
Abstract
The transition from a committed progenitor cell to one that is actively differentiating represents a process that is fundamentally important in skeletal myogenesis. Although the expression and functional activation of myogenic regulatory transcription factors (MRFs) are well known to govern lineage commitment and differentiation, exactly how the first steps in differentiation are suppressed in a proliferating myoblast is much less clear. We used cultured mammalian myoblasts and an RNA interference library targeting 571 kinases to identify those that may repress muscle differentiation in proliferating myoblasts in the presence or absence of a sensitizing agent directed toward CDK4/6, a kinase previously established to impede muscle gene expression. We identified 55 kinases whose knockdown promoted myoblast differentiation, either independently or in conjunction with the sensitizer. A number of the hit kinases could be connected to known MRFs, directly or through one interaction node. Focusing on one hit, Mtor, we validated its role to impede differentiation in proliferating myoblasts and carried out mechanistic studies to show that it acts, in part, by a rapamycin-sensitive complex that involves Raptor. Our findings inform our understanding of kinases that can block the transition from lineage commitment to a differentiating state in myoblasts and offer a useful resource for others studying myogenic differentiation.
Similar content being viewed by others


Introduction
The transition between a proliferating, committed progenitor cell and one that is actively differentiating represents a fundamental step in cellular and, ultimately, organism development. Skeletal muscle formation represents a particularly important example as a process choreographed by a limited number of transcription factors determining muscle lineage and driving the differentiation program1,2. Elegant mouse studies show that the lineage is determined by expression of MyoD or Myf5, two members in a family of basic helix-loop-helix (bHLH) transcription factors3,4. Certain extracellular signals guide early stages of lineage commitment: surface ectoderm-derived Wnts and Shh from the floor plate and notochord drive the initial expression of Myf5 and MyoD in the dermomyotome, whereas Notch- and BMP-derived signals repress their expression5,6,7,8. However, the mere expression of myogenic bHLH proteins is not sufficient to start the differentiation cascade in committed myoblasts. Hence, a major question remains: How is the differentiation program suppressed in proliferating myoblasts?
The function of myogenic bHLH proteins, which form heterodimers with E2a gene products to bind to “E-boxes” in regulatory elements of muscle-specific genes1, hinges largely on physical and functional interactions with a variety of cofactors including other sequence-specific transcriptional activators like Mef2 proteins9,10; cell cycle proteins including the retinoblastoma tumor suppressor, Rb11,12; and histone and chromatin remodeling proteins p300 and PCAF13,14. In many cases, these interactions are controlled by kinases. For example, consistent with the fact that functional Rb is critical for myogenic bHLH protein activity11,15, excess Cyclin D1-associated Cyclin-dependent kinase (Cdk) 4 or 6 can impede muscle differentiation16 and expression of a Cdk inhibitor16 or chemical inhibition of Cdk4/6 activity17 enhances muscle gene expression. Kinases also positively influence the activity of MyoD- and Mef2-related proteins. Activation of p38 Mapk provides the clearest example, as its inhibition or forced activation in cultured myoblasts impedes or augments the expression of differentiation markers, respectively18,19.
Regulation of the myoblast-to-myocyte transition holds substantial importance for muscle development and disease. Major muscle groups representing 30–40% of the adult human body mass are defined during embryonic and perinatal development, yet adult skeletal muscle retains a capacity to increase muscle mass by hyperplastic and hypertrophic processes6,20. Similarly, muscle mass diminishes in physiological aging and a variety of diseases and this is linked to loss of myonuclei and decreased synthesis of skeletal muscle proteins6,21,22. In another example, rhabdomyosarcoma represents a malignant soft tissue sarcoma composed of skeletal myoblast-like cells2,23. Although rhabdomyosarcoma expresses lineage-defining muscle proteins, the cells have lost the capacity to undergo terminal muscle differentiation24. This disease, in particular, may be viewed as an extreme example of derailed skeletal muscle differentiation.
To better understand the breadth of kinases that can block the differentiation program in proliferating myoblasts, we leveraged a well-characterized mammalian cell culture model in a high-content, cell-based screen. In contrast to other myogenic screens focusing on enhancers of muscle differentiation25,26 or muscle lineage specification27, we searched for kinases that block muscle gene expression in proliferating myoblasts. In this report, we describe our high-content screen and initial findings, bioinformatics analyses highlighting potential interactions between hit kinases and MRFs and functional validation and mechanistic studies of mTOR, one of the hits.
