atom–atom distance [72]. Both single-atom decoration and strain engineering have homogeneous chemical environments, which is therefore expected to increase the selectively of CO2RR to specific target products.
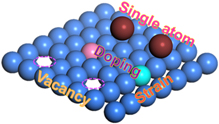
Figure 2. Schematic illustration of various atom manufacturing in a photocatalyst.
Download figure:
Standard image High-resolution image3.1. Characterizations of atom manufacturing
Until now, the identification and quantization of diverse engineered atoms can be performed by several advanced characterization techniques regarding atom alignment, electronic structure, or chemical bonds. Microscopic techniques at the atom level are a direct tool to identify the vacancies, heteroatom and single atom on the surface or subsurface of the photocatalyst. For example, according to high-resolution transmission electron microscopy (HRTEM) image and crystal structure, a dual vacancy of W and O atoms in the disordered WO3 can be clearly concluded (figure 3(a)) [73]. Through spherical aberration-corrected scanning TEM, the Au single atoms supported on PCN could be explicitly observed by high-angle annular dark-field STEM (HAADF-STEM) due to different atomic numbers (figure 3(b)) [74]. In addition, for thinner two-dimensional materials, the STEM image can also identify the vacancy with different types. As displayed in figure 3(c), according to the crystal structure of 1T MoS2, the concentric circle-type vacancies (red frame) can be clearly distinguished by STEM images [75].
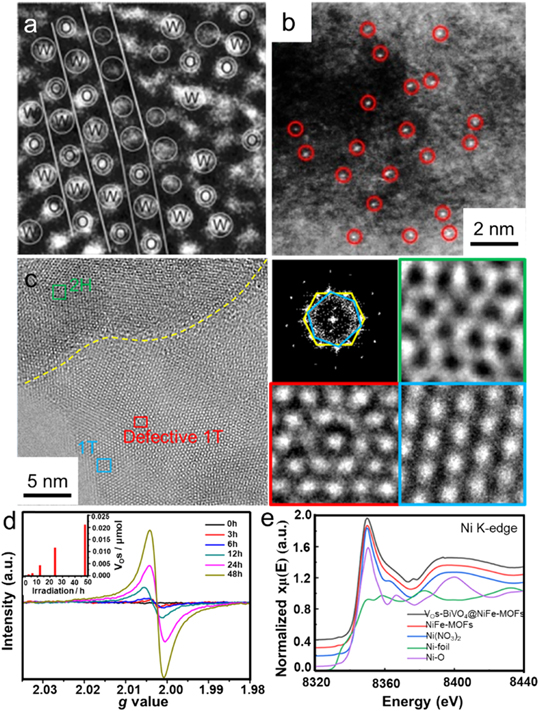
Figure 3. Characterizations of photocatalysts. (a) HRTEM image at the atomic scale of tungsten and oxygen vacancies modified WO3. [73] John Wiley & Sons. Copyright 2016, Wiley-VCH Verlag. (b) HAADF-STEM image of Au SACs supported on PCN. Reprinted with permission from [74]. Copyright (2020) American Chemical Society. (c) TEM image of 2H/1T-MoS2 and the corresponding fast Fourier transform pattern. [75] John Wiley & Sons. Copyright 2019, Wiley-VCH Verlag. (d) EPR spectra at room temperature. Reprinted with permission from [77]. Copyright (2018) American Chemical Society. (e) Ni K-edge XANES spectra. [78] John Wiley & Sons. Copyright 2021, Wiley-VCH Verlag.
Download figure:
Standard image High-resolution imageIn addition to the surface or subsurface characterizations by using various microscopic techniques, various electron spectroscopies also are powerful tools for bulk materials. For example, the extrinsic dopants or oxygen vacancies (VOs) in CeO2 can be reflected by the variations of chemical bonds in their Raman shifts [76]. Electron paramagnetic resonance (EPR) spectroscopy can also offer valuable information about unpaired electrons. For instance, Wang et al introduced VOs into amorphous zinc germanate (α–Zn–Ge–O) via ultraviolet irradiation, which could be reflected in the EPR signal at g = 2.003. As shown in figure 3(d), the EPR signal intensities were continuously enhanced by prolonging the irradiation time from 0 to 48 h, indicating that the VOs concentration was increased from 0 to 0.021 μmol of 0.1 g photocatalyst [77]. Compared to the electron structure characterizations in bulk materials, x-ray absorption spectroscopy (XAS), which can be divided into x-ray absorption near-edge structure (XANES) and extended x-ray absorption fine structure (EXAFS) based on the absorption threshold of energy, is a promising technique to identify the local chemical environment. The XAS can provide valence of elements, the coordination environment of the specific atom, and structure symmetries, etc. For instance, Pan et al conducted the XAS to characterize both the chemical states and coordination environment of Ni and Fe atoms in NiFe-metal–organic frameworks (MOFs) and VOs–BiVO4@NiFe-MOFs. As displayed in figure 3(e), the absorption energy of the Ni K-edge in VOs–BiVO4@NiFe-MOFs was slightly higher than that of NiFe-MOFs, suggesting that the Ni in VOs–BiVO4@NiFe-MOFs possessed a mildly higher oxidation state [78].
3.2. Vacancies
Vacancies can not only offer additional adsorption and active sites but also promoting the light harvest, which is beneficial for photocatalytic CO2RR [79–81]. In general, vacancies can be divided into cationic vacancies, anionic vacancies, and mixed vacancies. Among them, anionic vacancies are widely explored because of their facile engineering.
3.2.1. Anionic vacancies
Among various vacancies species, VOs are most easily formed in various oxygen-containing compounds due to their relatively low formation energies. Because the CO2 molecule can be preferentially adsorbed by these VOs by filling the oxygen atom of CO2 into VOs, therefore VOs are widely investigated to promote photocatalytic CO2 conversion over various photocatalysts [82, 83]. For instance, Zhao et alprepared VO-rich ZnAl-layered double hydroxide (ZnAl-LDH) nanosheets by reducing the lateral size of ZnAl-LDH from 5 μm to 30 nm, followed by lessening the nanosheet thickness into a few layers via a bottom-up process of inverse microemulsion and subsequently controllable hydrolysis (figure 4(a)) [84]. The stable reverse emulsion was composed by sodium dodecylsulfate, isooctane, and water with 1-butanol as a co-surfactant. Benefitting from the decreased lateral size, VOs were successfully introduced into ZnAl-LDH, which could not only result in abundant coordinatively unsaturated Zn ions (denoted as Zn+-VO) but also serve as electron trapping sites accompanying by offering a new defect energy level, thereby promoting the separation and transfer of photogenerated charge carriers. Particularly, the VO-rich ZnAl-LDH exhibited more favorable for CO2 and H2O adsorption because of the higher adsorption energy for both CO2 and H2O than those of defect-free LDH (figure 4(b)). In addition, density functional theory (DFT) calculations revealed a concentrated charge density at the Zn atoms, indicating that the active electrons were generated from Zn+-VO defective site in LDH, which was good for facilitating the interaction between active electrons and the reactant, thereby improving the efficiency of photocatalytic CO2RR. Based on the above virtues, VO-rich ZnAl-LDH (denoted as ZnAl-1) delivered a superior CO formation rate of 7.6 μmol g−1 h−1, much higher than that of the bulk counterpart (ZnAl-3, ∼0 μmol g−1 h−1) under the same operation condition (figure 4(c)). Besides bottom-up processing, top-down approach is an alternative to synthesis nanosheet architecture, which is susceptive to introducing vacancy-type defects. Using BiOBr-based lamellar precursors, Wu et al synthesized BiOBr atomic layers via ultrasonication for 24 h, which were further treated by ultraviolet irradiation to introduce VOs on BiOBr (figure 4(d)) [82]. The introduced VOs were favorable for improving both light harvesting and separation of photo-generated electron–hole pairs.
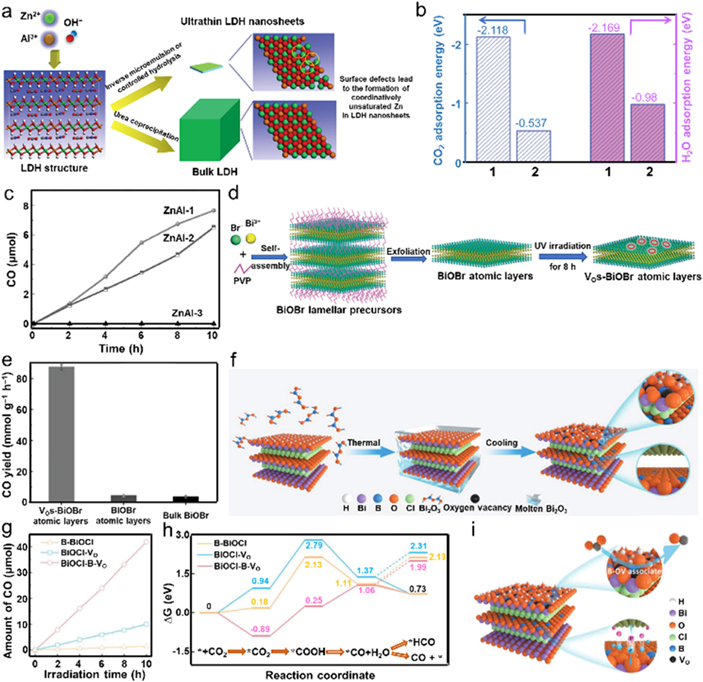
Figure 4. The fabrications of VO-modified photocatalysts with their photocatalytic CO2RR activities towards CO. (a) Schematic illustration of the preparation route of VO-rich ultrathin ZnAl-LDH. (b) DFT-calculated adsorption energies of CO2 and H2O molecules on VO-doped ZnAl-LDH and bulk ZnAl-LDH. ((1) VO-doped ZnAl-LDH, (2) bulk ZnAl-LDH). (c) Photocatalytic CO2RR toward CO with water vapor under UV–Vis light. [84] John Wiley & Sons. Copyright 2015, Wiley-VCH Verlag. (d) Illustration of the synthesis process of VO modified BiOBr atomic layers. PVP: polyvinyl propylene. (e) CO evolution of various photocatalysts. [82] John Wiley & Sons. Copyright 2018, Wiley-VCH Verlag. (f) Molten-salt fabrication of B-doped BiOCl associated with oxygen vacancies. (g) CO yield of the photocatalysts as a function of irradiation time. (h) Gibbs free energy diagrams of reaction intermediates during CO2-to-CO catalysis over various photocatalysts. (i) Schematic of photocatalytic CO2RR over BiOCl–B–VO. [85] John Wiley & Sons. Copyright 2021, Wiley-VCH Verlag.
Download figure:
Standard image High-resolution imageFurthermore, the negatively charged surface of VO modified BiOBr atomic layers was beneficial for CO2 adsorption compared with pristine BiOBr atomic layers and bulk sample. Benefited from the aforementioned advantages, the VO–BiOBr atomic layers exhibited a superior CO evolution rate of about 87.4 μmol g−1 h−1, which was much higher than those of pristine atomic layers and bulk counterpart (figure 4(e)). In situ Fourier-transform infrared spectroscopy was performed to identify photocatalytic CO2RR process, revealing that the presence of VOs was conductive to stabilize the COOH* intermediate according to the more COOH* groups on VO–BiOBr, which could effectively decrease the activation energy of CO2 and promote CO evolution.