Results
High content, cell-based screen to detect Myogenin induction in myoblasts
We designed a high throughput screen to uncover kinases that block muscle gene expression in committed, but undifferentiated, myoblasts. Specifically, we leveraged an siRNA library targeting much of the mouse kinome to test how knockdown of individual kinases influenced the expression of Myogenin, a myogenic bHLH protein induced by MyoD binding to the Myog promoter28. We utilized the well characterized, mouse C2C12 myoblast model, which has been used for many years to illuminate fundamental steps in myogenesis. “Hits” represented those kinases for which knockdown increased the fraction of cells expressing Myogenin, even though the cells were maintained in mitogen-rich growth medium (GM) (Fig. 1A). Myogenin expression was assessed by fluorescence staining and quantified using automated fluorescence microscopy in cells cultivated in GM versus serum-poor differentiation medium with insulin (DM + I)(Fig. 1B). We established a robust assay window based on both Myogenin expression and decreased cell number due to the cell cycle arrest that normally accompanies differentiation (Fig. 1C,D). Recognizing the capacity for Cyclin D-Cdk4/6 to promote proliferation and blunt differentiation, we included an experimental arm with cells sensitized using PD 0332991 (Fig. 1A). This Cdk4/6 inhibitor blocks proliferation in these myoblasts and slightly increases Myogenin17; however, that small increase was not detectable in this screen (see below).

High content siRNA screen for kinases negatively regulating myogenic differentiation.
(A) Schematic diagram showing morphological changes and important molecular events during skeletal myogenesis. (B) Immunofluorescence staining for Myogenin and DAPI in C2C12 cells grown in GM or DM + I (top) and digitized versions of images (bottom), used by MetaXpress software to count the nuclei. (C,D) Quantification of Myogenin-positive cells (C) producing a Z-factor of 0.527 and total cell number (D) in aggregate wells from 384-well plate from negative (GM) and positive (DM + I) controls. *p < 0.05 in GM vs. DM + I; error bars represent standard deviation.
In the screen, an individual well scored positively if the knockdown both a) significantly increased the average fraction of Myogenin-positive cells in the four sites automatically sampled by the immunofluorescence microscope and b) decreased the average number of cells in the triplicate wells to a value that is within one median absolute deviation (MAD) of the cell number for the DM control. In both cases, comparison was made to cells cultivated in GM and transfected with control siRNA. For cells receiving PD 0332991 as a sensitizer, total cell number was not included as a variable because this drug decreases myoblast cell number independently of siRNA knockdown17. “Hits” were called if two of the replicates (or all three when the sensitizer was included) were positive. To test these definitions, we applied them to the 64 negative and positive control wells containing myoblasts cultivated in GM or DM + I, respectively. None of the negative control wells scored positively whereas 89% and 98% of the positive control wells scored as such with either vehicle or PD 0332991, respectively (Fig. 2A).

High content siRNA screen identifies a subset of kinases that block myogenin induction in myoblasts.
(A) Chart showing experimental conditions and controls used in the screen and quantitation of hits under different conditions. (B,C) Charts showing total number of cells per well (B) or percent of cells expressing Myogenin (C) when cultivated in the indicated medium with or without PD 0332991 (PD) or or with siRNAs targeting individual kinases. (D) Chart showing average total cell number in experimental wells targeting each kinase. Gray and black bars signifying kinases scoring as negative or positive, respectively, for Myogenin induction. (E) Venn diagram showing the positively scoring “hits” with and without PD. PD, PD 0332991; GM, growth media; DM + I, differentiation media plus insulin; *p < 0.05; error bars represent standard deviation.
Skeletal muscle differentiation is intimately coupled to arrest in G0/G1 phase of the cell cycle2. We discovered, though, that decreased total cell number was not sufficient for an individual kinase to score positively. As indicated above, none of the wells scored positively when cells were in GM plus PD 0332991, even though cell number significantly decreased (Fig. 2B,C). Our panel included well-characterized cell cycle regulators like Cdk1, Aurkb and Plk1, the knockdown of which decreased cell number without increasing Myogenin (Fig. 2B,C). Indeed, focusing on the non-sensitized screen, the total cell number decreased by at least one median absolute deviation from the GM control in 138 wells, but only a small number of these displayed significantly increased Myogenin (Fig. 2D). Hence, merely arresting cell proliferation was not sufficient to initiate the muscle differentiation program in MyoD-expressing myoblasts.
Kinases identified as negative regulators of muscle gene expression in proliferating myoblasts
The siRNA screen included triplicate plates containing 571 experimental wells, each of which targeted a single mouse kinase. We observed that knockdown of 19 of those kinases increased the Myogenin-expressing fraction and decreased the total cell number, providing a hit-rate of 3.3% for siRNA alone and 7.4% for siRNA plus the Cdk4/6 inhibitor (Fig. 2A and Supplementary Figure S1). The magnitude of the effect ranged from a Z-score of 0.9 (Fyn) to 3.5 (Nek2) without the Cdk4/6 inhibitor and from 1.5 (Brsk2) to 4.8 (Mapkapk3) with the sensitizer (Supplementary Table S1). Six of the 19 hits without the sensitizer also scored positively when the sensitizer was included (Fig. 2E). Taking into account those “double hits” (Axl, Bub1b, Nek4, Peak1, Pkn2 and Src), 55 (9.6%) of the kinases scored positively.
We studied how hit kinases were related to each other and to MyoD- and Mef2-family myogenic regulatory factors (MRFs) by exploring known human genetic interaction networks using the HPRD and BioGRID databases (Fig. 3A,B). First, we observed that only one kinase, Csnk2a2, which scored positively with PD 0332991, had a direct connection with one of the myogenic regulators, Myf5 (Fig. 3B). Second, nine of the 19 hits from the primary siRNA screen (and 28 from the sensitized screen) were just one interaction node away from one of the MRFs; we considered them “semi-direct” regulators. Four of these semi-direct regulators – Src, Axl, Bub1b and Pkn2 – were identified in both screens. Src interacted semi-directly with the most MRFs (n = 5) in both screens. Without Cdk4/6 inhibition, Ep300 was the node interacting with the greatest number of MRFs, including Mef2a, Mef2c, Mef2d and Myod, whereas Myod was the MRF that interacted semi-directly with the most regulatory kinases (n = 8). These data point toward Src, Ep300 and MyoD as particularly important regulatory nodal points in the initial induction of Myogenin.

Hit kinases intersect with individual proteins in biological pathways that are relevant to skeletal muscle differentiation.
Genetic interaction network for hits without PD (A) and with PD (B) linking MyoD- and Mef2-family MRFs (pink) to selected kinases (yellow). Note that one hit kinase, CSNK2A2 (B, arrow), shows a direct connection to MYF5, while others represent all hit kinases interacting semi-directly through one interaction node (light blue). Asterisks (A) denote the hit kinase, MRF and interacting node with the greatest number of interactions. Several of these semi-direct regulators were identified in both the sensitized screen and non-sensitized screens (arrowheads).
ToppGene29 analysis revealed a number of pathways that were enriched in the hit kinases, including mTOR, Eph/ephrins and a variety of other receptor/kinase signaling pathways (Fig. 4A, top panel). As expected, including the Cdk4/6 inhibitor as a sensitizer revealed additional enriched pathways, including pyrimidine biosynthesis and Map kinase signaling (Fig. 4A, bottom panel). Many of the pathways identified in the sensitized and non-sensitized screen and the processes identified by gene ontology analysis of hit kinases (Fig. 4B), highlight factors that directly or indirectly couple extracellular environmental cues to cytoskeletal changes and other intracellular signaling processes.

Pathway and gene ontology analysis identify processes linked with triggering skeletal muscle differentiation.
Charts showing top 15 (A) biological pathways and (B) processes enriched with and without PD 0332991 as sensitizer. Fold enrichment is calculated as percentage of hits in the indicated pathway divided by the expected percentage based on the number of tested kinases in that pathway. Source indicates specific database: B, BioCarta; G, GenMAPP; K, KEGG; P, Pathway Interaction Database; R, Reactome; W, Wiki Pathways.
We considered whether the hit kinases were differentially expressed in myoblasts versus differentiated myocytes. We employed RNA-Seq and a protein microarray to test the mRNA and protein expression and phosphorylation status of the hit kinases. We found that mRNA encoding many of the tested kinases was similar in GM and DM + I, but some decreased to varying degrees (Fig. 5A). In most of those cases, there was a corresponding decrease in the protein and activating phosphorylation sites (Fig. 5B,C). In the case of JNK3, encoded by Mapk10, neither mRNA nor protein decreased, but we found relative loss of phosphorylation of kinase-activating amino acid residues (Fig. 5C). We conclude that decreased expression or decreased phoshorylation in a subset of the hit kinases may represent a mechanism to couple decreased kinase activity with the initiation of myogenic differentiation.

Decreased mRNA and protein/phosphoprotein expression of certain hit kinases correlates with muscle differentiation.
(A) mRNA levels of a subset of the hits in growth medium (GM) versus differentiation medium (DM + I), determined by RNA-Seq. (B) Relative expression of protein (B) and specific phospho-amino acids (C) in a subset of the hits in GM versus DM + I, relative to HSC70 as a loading control, determined using KAM-880 protein microarray. Bars represent standard error for duplicate measurements. Phosphosite activity is listed as stimulatory ( + ) or inhibitory (−) of kinase activity when phosphorylated. Asterisks (*) indicate that an antibody has known or potential cross reactivity with corresponding phosphosite(s) on closely related protein(s). Note: In this figure, data are shown as relative to GM (values in GM are set as 1). The absolute expression of mRNA (A), expression of protein (B) and phospho-amino acid (C) relative to Hsc70 are shown in Supplementary Fig. S5.
Secondary validation of mTOR and other kinases in myogenesis control
Many of the hits encode kinases that are well known negative regulators of muscle gene expression, such as Src30,31 and Rock132,33. Consistent with previous studies, ectopic expression of Src repressed the activation of a plasmid reporter driven by the mouse Ckm promoter and pharmacological inhibition of Src family kinases with dasatinib augmented the expression of muscle-specific genes in myoblasts maintained in GM (Supplementary Fig. S2). Some hits encode kinases that have not been well studied in myogenesis, such as Glyctk and Bub1b. We found that knockdown of Glyctk to about 60% of baseline significantly increased the mRNA expression of Myog and other muscle genes in C2C12 cells cultured in GM (Supplementary Fig. S3), revealing a role of Glyctk in repressing muscle differentiation genes.