The aforementioned methods suffer from tedious soft-template process, which goes against large-scale preparation of photocatalysts. In contrast, one-pot thermal treatment is a promising strategy to fabricate photocatalysts on mass production. Shi and coworkers developed a molten-salt way to synthesize B-doped BiOCl using B2O3 as both molten salt and doping precursor (figure 4(f)) [85]. The resultant sample possessed a homogeneous B-doping feature from the surface to bulk, accompanied by the formation of VOs. Surface B doping could induce surface reconstruction of BiOCl, resulting in VO formation by extracting adjacent hydroxyl, thereby bringing intimate B–VO associates (BiOCl–B–VO). Photocatalytic CO2RR test showed that the BiOCl presented the highest CO evolution rate of 83.64 μmol g−1 h−1 without any scavengers or cocatalysts (figure 4(g)), which was attributed to the synergistic effect between B doping and VO. More importantly, BiOCl–B–VO sample possessed a high selectivity of more than 98% for CO generation, much exceeding those of BiOCl and BiOCl–VO. According to corresponding DFT calculation, the B–VO associates could lengthen the C–O bond and contribute to a CO2 adsorption energy of −0.89 eV, resulting in efficient CO2 activation. For CO2 to CO conversion, the formation of *COOH typically requires high energy barriers and has been regarded as the rate-limiting step. Compared with an individual VO, B–VO associates delivered a much lower energy barrier of 1.14 eV to form *COOH, which was subsequently dissociated with a proton to develop *CO and H2O (figure 4(h)). Afterwards, *CO was released from BiOCl–B–VO surface rather than further hydrogenation to form CHO* for CH4 generation because of the exothermic of *CO desorption. This suggested CH4 formation was not a favorable pathway over BiOCl–B–VO sample. Additionally, the competitive hydrogen generation was also largely suppressed. Therefore, the introduced B–VO associates could mediate BiOCl for both enhancing photocatalytic CO2RR activity and selectivity, restrict the competitive hydrogen evolution, and improve the stabilization of *COOH for selective CO evolution (figure 4(i)).
Considering the less energetically demands of CH4 and CO, photocatalytic CO2RR into liquid products, such as methanol and formic acid, is more sustainable and valuable for CO2 reutilization. Rational atom manufacturing of suitable semiconductors can achieve CO2 activation towards liquid solar fuels. Hou et al proposed the VOs confined in phosphate-doped Bi2WO6 atomic layers (VO–PO4–BWO) as an ideal photocatalyst for triggering CO2 conversion, which was synthesized by thermal treatment of PO4 oxoanion doped Bi2WO6 under H2/Ar mixture gas [86]. Particularly, some pits could be distinctly observed in the HRTEM image (figure 5(a)), suggesting the successful elimination of the oxygen atom connecting with the Bi atom and demonstrating the formation of VOs confined in PO4–BWO layers (figure 5(b)). In addition, the stronger EPR signal of VO–PO4–BWO further confirmed the introduction of VOs, which was beneficial for reducing the bandgap and enhancing light absorption property. As a result, the VO–PO4–BWO delivered a methanol yield rate of 157 μmol g−1 h−1, outperforming those of VO–BWO, PO4–BWO, BWO atomic layers, and bulk BWO (figure 5(c)). The superior performance for photocatalytic CO2 conversion of VO–PO4–BWO could be ascribed to both the PO4 doping and introduced VOs. The presence of VOs could trap the photogenerated electrons in the CB, thereby bringing about the effective charge transfer and separation over the VO–PO4–BWO sample. In principle, photocatalytic CO2RR usually follows a multi-electron transfer pathway with proton assistance to overcome the high energies, thus suppressing the formation of carbonate radical (CO2 −) to generate long-chain chemicals with more than three carbons. Rational design of VOs also offers an alternative strategy to allow CO2 activation through a single-electron process to the formation of long-chain chemicals. For instance, Chen et al prepared Bi2O3 atomic layers through in situ oxidation of exfoliated Bi nanosheets by use of its high surface energy (figure 5(d)) [68]. After that, VOs could be successfully introduced by ultrasonication treatment. Interestingly, the short oxidation time promoted the formation of higher concentration of VOs than that of long oxidation time. The distinct lattice disorder originated from VOs induced by the unsaturated coordination of Bi can be clearly observed from HAADF-STEM image, confirming that VO defective structures were successfully developed in Bi2O3. Moreover, the VO-rich-Bi2O3 exhibited a lower photoluminescence (PL) emission intensity than that of VO-poor-Bi2O3, indicating that the VOs could suppress the recombination of photogenerated electrons and holes. Subsequently, the photocatalytic CO2 and CH3OH conversion to dimethyl carbonate were carried out and showed a conversion yield of above 18% over VO-rich-Bi2O3, which was nine-fold higher than that of VO-poor-Bi2O3, implying the VOs provided additional active sites for triggering CO2 reduction (figure 5(e)). The VOs confined in the surface of Bi2O3 nanosheets could supply abundant unsaturated coordination sites for CO2 adsorption, which was further activated by the localized electrons on the VOs to the generation of •CO2 −. Meanwhile, CH3OH was also inclined to be adsorbed on VO-rich-Bi2O3 nanosheets with more negative adsorption energy than that of vacancy-free counterpart (figure 5(f)). Benefiting from the above advantages associated with the improved separation efficiency of photogenerated carriers, the VO-rich-Bi2O3 nanosheets could give a superior CO2 conversion performance toward dimethyl carbonate with nearly 100% selectivity.
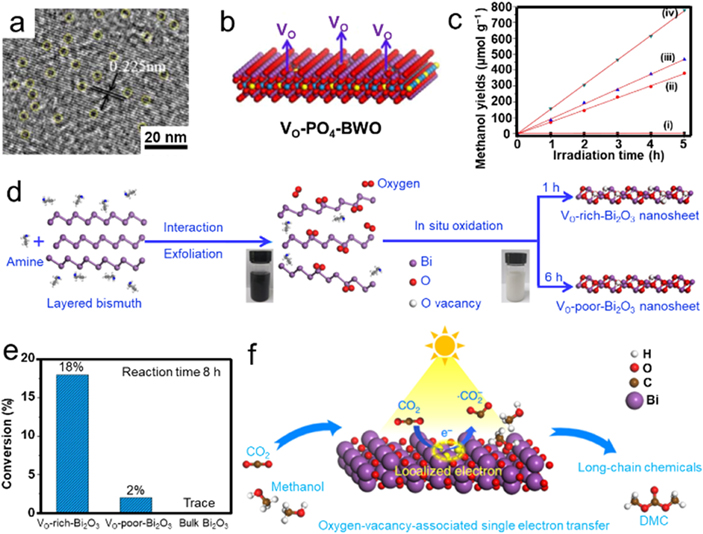
Figure 5. The fabrication of VO-modified photocatalysts with their photocatalytic CO2RR activities towards CO and liquid fuels. (a) HRTEM image of VO–PO4–BWO. (b) The corresponding schematic structure of VO–PO4–BWO. (c) Methanol yields over various photocatalysts. (i) Bulk BWO, (ii) BWO atomic layers, (iii) PO4–BWO layers and (iv) VO–PO4–BWO. Reprinted from [86], Copyright (2017), with permission from Elsevier. (d) Schematic preparation process of ultrathin Bi2O3 nanosheets with rich/poor VOs. (e) Photocatalytic CO2 conversion performances over various catalysts at 373 K. (f) Schematic illustration of CO2 photoreduction to long chain chemicals. Reproduced from [68]. CC BY 4.0.
Download figure:
Standard image High-resolution imageBesides VOs, other types of anionic vacancies have also been widely studied to enhance the corresponding photocatalytic performance toward CO2 conversions, such as nitrogen vacancies (VNs), sulfur vacancies (VSs), and carbon vacancies (VCs). For instance, Tu et al prepared VNs modified PCN nanosheets for photocatalytic CO2 conversion [45]. The density of VNs could be tuned by varying the post-treatment temperature at 475, 500, 525, and 550 °C. In comparison with the negligible EPR signal of pristine bulk PCN, the intensity of the EPR signal gradually enhanced as the post-processing temperature increased, indicating the improvement of two-coordinated VNs confined in PCN frameworks, which could offer unpaired electrons to adjacent sp2 carbon atoms with aromatic rings of PCN. The photocatalytic CO2RR tests demonstrated that the CO generation rate was enhanced as the increase of VNs from bulk pristine PCN to PCN-525, delivering a superior CO evolution rate of 6.21 μmol h−1 for PCN-525, which was nearly 4.2 folds higher than that of bulk PCN. Noticeably, an excessive VNs could result in a slightly inferior CO generation rate (PCN-550), which might be attributed to the recombination of photo-generated electrons and holes. Benefiting from the introduced VNs, the formed middle bandgap states could not only promote the excitation of more photo-generated electrons and holes, but also serve as a temporary reservoir for trapping photo-generated electrons, thereby accelerating the separation of photo-generated electrons and holes for favoring the remarkable photocatalytic activity.
Given the role of VNs as recombination centers for photo-generated carriers, the integration of dopants and VNs may be an effective alternative strategy for avoiding drawbacks of VNs. For this matter, Wang et al developed a B, K co-doped PCN associated with controllable VNs over thermal polymerization of dicyandiamide with KBH4 (KBH–PCN), in which the K doped in vacancy site could promote the electron transfer between CO2 and PCN [87]. Meanwhile, the B dopants were helpful for keeping the necessary reduction potential for triggering CO2 photoreduction. Moreover, the presence of VNs could lower the bandgap for boosting light-harvest capability. The CO2 temperature-programmed desorption results elucidated that the optimized 3% KBH–PCN delivered the superior CO2 adsorption capability with higher desorption temperature, which could be attributed to both the improved number and strength of Lewis basic sites. According to the density of states (DOS) of PCN and KBH–PCN, the K and B dopants did not significantly participate in developing the middle gap states. Photocatalytic CO2 reduction with H2O demonstrated that the 3% KBH–PCN offered the highest CO2 conversion rates of 5.93 and 3.16 μmol g−1 for CH4 and CO production, respectively, after 5 h irradiation, which were approximately 1.61 and 5.27 folds of pristine PCN (figure 6(a)).
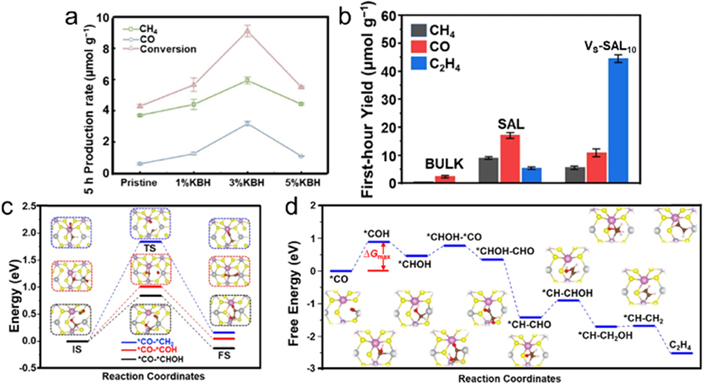
Figure 6. Other types of anionic vacancies for photocatalytic CO2RR. (a) The total conversion of CO2 to CH4 and CO over pristine PCN and KBH-modified PCN. Reprinted from [87], Copyright (2019), with permission from Elsevier. (b) Photocatalytic CO2RR activity at the first hour. (c) Three possible C–C coupling pathways over VS–AgInP2S6. (d) Gibbs free energy diagrams for CO2 reduction towards C2H4 on VS–AgInP2S6. Reproduced from [90]. CC BY 4.0.
Download figure:
Standard image High-resolution imageBesides VNs, VCs can be also engineered in PCN skeleton, which could serve as adsorption sites for capturing CO2 because of the left unsaturated nitrogen, enabling the activation of CO2. For instance, Yang et al introduced VCs on PCN using a constant temperature steam etching approach to selectively remove carbon atoms of PCN skeleton [88]. After that, the surface C/N ratio of PCN decreased from 0.859 to 0.705, indicating the successful removal of carbon atoms toward VC creation. The positron annihilation spectroscopy (PAS) was employed to reveal the surface and bulk defects in materials. In comparison with pristine PCN, the PCN-500-4 possessed an increased τ2 value, suggesting that numerous VCs were presented onto the PCN surface. Correspondingly, the PCN-500-4 sample also delivered an enhanced EPR signal originated from the unpaired electrons around the VCs. In addition, the time-resolved PL spectra revealed a longer lifetime of the photo-generated carriers after VCs introduction, implying that the VCs could capture the photo-generated electrons and thereby prolong the life-span of charges for participation in CO2 conversion. The optimized VC-modified PCN gave a CO evolution rate of 21.5 μmol h−1, which was much higher than that of pristine PCN (1.2 μmol h−1). DFT calculations demonstrated that *CO2 was preferentially trapped in the location near the two NH2 groups generated by carbon removal with no obvious geometric differences. The required energy for the rate-determining step of the hydrogenation of *CO2 to *COOH could be lowered by about 0.15 eV in VC modified PCN, thereby promoting photocatalytic CO2 reduction.