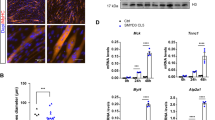
One of the hits, Mtor, caught our attention because mTOR signaling is widely viewed as a positive myogenesis regulator in cultured cells18,33,34,35,36. However, our screen identified it is a negative regulator. To confirm this role, we first tested whether transient knockdown of RNA encoding mTOR could increase Myog mRNA in cultured C2C12 myoblasts maintained in GM. Similar to the effects in the screen, Mtor knockdown increased Myog mRNA (Fig. 6A, left panels) and Myogenin protein (Fig. 6A, right panels). Mtor knockdown also increased the expression of several other muscle genes and in some cases, the magnitude of induction approximated that achieved in cells cultivated in DM + I (Fig. 6B, left panels). In addition, we demonstrated that Mtor knockdown had similar effects in early-passage, mouse skeletal muscle satellite cells (Fig. 6C).

Mtor knockdown increases the expression of Myogenin and other muscle-specific differentiation genes in myoblasts.
Charts showing mRNA or protein expression of indicated genes in (A,B) C2C12 cells and (C) primary mouse myoblasts cultivated in growth medium (GM) or differentiation medium (DM + I) with control (CTL) siRNA, Mtor knockdown, as indicated. In all cases, mRNA was quantified by qRT-PCR, normalized to Gapdh and shown relative to expression in GM. Of note, Myh7 was excluded from the primary myoblast studies (C) because it was not induced by DM + I. *p < 0.05 compared to GM; error bars represent standard deviation.
We then tested whether chemical inhibition of mTOR could drive the initial steps in myogenic differentiation. The mTOR kinase sits at the heart of pathways that integrate environmental signals with intracellular processes guiding critical cell biological processes, positioning it well to guide the myoblast-to-myocyte transition. Although mTOR acts in two well-characterized complexes, we focused on the mTORC1 complex because it is closely coupled to the regulation of cell proliferation and protein translation37,38. Pharmacological inhibition of mTORC1 using rapamycin increased muscle gene expression in myoblasts maintained in GM and also arrested myoblasts in G1 phase of the cell cycle (Fig. 7A,B). Importantly, the magnitude of muscle gene induction did not correlate with the dose-dependent cell cycle arrest by rapamycin (Fig. 7B), indicating that its pro-differentiation effects were not merely related to its capacity to arrest proliferation.

mTORC1 inhibition by rapamycin or Rptor knockdown increases muscle gene expression, independently from cell cycle arrest.
(A,B) Quantification of (A) mature mRNA expression and (B) G1 cell cycle fraction in C2C12 myoblasts cultivated in growth medium (GM), differentiation medium with insulin (DM + I), or GM with rapamycin for 48 hours at the indicated doses (nM). (C) Aggregate quantification of muscle-specific signature genes (left) or indicated genes (right) in myoblasts cultivated as above with Mtor or Rptor siRNA knockdown. In all cases, mRNA quantified by qRT-PCR, normalized to Gapdh and shown relative to expression in GM. CTL, scrambled siRNA control; *p < 0.05 compared to GM; error bars represent standard deviation.
The rapamycin findings suggested a specific role for Raptor in the mTORC1 complex. Interestingly, Rptor knockdown to approximately 60% of baseline only augmented expression of the five tested muscle genes by approximately 2.6-fold (range: 1.8 – 3.1) over baseline in GM, while parallel Mtor knockdown to ~53% increased their expression by ~18.8-fold (Fig. 7C). That the effects of rapamycin and Rptor knockdown are quantitatively smaller than Mtor knockdown suggests that Mtor knockdown likely does more to foster muscle gene expression than merely disrupt the mTORC1 complex. Further, mTORC1 is primarily thought to foster eIF-4E-dependent translation39,40. Our finding that knockdown of that translation factor was not sufficient to mimic the effects of Mtor or Rptor knockdown (Supplementary Fig. S4) suggests a role for additional mTOR effectors.
mTOR inhibition increases transcription of MyoD-driven skeletal muscle promoters
We investigated whether increased mRNA was due to increased transcription or post-transcriptional RNA stabilization. We first employed quantitative RT-PCR using primers specific for the nascent, unprocessed transcript. As was the case for the mature transcript, expression of the primary mRNA transcripts for the surveyed muscle genes increased when Mtor was knocked down (Fig. 8A). We also utilized transiently transfected plasmid reporters driven by the mouse Ckm promoter or a simplified reporter containing four, MyoD-specific, E-boxes (CANNTG) and a minimal promoter16; both of these reporters were induced in myoblasts cultivated in DM + I and Mtor knockdown also increased the expression of both the complex and simplified promoters (Fig. 8B). Taken together, these data indicate that Mtor knockdown activates transcriptional programs driven by MyoD or related bHLH MRFs in myoblasts even though they are maintained in a mitogen-rich growth medium.

mTOR inhibition increases bHLH MRF-driven transcription of muscle-specific promoters.
(A) Quantification of unprocessed RNA for four muscle-specific genes in C2C12 myoblasts cultivated in growth medium (GM) or differentiation medium (DM + I) and with control (CTL) siRNA or Mtor knockdown. mRNA was quantified by qRT-PCR, normalized to Gapdh and shown relative to expression in GM. (B) Quantitation of luciferase in C2C12 cells cultivated as above, following transfection with reporter plasmids containing the Muscle Creatine Kinase promoter (Ckm) or a minimal SV40 promoter driven by four MyoD-specific “E” boxes (4 R-SV). Average values presented relative to baseline in GM. CTL, scrambled siRNA control; *p < 0.05 compared to GM; error bars represent standard deviation.
Discussion
The molecular underpinnings of skeletal muscle lineage commitment and the consequent transcriptional induction of muscle specific genes are very well described41, but the factors that repress muscle differentiation genes in proliferating myoblasts are not as clear. Our findings provide new and largely unbiased insight into kinases that can accomplish this. As supported by our functional data, the inhibition of these individual kinases can represent a mechanism to initiate the muscle differentiation process, effectively guiding the transition between a proliferating, MyoD-expressing myoblast and a post-mitotic myocyte expressing Myogenin. Pathway and gene ontology analyses highlight the role that many of these kinases play to connect extracellular signals to intracellular events.
Many of our hits encode kinases known or suggested to play a role in skeletal muscle development. For example, the Src family kinases Src and Fyn were both identified in the non-sensitized screen. Ectopic expression of v-Src in quail and mouse myoblasts blocks the ability of myogenic bHLH proteins to induce Myogenin, whereas use of chemical inhibitors of Src or expression of a dominant negative form enhances Myogenin expression30,31. Pkn2 (also known as Prk2), identified in both sensitized and non-sensitized screens, can activate Fyn in keratinocytes42. The Rho kinase, Rock1, is known to negatively regulate later stages of myogenesis32,33 and has been implicated in signaling related to mTOR33, which we also identified as a negative regulator (see more below). Eph/ephrin signaling drives repulsive migration patterns in muscle satellite cells, but a direct role influencing muscle differentiation was not previously suspected43. Certain hit kinases also intersect with the aforementioned Wnt and Bmp signaling pathways, which positively and negatively influence muscle development, respectively5,6,7,8. For example, Bmp-dependent myogenesis suppression might be relieved by knockdown of Acvrl1, which encodes a Type I Tgfβ superfamily receptor44. Casein kinase I, encoded by Csnk1a1, can phosphorylate β-catenin and prime it for degradation in certain contexts45. Cskn1a1 knockdown in myoblasts, then, would be expected to activate the canonical Wnt pathway, fostering muscle differentiation. Additional validation and mechanistic studies are needed to confirm these possible connections and extend them to in vivo settings.
We also identified mTOR as a negative regulator of muscle gene expression in myoblasts and we confirmed that chemical inhibition of the mTORC1 complex is sufficient to foster the first steps in muscle differentiation. Positioning mTOR/mTORC1 activity as a negative regulator of the myoblast-to-myocyte transition is consistent with the role it plays to read extracellular cues from growth factors and nutrients that would be critical to a cellular “decision” to begin differentiation34. Our conclusion contrasts an elegant study from the Chen laboratory showing that Mtor knockdown decreases whereas Rptor knockdown increases muscle gene expression46. It is key to point out that their work focused on effects of knockdown in cells cultivated in differentiation medium, whereas our studies were performed in myoblasts maintained in GM. Ge and colleagues also demonstrate that Raptor governs phosphorylation of Serine-307 in IRS-146 and phosphorylation of the orthologous site on human IRS-1 of leads to its degradation47. Rapamycin-dependent blockade of this phospho-serine in proliferating myoblasts would be predicted to increase IRS-1 to potentiate muscle gene expression.
It is important to note that mTOR signaling is also recognized as a positive myogenesis regulator in cultured cells18,33,34,35,36 and in experimental animals with muscle-specific targeting of Mtor and Rptor48,49. Hence, viewing our findings with these other reports highlights the biphasic role mTOR can play in myogenesis. In proliferating myoblasts, mTOR and likely mTORC1 activity, helps to repress differentiation, whereas mTORC1 activation after differentiation facilitates the achievement of a terminally-differentiated state.