Transition metal sulfide is a broad class of layered structures that possess unique optical and thermoelectric properties, which can be exfoliated into atomically 2D nanosheet structure via atom manufacturing. Using Cu2O as a precursor, Wang et al synthesized sulfur vacancy modified Cu3SnS4 (VS–Cu3SnS4) via a simple hydrothermal treatment [89]. A series of characterizations confirmed the successful introduction of VS in Cu3SnS4, which promoted electron accumulation on surrounding Sn(II) atoms. Accordingly, the CB position negatively shifted with enhanced reduction capability of electrons. As a result, the optimized VS–Cu3SnS4 gave a CH4 generation rate of 22.65 μmol g−1 h−1 with about 83.1% selectivity. Compared with CH4, C2 products are more valuable for CO2 conversion, but still remains a great challenge. To this end, the AgInP2S6 single atomic layers (AgInP2S6 SAL) was fabricated using ultrasound exfoliation of the bulk counterpart, in which VSs could be successfully introduced by etching process in H2O2 solution [90]. Photocatalytic CO2RR was performed with water vapor under light irradiation. For both bulk sample and vacancy-free AgInP2S6 SAL, CO was the major product. A small amount of C2H4 was also detected for AgInP2S6 SAL, which was formed via a 12 electrons and 12 protons process. After the rational introduction of VSs, C2H4 became the dominant product with a yield of 44.3 μmol g−1 associated with an impressive selectivity of ∼73% (figure 6(b)). In the meantime, both CO and CH4 were also detected but at lower concentrations compared with vacancy-free SALs, suggesting that the presence of VSs in SALs preferentially enabled C–C coupling process between C1 intermediates toward C2 generation rather than isolated them into C1 gases. DFT calculations were carried out to investigate the catalytic selectivity mechanism toward C2H4 on VS–AgInP2S6 surface. It is well known that the CO2 hydrogenation to generate *COOH is the key step for CO evolution. Further coupling a pair of proton/electron with *COOH promoted the formation of CO and H2O. For vacancy-free AgInP2S6, the generated *CO suffered from a low adsorption energy of −0.07 eV, indicating a facile extraction of *CO from catalyst surface to become CO gas and thereby presenting a high CO selectivity. After introducing VSs, the exposed Ag atom around the VS exhibited charge accumulation, which could anchor the *CO via chemical adsorption way with an adsorption energy of −0.25 eV. On one hand, the adsorbed *CO could be further protonated to generate a series of intermediates to participate in the following reaction. On the other hand, other generated *CO on catalyst surface diffused to VSs and coupled with these intermediates to form C2H4. As shown in figure 6(c), three possible unsaturated reaction intermediates were proposed. In comparison with other two coupling processes, *CO–CHOH coupling possessed a relatively lower energy barrier of 0.84 eV. Therefore, C2H4 evolution would follow *CO–CHOH coupling associated with hydrogenation. Notably, the protonation of *CO toward *COH was the rate-determining step during C2H4 generation process based on the whole energy diagram (figure 6(d)).
3.2.2. Cationic vacancies
Similar to anionic vacancies, cationic vacancies can also bring about significant electronic structure variation with respect to perfect ones, which is beneficial for improving the corresponding catalytic activities. Particularly, cationic vacancies seem to be preferable compared with anionic vacancies because of the susceptible deactivation of anionic vacancies induced by oxygen species from the environment. Di et al prepared BiOBr ultrathin nanosheets with tunable Bi vacancies (VBi) using controllable ionic liquid-assisted synthesis under ambient conditions [91]. The aberration-corrected STEM-HAADF was employed to uncover the surface microstructure of VBi–BiOBr nanosheets (figure 7(a)), demonstrating that partial surface Bi atoms were missing, which confirmed the successful formation of VBi. On the contrary, the BiOBr nanosheets sample exhibited an ordered lattice and surface atomic configuration. VBi–BiOBr nanosheets delivered an apparently red-shifted absorption edge compared to BiOBr nanosheets. In particular, benefiting from the Bi vacancies, the VBi–BiOBr nanosheets displayed a tail peak range from 430 nm to 500 nm. The bandgaps of VBi–BiOBr and BiOBr nanosheets were estimated to be 2.65 eV and 2.77 eV, respectively. Combined with x-ray photoelectron spectroscopy (XPS) valence band (VB) spectra, the VB and CB potentials of VBi–BiOBr were shifted from 2.01 eV and −0.76 eV to 1.57 eV and −1.08 eV (vs NHE at pH = 7), respectively, compared with BiOBr nanosheets (figure 7(b)). The negatively shifted CB potential of VBi–BiOBr contributed to higher reducibility of photo-generated electrons and thus favored the photocatalytic CO2 conversion process. The VBi–BiOBr delivered a photocatalytic CO evolution rate of 20.1 μmol g−1 h−1 with a 98% selectivity, which was almost 3.8 folds higher than CO generation rate of BiOBr nanosheets (figure 7(c)). The enhanced photocatalytic activity could be attributed to the association of the tunable electronic structure, optimized CO2 adsorption/activation, and CO desorption process for VBi–BiOBr sample. It should be noted that the VBi–BiOBr still retained an efficient photocatalytic activity after fourth cycling tests, implying its superior stability.
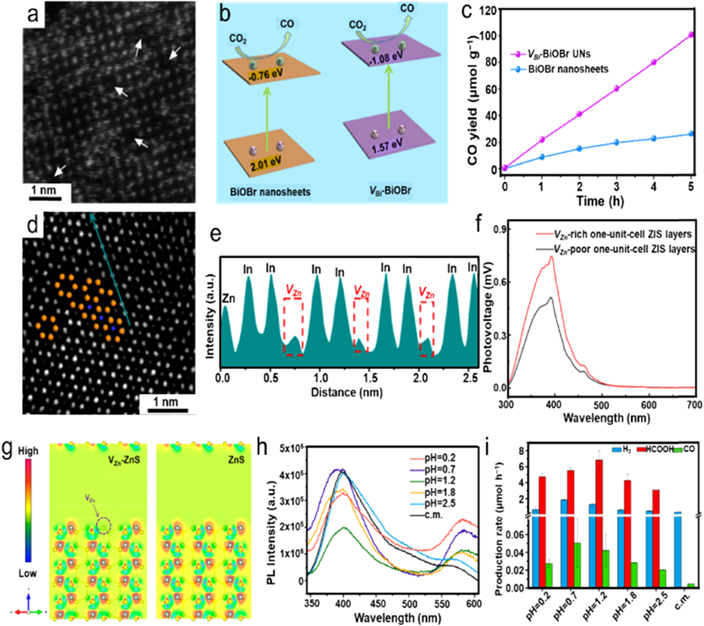
Figure 7. Various cationic vacancies over photocatalysts. (a) HAADF-STEM image of VBi–BiOBr, scale bar is 1 nm. (b) Schematic illustration of band structures of pristine BiOBr and VBi–BiOBr. (c) Photocatalytic CO2 conversion toward CO over pristine BiOBr and VBi–BiOBr. Reprinted with permission from [91]. Copyright (2019) American Chemical Society. (d) HAADF-STEM image. (e) Intensity profile of the dark cyan arrow in figure (d). (f) SPV spectra of ZnIn2S4 layer. Reprinted with permission from [92]. Copyright (2017) American Chemical Society. (g) Optimized cell structures of VZn modified ZnS (Zn53S54) and perfect ZnS (Zn53S54) with charge density. (h) PL spectra of the pristine ZnS and the VZn modified ZnS samples. (i) Product evolution rates over the as-prepared samples. Reprinted with permission from [93]. Copyright (2019) American Chemical Society.
Download figure:
Standard image High-resolution imageZn vacancies (VZn) are another case of common cationic vacancies. For instance, Jiao et al successfully synthesized one-unit-cell thick ZnIn2S4 layers with tunable vacancy concentrations through a simple hydrothermal way. TEM and atomic force microscope (AFM) results verified the sheet-like morphology with an average thickness of about 2.46 nm. The Zn vacancies could be well observed in the HAADF-STEM image shown in figure 7(d), and the corresponding line intensity profiles confirmed that the vacancies were VZn (figure 7(e)) [92]. EPR and PAS further manifested the successful preparation of one-unit-cell ZnIn2S4 atomic layers with distinct VZn concentrations. Benefiting from the presence of VZn, the VZn-rich one-unit-cell ZnIn2S4 layers possessed the distinctly enhanced carrier separation efficiency compared with the VZn-poor sample, as demonstrated by surface photovoltage (SPV) spectroscopy in figure 7(f). Furthermore, the abundant VZn also favored light-harvesting, CO2 adsorption capability, and improved surface hydrophilicity, which could promote the photocatalytic performance of ZnIn2S4. As a result, the VZn-rich one-unit-cell ZnIn2S4 layers delivered the photocatalytic CO2RR toward CO formation rate of 33.2 μmol g−1 h−1, which was about 3.6-fold higher than that of VZn-poor counterparts, signifying the vital role of VZn in fully optimizing the photocatalytic performance. The corresponding DFT calculations unveiled that the VZn could not only largely enrich charge density at the adjacent sulfur atoms to enable the electrons photoexcitation easier but also serve as electron capture centers to promote charge separation. Of note, the presence of abundant VZn could endow a more negatively charged surface, which potentially improved the CO2 adsorption capabilities. ZnS is also a promising photocatalyst for CO2 conversion with a relatively high CB position at −1.04 V (vs NHE at pH = 7). However, highly crystalline ZnS seriously suffers from insufficient active sites, resulting in a thwarted photocatalytic CO2 conversion capability. Pang et al created VZn on the surface of ZnS via an acid-etching strategy, which could effectively avoid the adverse influence on photo-generated electron transfer induced by the bulk defects [93]. As DFT calculation shown in figure 7(g), the S atoms in the VZn vicinity presented the lower charge density relative to pristine ZnS based on (001) surface slabs. The steady-state PL spectroscopy was also conducted and revealed that the peak intensities at 580 nm, which represented the donor–acceptor luminescence band associated with VZn, increased as the acid increasing, implying more VZn were formed by stronger acidic etching (figure 7(h)). Subsequently, photocatalytic CO2 conversion was explored without any cocatalyst. As shown in figure 7(i), for pristine commercial ZnS, only H2 and a small quantity of CO could be monitored without any liquid product. In contrast, the VZn modified ZnS processed by various sulfuric acid delivered significantly improved photocatalytic activity with formate as the main product when the pH of the acid solution was adjusted to 1.2. Particularly, a selectivity up to 86.6% for HCOOH could be achieved for acid-treated ZnS (pH = 0.2). In situ attenuated total reflection-infrared spectra were collected to identify CO2RR over VZn–ZnS, demonstrating the generation of HCOO* under light irradiation. And then HCOO− was formed after the second electron transfer, which subsequently desorbed from VZn–ZnS surface. DFT calculations clarified that the adsorption energy of CO2 was lower than that of the competitive proton, suggesting that the presence of VZn was not favorable for hydrogen evolution. Furthermore, compared with the higher adsorption energy of *CO intermediate that promoted CO generation, the photocatalytic CO2RR towards formate possessed a more facile energy barrier.
By varying the temperature of hydrothermal treatment, Gao et al successfully tuned the vanadium vacancy (VV) density on orthorhombic-BiVO4 (o-BiVO4) atomic layers with the single-unit-cell thickness (figure 8(a)) [94]. During the process, the BiVO4-based lamellar structures, which was formed by the reaction between the added VO4 3− and the Bi3+ of the BiCl4 −-CTA+ (cetyltrimethylammonium) lamellar hybrids, could spontaneously exfoliate into o-BiVO4 ultrathin nanosheets. The AFM image shown in figure 8(b) disclosed an average thickness of 1.28 nm, which was consistent with a unit cell thickness along the [001] direction. PAS was also conducted to relatively quantify the VV concentration. As shown in figure 8(c), the VV-rich o-BiVO4 sample possessed a longer positron annihilation time at VV and higher VV density than those of VV-poor o-BiVO4, resulting in a new defect level for enhancing light harvesting. In addition, SPV spectroscopy was performed to unveil the dynamic features of photogenerated carriers, which delivered an improved SPV intensity for VV-rich o-BiVO4, ensuring a higher separation efficiency of photogenerated electrons and holes, which was further verified by the time-resolved fluorescence spectra. Benefiting from the above virtues, the VV-rich o-BiVO4 ultrathin nanosheets showed a CH3OH generation rate of 398.3 μmol g−1 h−1, which was about 1.4 times higher than that of VV-poor o-BiVO4 (figure 8(d)), suggesting the pivotal role in promoting the photocatalytic performance of Vv. Furthermore, the VV-rich o-BiVO4 also exhibited a higher apparent quantum efficiency with a maximum of 5.96% at 350 nm compared with the VV-poor o-BiVO4 sample (figure 8(e)).
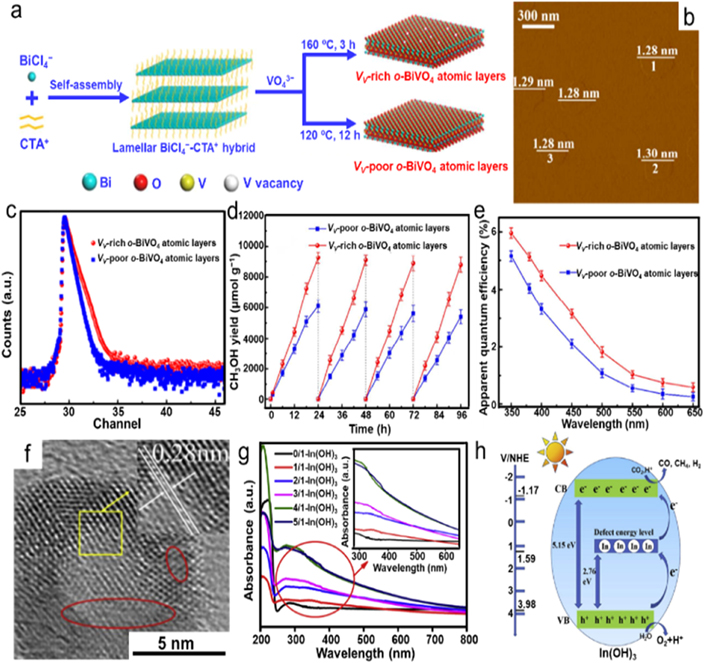
Figure 8. Vanadium and indium vacancies of photocatalysts. (a) Schematic illustration of the preparation process of the VV-rich and VV-poor o-BiVO4 atom layers. (b) AFM image. (c) PAS spectra. (d) Photocatalytic cycling stability tests. (e) Apparent quantum yields. Reprinted with permission from [94]. Copyright (2017) American Chemical Society. (f) HRTEM image of 4/1-In(OH)3. (g) UV–Vis absorption spectra of samples. (h) Schematic illustration of the mechanism for photocatalytic CO2 conversion with H2O vapor over VIn modified In(OH)3. Reprinted from [95], Copyright (2019), with permission from Elsevier.
Download figure:
Standard image High-resolution imageIn addition, taking advantage of the middle energy level created by vacancies, direct utilization of wide bandgap semiconductors as photocatalysts for CO2 conversion becomes possible. In this regard, Hu and coworkers prepared a series of In(OH)3 with a large number of indium vacancies (VIn) via a simple hydrothermal way using mixed solvent comprised of ethylenediamine and water, of which the volume ratio of ethylenediamine/water was varied from 0/1 to 1/1, 2/1, 3/1, 4/1 and 5/1 [95]. The HRTEM image shown in figure 8(f) obviously indicated indiscernible lattice fringes in the circle regions originated from the presence of vacancies. On the contrary, the In(OH)3 synthesized using water as the solvent delivered a smooth surface with a cubic structure. UV–Vis absorption spectra were collected to investigate the effects of vacancies on the light-harvesting capability. As depicted in figure 8(g), the 0/1-In(OH)3 sample showed single absorption at the ultraviolet region with a calculated bandgap of more than 5.2 eV. After introducing VIn, the In(OH)3 samples not only possessed improved light absorption in the range of 260–350 nm and presented a crucial tailing absorption at the wavelength ranging from 480 nm to 650 nm, but also possessed CO2 chemisorption capability, which was enhanced as the vacancy content increased. Furthermore, the photocatalytic CO2 conversion capabilities were determined under visible light irradiation at 200 °C using water vapor as a reducing agent, illustrating that all VIn-modified samples delivered much higher H2, CO and CH4 production rates than that of pristine In(OH)3. Among them, the 4/1-In(OH)3 sample processed the highest photocatalytic CO2 conversion activity with superior cycling stability. Benefiting from the middle energy level induced by VIn, the In(OH)3 could be excited by visible light over a two-step process. The electrons in VB could be excited to defect energy level, which was further excited to the CB of In(OH)3 for CO2 conversion, leaving photo-generated holes on the VB for water oxidation (figure 8(h)).
3.2.3. Dual vacancies
Similar to the multiple heteroatom dopants to offer more catalytic sites relative to single ones, the combination of two or more types of vacancies would be an intriguing way to promote photocatalytic performance owing to the existence of more active sites and the synergistic effects between multiatomic vacancies. Therefore, exploring the association of various vacancies is extremely significant and imperative. Li et al reported dual anionic vacancies of oxygen and chlorine in BiOCl (BiO1−x Cl1−y ) via a simple hydrolysis approach using the extracting solutions separately derived from green tea and spinach as reductant, denoted as BiO1−x Cl1−y -T and BiO1−x Cl1−y -S, respectively [96]. Figure 9(a) showed the Fourier transformed profile of x-ray absorption fine structure (XAFS), which revealed that BiOCl-T delivered a shorter Bi–O bond length relative to pristine BiOCl derived from the lattice local contraction of the VOs, while the Bi–Cl bond length was longer than that of BiOCl due to the Cl vacancies. Furthermore, the pristine BiOCl exhibited no EPR signals because of no defects in BiOCl. In contrast, both BiOCl-T and BiOCl-S samples exhibited an obvious signal peak at g = 2.0, which was attributed to VOs. Especially, the peak intensity of BiOCl-S was stronger than that of BiOCl-T, validating a higher VOs content in BiOCl-S. UV–Vis absorption spectroscopy was performed to reveal the optical harvest capabilities of samples, displaying that BiOCl-T and BiOCl-S gave visible responses extending to 500 nm, whereas the pristine BiOCl only possessed slight absorption in the visible region (figure 9(b)). It should be noted that both BiOCl-T and BiOCl-S had two new absorption peaks centered at 430 nm and 660 nm due to the formation of two defect energy levels. Base on the above, the calculated bandgaps of BiOCl, BiOCl-T, and BiOCl-S were 3.07 eV, 2.66 eV, and 2.31 eV, respectively. With dual vacancies introduced, both BiOCl-T and BiOCl-S samples presented largely boosted photocatalytic CO2RR toward CO, even the CH4 production rate decreased. The CO selectivity for BiOCl-T and BiOCl-S were 2.2 and 2.5 folds higher than that of pristine BiOCl, which could be attributed to the presence of VOs and Cl vacancies (figure 9(c)). DFT calculation demonstrated that the introduced dual vacancies could efficiently tune the electronic structure of BiOCl, resulting in downward shifted position of CB and VB with a decreased bandgap.
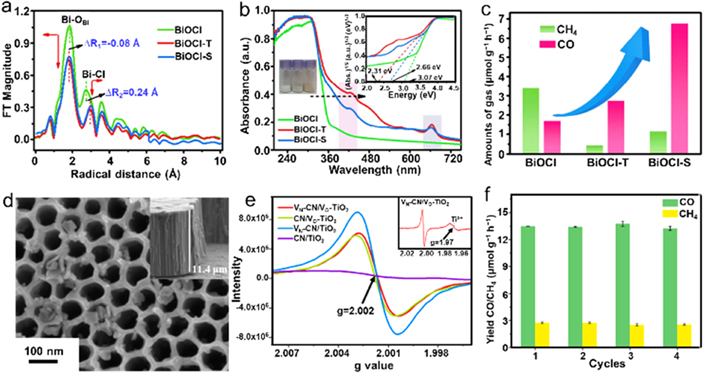
Figure 9. Dual anionic vacancies of photocatalysts. (a) The Fourier transformed profiles of Bi coordination environments. (b) UV–Vis diffuse reflectance spectra of samples; inset exhibited the bandgaps. (c) Photocatalytic CO2 conversion under stimulated solar light. Reprinted with permission from [96]. Copyright (2018) American Chemical Society. (d) SEM image of VNs–PCN/VOs–TiO2. (e) ESR spectra. (f) Cycling stability of VNs–PCN/VOs–TiO2. Reprinted from [97], Copyright (2021), with permission from Elsevier.
Download figure:
Standard image High-resolution imageIn another study, Yang et al constructed a Z-scheme heterostructure photocatalyst with interfacial dual vacancies, of which the VNs and VOs were originated from PCN and TiO2, respectively [97]. The SEM image shown in figure 9(d) clearly revealed VOs–TiO2 nanotube arrays with VNs–PCN intimate deposition. The room temperature ESR spectra demonstrated an apparently enhanced ESR signal for VNs–PCN/VOs–TiO2 compared with PCN/TiO2 without vacancies (figure 9(e)). Of note, the interaction between VOs and VNs might deliver a slightly reduced ESR signal with respect to VNs–PCN/TiO2. Furthermore, the low-temperature ESR spectra exhibited a distinct signal at g = 1.97 (inset in figure 9(e)), confirming the existence of VOs in VNs–PCN/VOs–TiO2. Benefits from the interfacial dual vacancies, the VNs–PCN/VOs–TiO2 possessed a broad light adsorption band edge in the range of 300–800 nm, accompanied by a superior visible light absorption intensity, indicating the alterant band structure of PCN and TiO2. With the emergence of dual vacancies, the as-prepared sample achieved visible light yields of 13.4 and 3.0 μmol g−1 for CO and CH4, respectively, significantly outperforming the performance of pristine PCN/TiO2. In addition, the dual vacancies modified photocatalyst also possessed good stability after four cycling tests (figure 9(f)).
In contrast to dual anionic vacancies, Di et al prepared freestanding Bi2MoO6 ultrathin nanosheets with surface 'Bi–O' vacancy pairs using hydrothermal way assisted with polyvinyl pyrrolidone as a surfactant [98]. During the process, layered BiOBr ultrathin nanosheets were initially formed, which served as a sacrificial template to guide the fabrication of Bi2MoO6 nanosheets over the ion-exchange approach (figure 10(a)). The aberration-corrected STEM-HAADF image clearly showed the dim sites of Bi associated vacancies. Surface VOs could be confirmed by low-temperature EPR analysis with the typical signal at g = 2.0. It is well accepted that the vacancies could serve as centers for reserving photo-generated electrons, thereby promoting the separation of photo-generated charge carriers. As displayed in figure 10(b), the time-resolved fluorescence spectra revealed that the Bi2MoO6 ultrathin nanosheets with 'Bi–O' vacancy pairs possessed a longer radiative lifetime of photo-generated charge carriers than that of bulk Bi2MoO6, which could be ascribed to the faster charge transfer derived from the atomically ultrathin framework as well as dual vacancies. Profiting from the successful introduction of dual vacancy, the Bi2MoO6 ultrathin nanosheets with 'Bi–O' vacancy pairs showed an absorption tail that extending to ∼520 nm, which was further verified by DFT calculations. As shown in figure 10(c), the enhanced DOS at the VB edge, as well as newly formed DOS in the forbidden band, could be observed in 'Bi–O' vacancy modified Bi2MoO6, implying the higher carrier concentration and much easier excitation of electrons into CB. As a result, the 'Bi–O' vacancy modified Bi2MoO6 delivered a CO evolution rate of 3.62 μmol h−1 g−1, which was much higher than that of the bulk counterpart (1.42 μmol h−1 g−1). In addition, the defective sample also had excellent cycling stability originated from no obvious deterioration after three cycle tests.