Mechanistically, mTOR/mTORC1 activity limits early muscle gene expression by blocking the capacity for myogenic bHLH proteins to augment the transcription of muscle specific promoters driven by reiterated MyoD-specific E-boxes. This is not likely to be a direct effect on MRFs because we were not able to identify the mTOR phosphorylation motif38 in human and mouse myogenic bHLH and Mef2 proteins (LX and SXS, unpublished data). The fact that eIF-4E knockdown does not mimic mTOR or Raptor knockdown speaks against a model in which mTORC1 acts by fostering eIF-4E-dependent protein translation34,40. Coupled with the fact that the observed pro-myogenic effects of rapamycin and Rptor silencing were relatively small when compared to Mtor knockdown, additional mechanistic studies should focus on other mTORC1 effectors as well as mTORC1-independent activities that may block MRF actions.
Finally, our results highlight the potential value of cell-based, high content screens to identify regulators of skeletal myogenesis. This type of approach has been previously leveraged using cultured myoblast lines25,26,50 and zebrafish embryos27 to study differentiation and lineage specification, respectively. Our screen differs from these prior efforts in two important ways. First, we have focused on identifying kinases that repress muscle differentiation in proliferating myoblasts. Second, we have focused on what could be construed as the initial step in the transition between a lineage-committed myoblast and one that has begun to differentiate: the induction of Myogenin. Elegant work from the Weintraub laboratory showed the transcriptional induction of Myogenin mRNA to be directly mediated by MyoD28 and this event is closely coupled to cell cycle arrest51,52. Others have successfully utilized myosin heavy chain, a later muscle differentiation marker26,50. Conceivably, that endpoint could be utilized in a pro-differentiation screen like ours to identify the subset of kinases that foster more complete differentiation when knocked down. A laudable goal for these types of screens could be to identify proteins whose activity might be manipulated therapeutically for skeletal myopathies or even to foster differentiation in rhabdomyosarcoma, a malignancy composed of myoblast-like cells. Given that multiple, forward-feeding signals are needed to achieve a terminally differentiated state, targeting multiple proteins is likely to be required.
Methods
Cell lines and reagents
Mouse skeletal myoblast C2C12 cells, purchased from American Tissue Culture Collection (Manassas, VA), were maintained as previously described17. FACS sorted mouse skeletal muscle satellite cells, obtained from Eric Olson (UT Southwestern Medical Center) and prepared as previously described53, were maintained with daily replenishment of medium consisting of Ham’s F-10 containing 20% FBS with 0.1 mg/ml Primocin (Invitrogen) and 5 ng/mL bFGF (Gibco) on Matrigel (BD Biosciences)-coated plates. To induce differentiation, myoblasts were cultivated in DMEM containing 2% horse serum (with 0.1 mg/ml Primocin for mouse muscle satellite cells) with or without 10 μg/mL insulin (Sigma Aldrich, St. Louis, MO) (DM or DM + I). All cell culture products were purchased from Gibco (Grand Island, NY) unless otherwise noted. Rapamycin and PD 0332991 (Selleckchem, Houston, TX) were resuspended in DMSO and used at concentrations as indicated.
siRNA screen
The siRNA screen was carried out at the Quellos High Throughput Screening Core at the University of Washington, using the mouse kinase Silencer siRNA library (Ambion) targeting 571 kinases. It was arrayed onto two 384-well plates with 32 wells per plate designated for GM, 32 wells for DM + I, 32 or 37 wells for mock transfection, universal control and Kif11 positive knockdown control and 288 or 283 wells for experimental transfection. Three different siRNA targeted against the same gene were pooled together in each well (17 nM final concentration for each siRNA, 50 nM final concentration total). Each condition, with and without sensitizer, was performed in triplicate.
C2C12 myoblasts were plated at 300 cells per well in 50 μL of DMEM with 10% FBS in poly-D-lysine coated 384-well plates (Corning, Corning, NY). Sixteen hours later, medium was removed and the cells were transfected with Dharmafect 2 (Thermo Scientific, Lafayette, CO) in OptiMEM using a Biomek FX liquid handler. After 24 hours, transfection medium was replaced with GM in the experimental and negative control wells or DM + I in the positive control wells. Of note, half of the plates also received the sensitizer: PD 0332991 (1 μM) in 0.1% DMSO final concentration, which does not measurably promote differentiation in these cells17. Forty-eight hours later, the cells were fixed using paraformaldehyde (4% in PBS) prior to staining.
The fixed cells were stained using mouse anti-myogenin (F5D) antibody (BD Biosciences, San Jose, CA, Cat. No. 556358), which was detected using biotinylated anti-mouse antibody (Jackson ImmunoResearch, West Grove, PA, Cat. No. 715-065-150), Cy3-Streptavidin (Jackson ImmunoResearch, Cat. No. 016-160-084) and DAPI (Sigma Aldrich), essentially as described17. The stained plates were imaged using an ImageXpress Micro (Molecular Devices, Sunnyvale, CA) automated fluorescence microscope. Four sites were imaged per well using both DAPI and TRITC filters and images were analyzed using MetaXpress 3.1 software (Molecular Devices).