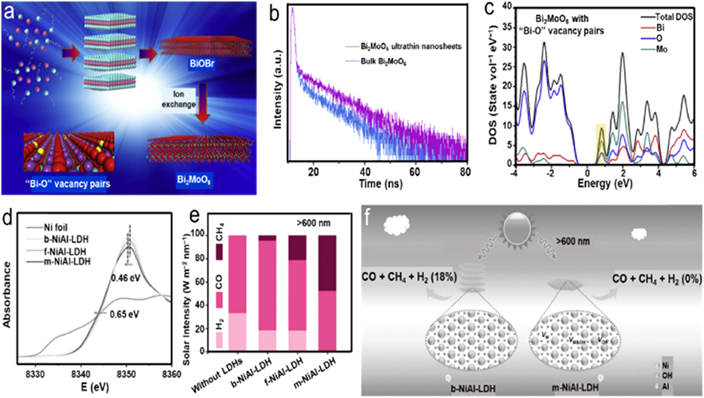
Figure 10. Mixed vacancy pairs of photocatalysts. (a) Schematic illustration of the synthesis process for 'Bi–O' vacancy modified Bi2MoO6 ultrathin nanosheets. (b) Time-resolved transient fluorescence decay. (c) The calculated DOS of Bi2MoO6 ultrathin nanosheets with 'Bi–O' vacancy pairs. Reprinted from [98], Copyright (2019), with permission from Elsevier. (d) Ni K-edge XANES spectra. (e) Photocatalytic CO2RR under irradiation above 600 nm of NiAl-LDH. (f) Schematic illustration for the selectivity of photocatalytic CO2RR over NiAl-LDH with different thicknesses under irradiation above 600 nm. [99] John Wiley & Sons. Copyright 2019, Wiley-VCH Verlag.
Download figure:
Standard image High-resolution imageThe accurate control of dual vacancies concentration is also an effective strategy to mediate the selectivity of photocatalytic CO2RR. Tan et al synthesized a series of Ni–Al layered double hydroxide (NiAl-LDH) with various thicknesses ranging from 27 nm (b-NiAl-LDH) to 5 nm (f-NiAl-LDH) and 1 nm (m-NiAl-LDH) [99]. The XAFS displayed in figure 10(d) revealed the decreased energy locations of Ni K-edge in the arrangement of b-NiAl-LDH, f-NiAl-LDH, and m-NiAl-LDH, showing a reduced average oxidation state of +1.6 for Ni in m-NiAl-LDH, which was further confirmed by soft XAS tests. The coordination number of Ni was analyzed and exhibited decrease of the first Ni–O shell along the sequence of b-NiAl-LDH, f-NiAl-LDH, and m-NiAl-LDH, manifesting a slightly greater degree of structural distortion as the thickness decreases, which might be induced by the hydroxyl defect. In addition, the PAS was also performed and further verified that the m-NiAl-LDH possessed the most surface defects in three samples. Resulted from the abundant surface vacancies, the m-NiAl-LDH exhibited a lowered bandgap of 1.78 eV, which promoted light harvesting in a wider spectrum. By using NiAl-LDH samples as photocatalysts with the addition of Ru(bpy)3Cl2•6H2O under visible light above 400 nm, the selectivity of CH4 could reach 16.54% for m-NiAl-LDH compared with the 0.11% of b-NiAl-LDH. While the H2 evolution was validly suppressed from a selectivity of 43.84% for b-NiAl-LDH to 13.26% for m-NiAl-LDH. Furthermore, the m-NiAl-LDH possessed CH4 and CO yields of 168 μmol h−1 g−1 and 712 μmol h−1 g−1, respectively, greatly surpassing the performance of b-NiAl-LDH. More intriguingly, when the light irradiation was extended to above 600 nm, the CH4 selectivity can be enhanced to 70.3% with the completely suppressive H2 evolution (figures 10(e)–(f)). Whereas, the selectivity of H2 was still about 18% for b-NiAl-LDH. To unveil the correlation between the selectivity and light wavelength of m-NiAl-LDH, DFT with Hubbard corrected calculations were conducted and demonstrated that the synergistic effect derived from metal and hydroxyl defects could effectively reduce the Gibbs free energy barrier of CO2RR toward CH4 (0.127 eV) with a driving force of 0.313 eV.
In summary, vacancy engineering is an effective strategy to tune photocatalytic performance by offering abundant active sites, altering reaction mechanisms or pathways to boost the catalytic activity. There are mainly three types of vacancies, anionic, cationic, and dual vacancies, which could be developed to optimize the electronic structure and effectively accelerate photocatalytic CO2 conversion rate. Although many efforts have been devoted to engineering vacancies for enhancing photocatalytic CO2, the current challenges and barriers, such as effectively engineering strategies and remarkably catalytic stability, are still urgent to be dealt with. In addition, the relationship between vacancies and photocatalytic activities remains ambiguous, which limits the further enhancement of the photocatalytic performance. Therefore, developing efficient vacancy engineering strategies and advanced characteristic techniques associated with theoretical simulation are significantly required to further unveil the key roles of vacancies during the photocatalytic process and promote practical applications.
3.3. Heteroatom doping
Typical metal-free dopants including N, B, and S atoms possess different electronegativity, resulting in variable surface charge distribution for affording extra active sites to optimize photocatalytic performance. For instance, Shan et al synthesized a boron-doped layered polyhedron SrTiO3 with controllable boron contents via varying used TiB2, delivering a three-times improved photocatalytic CO2RR activity compared with pristine SrTiO3 [100]. Introducing boron could not only enhance the electrostatic potential variation to improve the internal electric field of SrTiO3 for accelerating the photogenerated charge separation, but also offer additional necessary sites for CO2 adsorption, thereby giving a superior photocatalytic CO2RR performance (figure 11(a)). In addition, metal-free heteroatom doping is also a potential strategy to optimize CO2RR pathway. Based on Azofra and coworkers' reports, the pathway of CO2RR towards CH3OH was CO2 → COOH* → CO → HCO* → HCHO → CH3O* → CH3OH using PCN as a photocatalyst [101]. After sulfur doping, the rate-determining step could be adjusted to process a lowered Gibbs free energy. For pristine PCN, the highest occupied molecular orbital (HOMO) mainly distributed on carbon atoms accompanied with moderate nitrogen atoms, while the corresponding LUMO located on the nitrogen atom (figure 11(b)) [102]. On the contrary, the HOMO was mainly located on N atoms while the LUMO mainly distributed on the S atom for S-doped PCN, which would possess a better photogenerated charge separation capability. Conceivably, heteroatom doping can also accommodate product selectivity of photocatalytic CO2RR. In this aspect, Liu et al proposed phosphorus-doped PCN nanotubes via thermal polymerization of melamine with NaH2PO2•H2O addition [103]. The phosphorus doping was beneficial for lowering the CB and VB positions associated with enhanced CO2 adsorption capability. Furthermore, the photocatalytic CO2RR towards CO and CH4 increased for 3.10 and 13.92 times, respectively, compared to pristine PCN. Notably, the product ratio of CO/CH4 significantly down shifted to 1.30 compared to 6.02 of pure PCN, suggesting a higher CH4 selectivity after doping phosphorus (figure 11(c)).
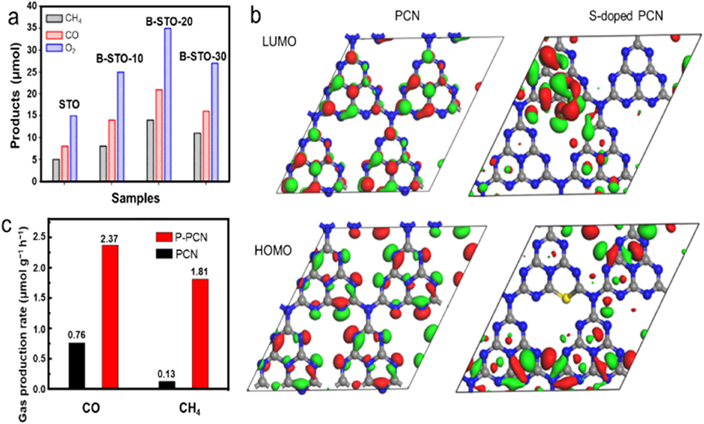
Figure 11. Metal-free dopants of photocatalysts. (a) The yield rates of CH4 during the photocatalytic CO2RR with H2O on different samples. Reprinted from [100], Copyright (2017), with permission from Elsevier. (b) LUMOs and HOMOs of pure PCN and S-doped PCN. Reprinted with permission from [102]. Copyright (2018) American Chemical Society. (c) The CO and CH4 production rate over pure PCN and P-doped PCN. Reprinted with permission from [103]. Copyright (2018) American Chemical Society.
Download figure:
Standard image High-resolution imageBenefiting from the variable valence of metal atoms, metal atom doping can be achieved by self-doping, which provides a well-matched degree of ionic radius. For instance, Sasan et al synthesized a Ti3+ self-doped rutile TiO2 (Ti3+–TiO2) via a hydrothermal process, which exhibited an apparent red shift based on the UV–Vis absorption spectra (figure 12(a)), resulting in a remarkable enhanced photocatalytic performance for CO2RR to CH4 with respect to commercial TiO2 (P25) [104]. In addition, heteroatom doping with a similar radius can also provide desired defects and electronic effects without noticeably altering the intrinsic crystal structure. Olowoyo et al introduced Mg into TiO2 via a sonothermal method because of the analogical radius between Mg and Ti, which can promote the CO2 chemisorption [105]. In particular, the surface Mg atoms preferred to locate on the (101), junctions of (101)/(101) and (001)/(101) facets, showing a synergetic effect between Mg reaction sites and the photo-generated electrons on (101) facets, thereby enhancing the corresponding photocatalytic CO2RR activities. Compared to TiO2, BiOX (X = Cl, Br, I) semiconductors show superior visible light response. Within this regard, Wu et al engineered the band structure of BiOBr microspheres composed of nanosheets via Gd3+ doping through a simple hydrothermal way [106]. The incorporation of Gd3+ was beneficial for both broadening visible light response and optimizing bandgap structure for photocatalytic CO2RR. As a result, the optimized BiOBr/Gd-0.05 sample gave a high CH3OH yield of 123.711 μmol g−1 in 3 h irradiation, which was nearly 5.2 times higher than that of pristine BiOBr (figure 12(b)).
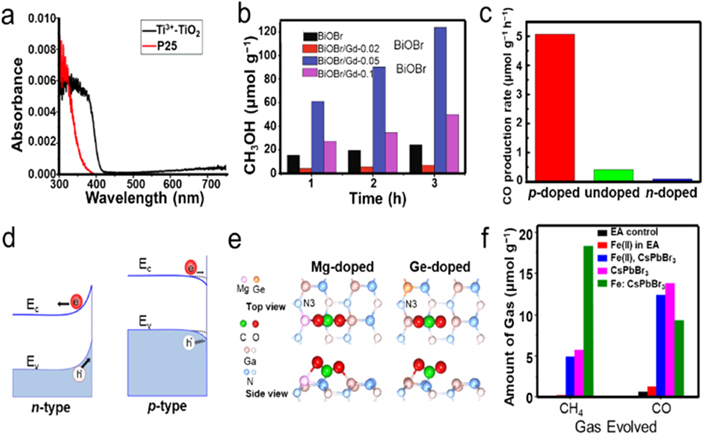
Figure 12. Metal-free dopants of photocatalysts. Metal atom doping samples. (a) UV–Vis diffuse reflectance spectra for commercial P25 (red) and Ti3+–TiO2 (black). Reproduced from [104] with permission of The Royal Society of Chemistry. (b) Photocatalytic CO2RR over various samples. Reprinted from [106], Copyright (2020), with permission from Elsevier. (c) CO evolution rates on Pt-decorated Mg-doped, undoped, and Ge-doped InGaN/GaN. (d) Schematic illustration of charge transfer on n- and p-doped semiconductors with different band bending. (e) The most stable structures of CO2 adsorbed on Mg- and Ge-doped GaN m-plane surfaces. Reprinted with permission from [108]. Copyright (2016) American Chemical Society. (f) Relative ratios of CH4 and CO evolved using Fe(II) acetate to CsPbBr3, CsPbBr3, and Fe(II)-doped CsPbBr3, only Fe(II) acetate without CsPbBr3 and the control reaction without any additive. Reprinted with permission from [110]. Copyright (2019) American Chemical Society.
Download figure:
Standard image High-resolution imageLowering semiconductor thickness accompanied with heteroatom doping can endow the exposure of unsaturated coordination surface atoms that act as highly catalytic sites. Liu et al synthesized 2.5 nm thickness Ni-doped ZnCo2O4 nanosheets, of which the introduced Ni dopants provided a new energy level and a higher DOS near the CB minimum, favoring a boosted separation efficiency of photo-generated carriers [107]. Particularly, after Ni doping, the chemisorption temperatures of CO2 increased from 47 °C to 69 °C, suggesting its favorable CO2 photoreduction process. Furthermore, CO desorption was preferable compared to CH4 desorption for Ni-doped ZnCo2O4, while the reverse case occurred on the pure ZnCo2O4. Accordingly, the Ni-doped ZnCo2O4 exhibited obvious improvement for the selective production of CO/CH4 during photocatalytic CO2RR.
In addition to metal oxides, metal nitride semiconductors, such as InGaN, have also emerged as promising catalysts in photocatalysis fields, of which the energy level can be tailored to harvest nearly the entire solar spectrum. Especially, the nonpolar metal nitride surfaces have also been validated to be highly reactive for photocatalytic H2 evolution, which is extremely significant during CO2 hydrogenation. Within this context, Alotaibi and coworkers demonstrated the multiband InGaN/GaN nanowire arrays grown on Si substrate via molecular beam epitaxy, which exhibited average photocatalytic CO2RR rates of ∼0.5, 0.1, and 0.25 mmol g−1 h−1 into CH3OH, CO, and CH4, respectively, and could be highly enhanced by nearly 50-folds after the incorporation of p-type Mg (figure 12(c)) [108]. The introduction of Mg could not only lower the surface potential barriers and improve the light harvesting ability, but also deform the adsorbed CO2 molecules. As shown in figure 12(d), the downward band bending created by p-type doping resulted in a hole depletion in the near-surface region, thereby promoting the photo-generated electrons transport for boosting reduction reactions. While the n-type doping could induce the upward band bending for offering an energy barrier to suppress the transfer of photo-generated electrons to semiconductor surface, thus inhibiting CO2RR. The corresponding DFT calculation results illustrated that the p-type Mg doping could further improve the CO2 activation because of the lower CO2 adsorption energy and higher CO2 deformation energy compared to the undoped sample (figure 12(e)). In contrast, the p-type Ge-doped surface cannot provide superiority to the undoped surface.
Perovskite is another one of the most efficient light-harvesting materials, which has been widely studied in photocatalytic applications but suffers from a high degree of photo-generated charge recombination. Recently, Tang et al explored the photocatalytic CO2RR of pure cesium lead halide (CsPbBr3) perovskite and Co- or Fe-doped CsPbBr3 perovskite using DFT calculations, predicting that the doped CsPbBr3 possessed a better CO2RR performance than that of CsPbBr3 benefited from the broadened absorption capability of visible light as well as facilitated photo-generated electron transport (design of doped cesium lead halide perovskite as a photo-catalytic CO2 reduction catalyst) [109]. In another study, Shyamal et al synthesized the Fe(II)-doped CsPbBr3 perovskite nanocrystals for photocatalytic CO2RR, in which the Fe(II) replaced the Pb (II) in the lattice [110]. The photocatalytic CO2RR in an ethyl acetate/water was explored using pure CsPbBr3 and Fe(II)-doped CsPbBr3. For pure CsPbBr3, the production rate of CO (4.6 μmol g−1 h−1) was clearly higher than that of CH4 (1.9 μmol g−1 h−1). On the contrary, the optimized Fe (II)-doped CsPbBr3 gave a predominant CH4 production rate (6.1 μmol g−1 h−1) over CO (3.2 μmol g−1 h−1). The favorable CH4 generation could be attributed to the more positive adsorption energy of CH4, which signified a faster desorption of active CH4 molecules on Fe (II)-doped CsPbBr3 surface (figure 12(f)).
3.4. Single-atom catalysts
Atomically dispersed metal atoms, referred to as single-atom catalysts (SACs), have been popularly studied in heterogeneous catalysis as one of the most active research frontiers [111, 112]. In comparison with conventional nanocatalysts, SACs possess potential in maximizing atom utilization efficiency, increasing reactivity and selectivity, bridging homo- and heterogeneous catalysis, and sometimes enhancing stability relative to their nanoparticle counterparts. Since the terminology of SACs was introduced in 2011, various SACs associated with metal atoms anchored at defective sites of reducible oxide supports have been developed for a variety of heterogeneous reactions including photocatalytic CO2RR [113–116]. For instance, Xiong et al developed a simple protocol to synthesize Ni SACs supported on an OV-rich ZrO2 substrate (Ni-SA/ZrO2) using a solvothermal process followed by calcining and subsequent selective etching (figure 13(a)) [117]. The HAADF-STEM image confirmed the isolated Ni atoms. And the EXAFS results suggested that the Ni atoms were coordinated with both O and Zr. The Ni in Ni-SA/ZrO2 was positively charged with close to +2 valence state due to the depletion of Ni free electrons toward neighboring O and Zr atoms. Benefiting from accessible Ni SACs immobilized by the surface defects, the optimized photocatalyst presented a high rate of 11.8 μmol g−1 h−1 with a selectivity of 92.5% toward CO2 conversion to CO, which was six- and 40-folds higher than the activities of defective ZrO2 and pristine ZrO2 (figure 13(b)). In situ diffuse reflection infrared Fourier transform (DRIFT) spectroscopy was also performed to reveal the reaction intermediates over Ni-SA-5/ZrO2 surface during photocatalytic CO2RR (figure 13(c)), which showed the peaks of monodentate bicarbonates (m-HCO3 −) and bidentate bicarbonates (b-HCO3 −), indicating a successive CO2RR with proton assistance. It should be noted that the surface COOH was clearly observed at 1615 cm−1, which has been regarded as an essential intermediate towards CO generation. Furthermore, Ni-SA-5/ZrO2 delivered the lowest CO desorption temperature, suggesting the most facile release of CO over the catalyst. Accordingly, DFT calculations revealed that the Ni SACs could dramatically lower the required energy barriers for CO2 conversion to CO via an adsorbed •COOH intermediate, which subsequently promoted •CO formation after further electron injection. For the competitive H2 evolution, the desorption of H2 was remarkably suppressed because of the highly endothermic process, indicating an extremely high CO/H2 production ratio during photocatalytic CO2RR on Ni-SA/ZrO2. In another study, Shi et al demonstrated that Co SACs immobilized on 2D tellurium nanosheets can serve as a remarkably active photocatalyst for enhancing CO2RR with a CO formation rate of 52.3 μmol h−1, which could be attributed to the introduced intermediate energy states derived from the strong mutual effects between Te nanosheets and Co SACs [118]. The generation of Co–Te bonding provided a higher charge density than the pristine Te nanosheets, resulting in the efficient charge transfer from Te nanosheets to Co SACs and accommodating the photo-generated electrons for promoting the separation and transfer of photo-generated charge carriers.
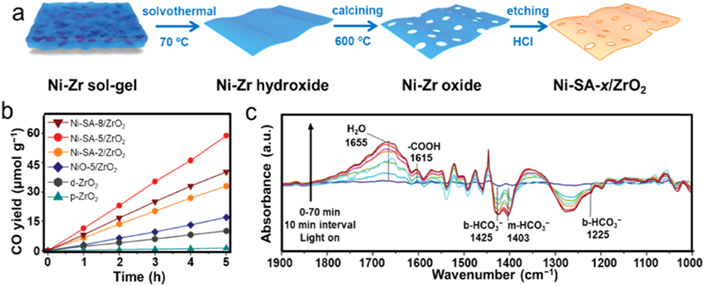
Figure 13. Ni SACs supported on ZrO2. (a) Schematic illustration of the synthesis of Ni-SA/ZrO2. (b) CO yields as a function of reaction time for various samples. Reaction conditions: 0.08 MPa humidified CO2, full-spectrum irradiation, and 10 mg of catalyst. (c) In situ DRIFT investigation for photocatalytic CO2RR over Ni-SA-5/ZrO2. The DRIFT spectra were collected at 10 min intervals after switching on the Xe lamp. [117] John Wiley & Sons. Copyright 2020, Wiley-VCH Verlag.
Download figure:
Standard image High-resolution imageThe presence of strong metal–support interaction between metal oxides and metal single atoms always brings about large net electron transfer from SACs to supports. This is detrimental to CO2 activation because of the suppressed π-back process for the d-orbital electrons of metal atoms to π* antibonding orbital of CO2, resulting in partially oxidized SACs with diminished activity. In contrast, the metal-free semiconductor support, such as PCN, could not only directly offer the required hydrogen source for CO2 hydrogenation, but also retain the neutral state of noble metal atoms. Using DFT calculations, Gao et al investigated single Pd and Pt atoms supported on PCN (Pd/PCN, Pt/PCN), respectively, showing that the obvious charge transfer between metal atoms and neighboring pyridinic N atoms occurred for both Pd/PCN and Pt/PCN [119]. It can be seen that the charge depletion of d-orbitals was dominated for Pd/PCN, while both charge accumulation and depletion emerged around the Pt atom of Pt/PCN (figure 14(a)). The oxidation state of Pd and Pt on PCN were +0.40 and +0.27, respectively, which were lower than the values of Pd and Pt supported on Fe2O3, thereby promoting the activation of Lewis acid CO2. In another study, using nitrogen atoms as coordinate sites, Huang et al obtained a single Co2+ on PCN photocatalyst via a simple deposition way, which presented a remarkable activity and selectivity for CO2 conversion to CO [120]. They also investigated the effect of Co loading on the activity of photocatalytic CO2RR, showing that significant CO production could be observed even with a small Co loading amount of 0.01 μmol mg−1.