Secondary validation studies
Small interfering RNA (siRNA) used for secondary validation of Mtor was purchased from Ambion (Austin, TX). Non-targeting siRNA #3 (Thermo Scientific, Lafayette, CO) was used as a negative control. siRNAs (50 nM) were used with Dharmafect 2 to transfect 1.8 x 104 C2C12 myoblasts/well seeded on 6-well plates. Sixteen hours after transfection, medium was changed to GM or DM + I and cells were harvested for cell cycle analysis or qRT-PCR 48 hours later. The expression of five skeletal muscle mature mRNA transcripts (or four primary, unprocessed transcripts) was quantified by qRT-PCR using gene-specific primers (Supplementary Table S2) and stained with SYBR green, using the 7900HT Fast RealTime PCR System (Applied Biosystems), essentially as previously described54. Gene expression was normalized to Gapdh using the ΔΔCt method and compared to that in cells transfected with non-targeting siRNA.
Protein and mRNA analyses
C2C12 cells were grown in GM or DM + I for 72 hours and harvested for either RNA-Seq or protein expression. RNA-Seq was carried out at the McDermott Next Generation Sequencing Core (UT Southwestern). Library preparation was performed using the TruSeq Stranded Total RNA LT Sample Prep Kit (Illumina, San Diego, CA), according manufacturer’s specifications. The library was quantified using a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) and the samples were sequenced on a HiSeq 2500 (Illumina) with 100 bp paired-end reads. RNA-Seq read quality was evaluated based on the Illumina purity filter and distribution of base quality scores at each cycle. Sequence reads for each sample were aligned to the hg19 version of the human reference genome assembly using Bowtie 2.1.055 and the splicing-aware aligner TopHat 2.1.056. The alignment allows only uniquely aligned reads and up to two mismatches per read. All other parameters were set to the default values. The quality of the RNA-Seq data was evaluated by FastQC (v0.10.1) and custom Perl (v5.16.1) and R (v3.0.1) scripts. Normalized gene expression level was calculated as fragments per kilobase per million fragments mapped (FPKM) by Cufflinks 2.0.257 with default settings.
Expression of “hit” proteins or phosphorylation of specific residues on these proteins was accomplished using the Kinex KAM-880 Antibody Microarray (Kinexus Bioinformatics Corp., Vancouver, B.C.), according to manufacturer specifications. Briefly, the cells were lysed in Kinexus Lysis Buffer and protein concentration was measured by BCA assay (Thermo Scientific), 50 μg of total protein for each sample was dye-labeled, chemically cleaved using the Kinexus Chemical Cleavage Kit and incubated on the microarray slide with the GM sample added to the control chamber and the DM + I sample added to the experimental chamber. The processed slide was returned to Kinexus for quantification and normalization. The corresponding proteins for hits from the siRNA screen were then identified and their expression values divided by the expression of HSC70, a loading control, to calculate fold change in GM and DM + I.
Cell cycle and accumulation analyses
How chemical inhibition or knockdown of specific kinases influenced cell proliferation or accumulation was assessed using C2C12 myoblasts. Cell cycle analyses were performed using promidium iodide (PI) staining with cells fixed in 70% ethanol, processed as previously described58. Cell accumulation was assessed using CellTiter-Blue cell viability assay (Promega) with analysis performed using a POLARstar Omega microplate reacher.
Muscle-specific promoter analyses
Plasmid DNA reporters, transiently transfected into C2C12 myoblasts using Lipofectamine (Invitrogen) according to manufacturer’s recommendations, were used to measure muscle specific promoter activity in cultured myoblasts. Plasmids included p3300MCK-Luc, containing 3300 bp from the muscle creatine kinase (Ckm) promoter, or p4R-SV-Luc, containing four MyoD-binding E-boxes and a minimal SV40 promoter – both analogous to previously described plasmids containing the chloramphenical acetyl transferase (CAT) reporter12,16 – as well as a control vector encoding Renilla luciferase driven by the thymidine kinase promoter (Promega). Transient transfection and luciferase assays were performed as previously described and expressed relative to internal control54.
Gene ontology and pathway analysis
The lists of positively scoring kinase “hits”, with and without PD 0332991 as a sensitizer, were analyzed using ToppGene software29 ( https://toppgene.cchmc.org) to identify significantly enriched pathways and biological processes. A binomial test identified the pathways enriched in the hits as compared to their representation in the library; enriched pathways were identified if the probability of enrichment solely due to library content was less than 0.05. FDR corrected p-values were calculated using the Benjamini-Hochberg method.
The Human Protein Reference Database (HPRD) ( http://www.hprd.org) (accessed on 9/27/2012) and the Biological General Repository for Interaction Datasets (BioGRID) ( http://thebiogrid.org) (release 3.1.92) were utilized to explore potential interactions between hit kinases and certain myogenic regulators (MyoD, Myogenin, Myf5, Myf6 and Mef2A, C and D). Maps showing the interactions were developed using Cytoscape59 as previously demonstrated60,61. Several hits – Nek4, Epha10 and Peak1 without PD 0332991 and Nek4, Peak1, Tk2 and Ror1 with PD 0332991 – displayed no connections in either database and were not included in the interaction maps.