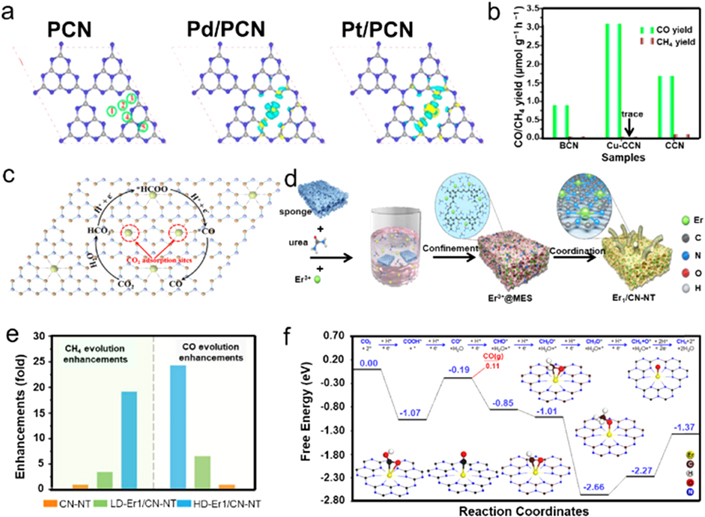
Figure 14. SACs modified metal-free photocatalysts. (a) Optimized structure of pure PCN (left), Pd/PCN (middle), and Pt/PCN (right). Positions: (1) the center of the sixfold cavity; (2) corner of the sixfold cavity; (3) top of the five-membered ring; (4) edge of the sixfold cavity; (5) top of PCN; yellow, charge accumulation; blue, charge depletion. Reprinted with permission from [119]. Copyright (2016) American Chemical Society. (b) Photocatalytic activity of CO2 reduction of Cu–CCN, CCN, and BCN samples. (c) Schematic diagram of proposed photocatalytic mechanisms and reduction pathway of Cu–CCN samples for photocatalytic CO2 reduction. Reprinted with permission from [33]. Copyright (2020) American Chemical Society. (d) Illustration of the synthesis process of single-atom Er1/CN-NT catalysts. (e) Improvement of product evolution over the LD-Er1/CN-NT, HD-Er1/CN-NT, and CN-NT photocatalysts. (f) Calculated free-energy diagram for CO2 reduction to CO and CH4 on the Er1/CN-NT, and the adsorption configuration of key intermediates. The Er, C, H, O, and N atoms are marked in yellow, brown, white, red, and blue, respectively. [121] John Wiley & Sons. Copyright 2020, Wiley-VCH Verlag.
Download figure:
Standard image High-resolution imageAlthough the inferior crystalline PCN prepared by traditional thermal polymerization has been widely investigated as a support to anchor SACs, the improvement of photocatalytic activity of a SAC supported on PCN is seriously restrained due to the excessive defects of low crystalline PCN. Therefore, increasing crystallinity associated with the introduction of SACs exhibits huge potential to improve the capacity of CO2 conversion to CO. To this end, Li et al reported Cu SACs supported on crystalline PCN nanorod (Cu–CCN) over molten salts combined with the reflux method [33]. The interplanar spacing of 0.33 nm was ascribed to PCN, suggesting enhanced crystallization. Further introduced single Cu atoms could serve as the CO2 adsorption sites, which was beneficial for photocatalytic CO2RR. The corresponding photocatalytic results showed that the Cu–CCN gave an improved CO2RR rate of 3.086 μmol g−1 h−1 with nearly 100% selectivity for CO2 conversion to CO (figure 14(b)), much exceeding the crystalline PCN and bulk sample (BCN). In situ Fourier transform infrared spectroscopy was applied and revealed that the content of HCO3 − intermediate reduced rapidly while HCOO− improved under light irradiation, illustrating that the adsorbed HCO3 − on Cu–CCN surface was quickly consumed with the formation of HCOO−. DFT calculation further revealed that HCO3 − was formed after CO2 adsorption over Cu–CCN with HCOO− as the main intermediates for photocatalytic CO2RR, which was finally converted into CO. During the process, the conversion from •HCOO to •CO was thermodynamically favorable with 1.27 eV of energy emission, whereas 0.37 eV of energy expenditure was required for •HCOO conversion to •CHO, indicating the easier pathway to the formation of CO over Cu–CCN (figure 14(c)).
Up till now, many efforts have been contributed to develop various SACs for photocatalytic CO2RR, but still suffers from low activities. Relative low loading of single atoms is one of the highly intractable problems confronted in a variety of SACs. In view of this, Ji et al proposed an atom-confinement and coordination method to synthesize Er SACs supported on carbon nitride nanotubes (Er1/CN-NT) with a tunable metal content using a sponge with porous structures and high specific surface areas, which could provide sufficient spaces and sites for anchoring Er atoms (figure 14(d)) [121]. The Er single atom contents varied from a low density of 2.5 wt% (LD-Er1/CN-NT) to a high density of 20.1 wt% (HD-Er1/CN-NT). Especially, the Er 4d spectrum in HD-Er1/CN-NT and LD-Er1/CN-NT located at 169.1 eV and 168.7 eV according to XPS results, respectively, indicating the different intrinsic activities of Er toward photocatalytic CO2RR in both samples. LD-Er1/CN-NT possessed a CH4 production rate of 0.45 μmol g−1 h−1 and a CO evolution rate of 12.75 μmol g−1 h−1 with turnover frequency of 0.0030 and 0.085, respectively, while HD-Er1/CN-NT gave a superior CH4 production rate of 2.5 μmol g−1 h−1 and CO generation rate of 47.1 μmol g−1 h−1 but with lower turnover frequency values compared with low density one. This result suggested the critical role of Er SACs in enhancing photocatalytic CO2RR (figure 14(e)). Figure 14(f) described the energy diagram of the favorable reaction pathways for CO and CH4 production from CO2RR over Er1/CN-NT, showing that −0.88 eV of energy was necessary for the transformation of –COOH to surface CO. The formation of CO had a lower theoretical limiting potential of 0.3 V than that of CH4 (0.39 V), suggesting the preferable formation of CO on Er1/CN-NT. It was also noticeable that the energy barrier for H2 evolution reaction was calculated to be 0.82 V, which was much higher than those of CO and CH4 formation, indicating the unfavorable for H2 evolution over Er1/CN-NT.
The above-mentioned attempts of photocatalytic CO2RR have mainly focused on CO and CH4 generation. The photocatalytic CO2 conversion to liquid solar fuels would be of significant importance in the pursuit of a sustainable CO2-based economy [48]. The emerging SACs possess a great potential for promoting photocatalytic CO2RR toward CH3OH with remarkable efficiency. Recently, Sharma et al introduced Ru single atoms into mesoporous polymeric carbon nitride (RuSA–mPCN) framework via an impregnation process followed by microwave heating, of which mPCN was synthesized by a hard template method (figure 15(a)) [122]. The RuSA–mPCN exhibited a similar XANES spectrum with RuO2, indicating that Ru4+ existed in RuSA–mPCN. However, RuSA–mPCN delivered a lower coordination number compared with RuO2 because of its only first shell Ru–N/C scattering in EXAFS spectrum (figure 15(b)). The corresponding HAADF-STEM ascertained the Ru was atomically dispersed with most Ru atoms located at the edge of mPCN originated from the easily accessible terminal –NH2 group. The optical feature of RuSA–mPCN was also investigated via UV–Vis absorption spectroscopy, which delivered obviously improved visible light absorption compared with PCN, indicating a reduced bandgap after RuSA incorporation (figure 15(c)). Generally, the introduction of foreign atoms is an efficient route to accommodate the lifetime of photo-generated electrons and holes, which can be reflected by PL spectra. RuSA–mPCN exhibited a less intense PL peak centered at 440 nm than that of PCN, signifying the suppression of the recombination rate of photo-generated electrons and holes. It is well known that the above advantages significantly contribute to improving the catalytic activities of photocatalysts. As a result, the photocatalytic CO2RR toward CH3OH of RuSA–mPCN achieved a high yield of 1500 μmol g−1 within 6 h using H2O as an electron donor.
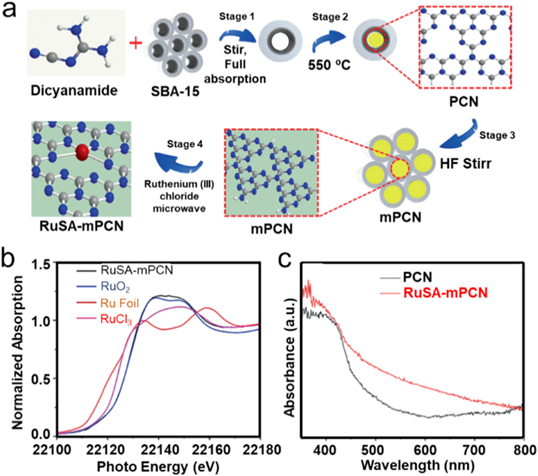
Figure 15. Ru SACs supported on mPCN. (a) Schematic illustration of the preparation process of RuSA–mPCN. (b) Ru K edge XANES of samples. (c) UV–Vis absorption spectra. [122] John Wiley & Sons. Copyright 2021, Wiley-VCH Verlag.
Download figure:
Standard image High-resolution image3.5. Strain defects
Up to now, abundant efforts have been contributed to engineering the atomic structure of catalysts to optimize the photocatalytic behavior, such as aforementioned heteroatom doping and vacancy creation. To further improve the photocatalytic CO2RR capabilities, strain engineering emerges as a potential alternative based on theory analysis, which can break linear scaling relationships for accelerating catalytic CO2RR [123]. Profiting from the deviation from the equilibrium space between catalyst atoms caused by strain, the binding energy of adsorbate exhibits a change in increase or decrease. In this respect, Di et al engineered surface atomic tensile strain via curving 2D Bi12O17Br2 materials into nanotubes to promote CO2 adsorption and activation, which could result in the stretched interatomic distance along with the curved orientation [124]. The HAADF-STEM image delivered a freestanding tube-like morphology with a recognizable hollow structure (figure 16(a)). Compared with sheet materials, the curved Bi12O17Br2 nanotubes remained the same Bi lattice spacing of 0.274 nm along the perpendicular to the tube wall, while the lattice distance of Bi atoms increased to 0.289 nm along the paralleled to the tube wall, confirming an anisotropic lattice tensile strain of the Bi12O17Br2 nanotubes, which could drive the significant variation of electronic structure. The UV–Vis diffuse reflectance spectra illustrated that Bi12O17Br2 nanotubes possessed a blue shift of adsorption edge relative to the nanoplate sample due to the quantum confinement effect. It was worth noting that in the range of 550–800 nm Bi12O17Br2 nanotubes presented higher adsorption capability, which could be attributed to the introduced vacancies by tensile strain. According to the ultraviolet photoemission spectroscopy (UPS) analysis (figure 16(b)), the VB positions of Bi12O17Br2 nanotubes and nanoplates were determined to be 1.72 eV and 2.1 eV (vs NHE), respectively. Combined with the values of bandgaps, the CB positions of Bi12O17Br2 nanotubes and nanoplates were calculated to be −0.97 eV and −0.24 eV (vs NHE), respectively. Finally, photocatalytic CO2RR tests were conducted and showed that CO was the main product accompanied by a trace amount of CH4 without any cocatalysts and sacrificial agents. By prolonging the irradiation time, the CO products could increase around 566.4 μmol g−1 in 20 h for the nanotube sample, which was almost 14.4 folds than that of nanoplates (figure 16(c)). The superior photocatalytic behavior could be ascribed to the enhanced specific surface areas, higher CO2 adsorption capability and better hydrophilicity of Bi12O17Br2 nanotubes, and the promoted photo-generated charge separation and transfer during the CO2RR process. In addition, the DFT calculations also verified that CO2 was inclined to be activated over tensile strain surfaces relative to perfect ones, suggesting the Bi12O17Br2 nanotubes with strain vacancies could efficiently accelerate the photocatalytic CO2RR process (figure 16(d)).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 16. Strain engineering of Bi12O17Br2 nanotubes. (a) HAADF-STEM image of Bi12O17Br2 nanotubes. (b) UPS spectrum of Bi12O17Br2 nanotubes. (c) Photocatalytic CO2RR toward CO over Bi12O17Br2 nanotubes. (d) Schematic illustration of the reaction mechanism over Bi12O17Br2 nanotubes for CO2 photoreduction. Reprinted with permission from [124]. Copyright (2020) American Chemical Society.
Download figure:
Standard image High-resolution image{kind=link}
4. Conclusions
Atom manufacturing of photocatalysts can remarkably boost the photocatalytic activity of CO2RR. In this review, we have primarily highlighted the fundamental understanding of photocatalytic CO2RR. Subsequently, the roles of atom manufacturing on heterogeneous photocatalysts have also been summarized, not only including provided active sites, and enhanced light absorption capability, but also gathering the atom manufacturing effects during the photocatalytic process. In addition, various types of atom manufacturing, mainly including heteroatom doping, SACs, vacancies engineering, and strain defects are summarized, along with their effects in improving the performance toward photocatalytic CO2RR. In principle, atom manufacturing is favorable for regulating the electronic structures of active sites, which tunes the adsorption and activation behaviors of CO2 and H2O, and then the CO2 conversion efficiency and product selectivity are altered. Furthermore, the varying desorption and transfer manners of intermediates caused by atom manufacturing also serve as a pivotal role in determining the overall photocatalytic CO2RR activity.
Despite the substantial developments of atom engineering to improve photocatalytic CO2 conversion performance, there are still several challenges and barriers limiting the design, fabrication, and applications of the modified photocatalysts, which should be carefully addressed. Firstly, it is still urgent to develop highly efficient preparation approaches for controllable atom manufacturing in materials on the large scale, even many methods have already been employed. On one hand, the current preparation strategies, such as chemical reduction and plasma treatments, still suffer from inaccurate atom engineering at specific sites with tunable concentrations. On the other hand, the mass production of photocatalysts with specific engineered atoms is highly desired.
Secondly, the insightful understanding of the relationship between atom manufacturing and photocatalytic performance is still in the preliminary stage, thereby further endeavors are required to investigate the corresponding structure–activity relationship. In view of the CO2RR that occurred on the surface of a photocatalyst, the surface or interface engineered atoms instead of bulk atoms are reasonable to be studied for unveiling their effects. Unfortunately, the presence of multiple types of engineered atoms often seriously complicates the clarification of defect effects in terms of concentrations and types. As the electronic structures induced by atom manufacturing play a significant role in determining the adsorption and desorption behaviors of CO2, H2O and reaction intermediates, it should be pointed out that precise theoretical simulations in real reaction conditions are highly recommended for a better understanding of the CO2 photoreduction pathway. In addition, integrating experiments and computation models is essential in both disclosing the structure–performance correlations and helping photocatalyst construction.
Thirdly, the engineered atoms always exhibit a dynamic variation over time under operation conditions, which cannot commendably be reflected using ex situ characterization techniques. In this context, in situ and operando techniques are extremely needed to reveal the situation of atom manufacturing of photocatalysts under reaction conditions. These in situ techniques, such as in situ STEM and XAS, are highly encouraged to study the real-time variation of catalysts in terms of morphological and electronic structures for better understanding the mechanism of CO2 photoreduction. Further, in situ XPS, in situ surface-enhanced IR absorption spectroscopy, as well as Raman, are useful for revealing the related intermediates in surface engineered atoms, allowing for catalyst design with superior performance and stability. Notably, the current characterization techniques can only give limited information of atom manufacturing, which is an existing problem in identifying the structure–activity relationship (table 1).
Table 1. Photocatalytic CO2RR systems with defective semiconductors.
| Photocatalysts | Types | Reaction medium | Major product | Selectivity (%) | Yield | Ref. |
|---|---|---|---|---|---|---|
| VO–BiOBr atomic layers | VO | CO2-bubbled solution | CO | — | 87.4 μmol g−1 h−1 | [82] |
| ZnAl-1 | VO | CO2-bubbled solution | CO | — | 7.6 μmol g−1 h−1 | [84] |
| BiOCl–B–OV | VO/doping | CO2-purged solution | CO | 98 | 83.64 μmol g−1 h−1 | [85] |
| VO–PO4–BWO | VO/doping | CO2-filled solution | CH3OH | — | 157 μmol g−1 h−1 | [86] |
| VO-rich-Bi2O3 | VO | CO2-filled CH3CN with CH3OH presence | Dimethyl carbonate | ∼100 | Coversion yield of ∼18% | [68] |
| PCN-525 | VN | CO2-filled solution | CO | — | 6.21 μmol h−1 | [45] |
| 3% KBH–PCN | VN/doping | CO2-purged solution | CH4 | — | 5.93 μmol g−1 | [87] |
| MP-500-4 | VC | CO2-filled CH3CN | CO | — | 21.5 μmol h−1 | [88] |
| CTS-1 | VS | CO2-pumped solution | CH4 | 83.1 | 22.65 μmol g−1 h−1 | [89] |
| VS–SAL10 | VS | CO2/H2O vapor | C2H4 | ∼73 | 44.3 μmol g−1 | [90] |
| VBi–BiOBr UNs | VBi | CO2-purged H2O | CO | 98 | 20.1 μmol g−1 h−1 | [91] |
| VZn-rich one-unit-cell ZnIn2S4 layers | VZn | CO2-filled H2O | CO | — | 33.2 μmol g−1 h−1 | [92] |
| VZn–ZnS | VZn | KHCO3 + K2SO3 | HCOOH | 86.6 | ∼7 μmol h−1 | [93] |
| VV-rich o-BiVO4 | VV | CO2-bubbled H2O | CH3OH | — | 398.3 μmol g−1 h−1 | [94] |
| 4/1-In(OH)3 | VIn | CO2/H2O vapor | CH4 | — | 28.2 μmol g−1 h−1 | [95] |
| BiOCl-S | VO/VCl | H2SO4 + NaHCO3 | CO | 85 | 3.94 μmol g−1 h−1 | [96] |
| VNs–PCN/VOs–TiO2 | VN/VO | CO2-filled H2O | CO | — | 13.4 μmol g−1 | [97] |
| 'Bi–O'–Bi2MoO6 | VBi/VO | CO2-filled H2O | CO | — | 3.62 μmol g−1 h−1 | [98] |
| m-NiAl-LDH | VOH/VM | CO2-filled CH3CN + H2O + TEOA | CH4 | 70.3 | 77.2 μmol g−1 h−1 | [99] |
| B-STO-20 | B-doping | CO2-filled H2O | CO | — | 21 μmol g−1 h−1 | [100] |
| P-PCN | P-doping | NaHCO3 | CO | — | 9.48 μmol g−1 | [103] |
| Mg–TiO2-2 | Mg-doping | CO2-purged H2O + CH3CN | CH3OH | — | 5910.0 μmol g−1 h−1 | [105] |
| BiOBr/Gd-0.05 | Gd-doping | CO2-filled H2O | CH3OH | — | 123.711 μmol g−1 | [106] |
| Ni-doped ZnCo2O4 | Ni-doping | CO2-purged H2O | CO | — | 31.4 μmol g−1 h−1 | [107] |
| Mg-doped InGaN/GaN | Mg-doping | CO2-purged H2O | CH3OH | — | ∼25 μmol g−1 h−1 | [108] |
| Fe (II)-doped CsPbBr3 | Fe-doping | CO2/H2O vapor | CH4 | — | 6.1 μmol g−1 h−1 | [110] |
| Ni-SA-5/ZrO2 | Ni SACs | CO2/H2O vapor | CO | 92.5 | 11.8 μmol g−1 h−1 | [117] |
| Co@Te nanosheets | Co SACs | CO2-filled CH3CN + H2O + TEOA | CO | — | 52.3 μmol h−1 | [118] |
| Cu–CCN | Cu SACs | CO2-filled H2O | CO | ∼100 | 3.086 μmol g−1 h−1 | [33] |
| HD-Er1/CN-NT | Er SACs | CO2-purged H2O | CO | — | 47.1 μmol g−1 h−1 | [121] |
| RuSA–mPCN | Ru SACs | CO2-purged DMF + H2O | CH3OH | — | 1500 μmol g−1 (24 h) | [122] |
| Bi12O17Br2 nanotubes | Strain | CO2-purged H2O | CO | 34.5 μmol g−1 h−1 | [124] |
Fourthly, the construction of effective photocatalytic CO2 conversion systems without any sacrificial agents is indispensable for practical applications of photocatalytic CO2RR, in which atom manufacturing serves as one of the key ingredients for accelerating the practical process. During the photocatalytic CO2RR process, limiting the hydrogen evolution should be taken into account to achieve the ideal CO2RR selectivity toward specific product formation. In this regard, the proton density can retain at a balanced level in the local regions of active sites with a lower diffusion rate to the catalyst surface compared to the CO2RR rate. In addition, considering the very low concentration of CO2 in the atmosphere, more endeavors should be widely contributed to explore photocatalytic CO2RR at low CO2 concentrations. As pointed out above, the atom manufacturing features as a double-edged sword in enhancing photocatalytic CO2 conversion efficiency, which can also serve as the recombination centers of photo-generated electrons and holes, thereby suppressing the corresponding photocatalytic activity. In this context, more efforts should be contributed to develop experimental techniques associated with theoretical efforts to reveal related parameters in the optimization of catalytic systems.
Fifthly, although atom manufacturing can significantly improve CO2RR performance via precisely modifying the local coordination environments of solid photocatalysts, the isolate active sites failed to stimulate interactions between intermediates of CO2RR because of the long interatomic distance, resulting that the products are mainly fasten on C1 species. Given that photocatalytic CO2RR to CH4 is a less energetically demanding pathway, the formation of ambitious long-chain hydrocarbons or liquid fuels is a preferably clean and sustainable path for CO2 reutilization in alleviating the depletion of fossil fuels. To date, researches on CO2 photoconversion to selective formation of both long-chain hydrocarbons and liquid fuels using atom manufacturing photocatalysts are still at extremely initial stages. Accordingly, releasing synergistic effects between active sites via lowering the interatomic distance play a vital role in enabling C–C coupling probability to generate C2+ products. In view of this, precise engineering stable atom dimers, dual vacancies or several sites may be intriguing strategies to tolerate the formation of long-chain hydrocarbons.
Overall, atom manufacturing is a promising method to construct high-efficiency photocatalysts toward CO2RR. It is unquestionable that the collaborative developments of atom manufacturing strategies and their characterizations can significantly contribute to the design of efficient, stable, and scalable photocatalytic CO2RR systems, which still have a long way to achieve practical applications. Moreover, the mechanistic understanding of the roles of engineered atoms in CO2 photoreduction is still at an initial stage, and more endeavors toward studying relative mechanisms should be undertaken to allow the boosted capabilities of novel photocatalysts. It is anticipated that the breakthrough in atom manufacturing of photocatalysts will emerge in the near future, and all influence factors will be integrated into account and investigated to extremely enhance the corresponding photocatalytic performance toward CO2 conversion at that time.
Acknowledgments
This work was supported by the National Key Projects for Fundamental Research and Development of China (2018YFB1502002), the National Natural Science Foundation of China (21902104, 51825205, 21902168, 22172077), the Natural Science Foundation of Jiangsu Province of China (BK20211573), and the Fundamental Research Funds for the Central Universities (30919012107).
Data availability statement
No new data were created or analyzed in this study.
Biographies
Kan Zhang obtained his Ph.D. degree in SKKU Advanced Institute of Nano-technology (SAINT) from Sungkyunkwan University, Korea (2015). Then, he was a post-doc researcher in Department of Chemical and Molecular Engineering (2015-2018). He is currently professor working at School of Materials Science and Engineering in Nanjing University of Science and Technology. His research interests involve photoelectrocatalytic synthesis of high value-added chemicals and photoelectrocatalytic system design towards energy conversion and environmental treatment.
Tierui Zhang is a full professor at Technical Institute of Physics and Chemistry, Chinese Academy of Sciences. He obtained his Ph.D. degree in Chemistry in 2003 at Jilin University, China. He then worked as a postdoctoral researcher in the labs of Prof. Markus Antonietti, Prof. Charl F. J. Faul, Prof. Hicham Fenniri, Prof. Z. Ryan Tian, Prof. Yadong Yin, and Prof. Yushan Yan. His current scientific interests focus on catalysts and nanomaterials for energy conversion.