Additional statistical analyses
For the siRNA screen, data were normalized using the robust Z-score method62 and tested for significance using a t-test comparing the average percent Myogenin positive cells in three replicates for each siRNA target with that in the negative control (GM) wells on the same plates. An individual well scored positive if the knockdown caused a) the average percent Myogenin staining to increase significantly (p < 0.05 by Student’s t-test) and b) the average total number of cells in the triplicate wells to decrease by at least one median absolute deviation (MAD) from the controls. For cells receiving PD 0332991 as a sensitizer, total cell number was not included as a variable because this drug decreases myoblast cell number independently of siRNA knockdown17. “Hits” were only called if two of the replicates (or all three when the sensitizer was included) were positive.
For all other experiments, quantitative data were expressed as mean ± SD pooled from either multiple biological replicates from within one representative experiment or from multiple experiments, with statistical differences between two populations determined by Student’s t-test.
Additional Information
How to cite this article: Wilson, R. A. et al. Negative regulation of initial steps in skeletal myogenesis by mTOR and other kinases. Sci. Rep. 6, 20376; doi: 10.1038/srep20376 (2016).
References
Tapscott, S. J. The circuitry of a master switch: Myod and the regulation of skeletal muscle gene transcription. Development 132, 2685–2695 (2005).
Saab, R., Spunt, S. L. & Skapek, S. X. Myogenesis and rhabdomyosarcoma the Jekyll and Hyde of skeletal muscle. Curr.Top.Dev.Biol. 94, 197–234 (2011).
Rudnicki, M. A. et al. MyoD or Myf-5 is required for the formation of skeletal muscle. Cell 75, 1351–1359 (1993).
Bergstrom, D. A. & Tapscott, S. J. Molecular distinction between specification and differentiation in the myogenic basic helix-loop-helix transcription factor family. Mol Cell Biol. 21, 2404–2412 (2001).
Munsterberg, A. E. & Lassar, A. B. Combinatorial signals from the neural tube, floor plate and notochord induce myogenic bHLH gene expression in the somite. Development 121, 651–660 (1995).
Bentzinger, C. F., Wang, Y. X. & Rudnicki, M. A. Building muscle: molecular regulation of myogenesis. Cold Spring Harbor perspectives in biology 4, doi: 10.1101/cshperspect.a008342 (2012).
Brack, A. S., Conboy, I. M., Conboy, M. J., Shen, J. & Rando, T. A. A temporal switch from notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell stem cell 2, 50–59, doi: 10.1016/j.stem.2007.10.006 (2008).
Polesskaya, A., Seale, P. & Rudnicki, M. A. Wnt signaling induces the myogenic specification of resident CD45+ adult stem cells during muscle regeneration. Cell 113, 841–852 (2003).
Molkentin, J. D., Black, B. L., Martin, J. F. & Olson, E. N. Cooperative activation of muscle gene expression by MEF2 and myogenic bHLH proteins. Cell 83, 1125–1136 (1995).
Olson, E. N., Perry, M. & Schulz, R. A. Regulation of muscle differentiation by the MEF2 family of MADS box transcription factors. Developmental Biology 172, 2–14 (1995).
Gu, W. et al. Interaction of myogenic factors and the retinoblastoma protein mediates muscle cell commitment and differentiation. Cell 72 309–324 (1993).
Skapek, S. X., Rhee, J., Kim, P. S., Novitch, B. G. & Lassar, A. B. Cyclin-mediated inhibition of muscle gene expression via a mechanism that is independent of pRB hyperphosphorylation. Mol.Cell Biol. 16, 7043–7053 (1996).
Puri, P. L. et al. Differential roles of p300 and PCAF acetyltransferases in muscle differentiation. Mol Cell 1, 35–45 (1997).
Yuan, W., Condorelli, G., Caruso, M., Felsani, A. & Giordano, A. Human p300 protein is a coactivator for the transcription factor MyoD. J.Biol.Chem. 271, 9009–9013 (1996).
Schneider, J. W., Gu, W., Zhu, L., Mahdavi, V. & Nadal-Ginard, B. Reversal of terminal differentiation mediated by p107 in RB -/- muscle cells. Science 264, 1467–1471 (1994).
Skapek, S. X., Rhee, J., Spicer, D. B. & Lassar, A. B. Inhibition of myogenic differentiation in proliferating myoblasts by cyclin D1-dependent kinase. Science 267, 1022–1024 (1995).
Saab, R. et al. Pharmacological inhibition of cyclin-dependent kinase 4/6 activity arrests proliferation in myoblasts and rhabdomyosarcoma-derived cells. Molecular Cancer Therapeutics 5, 1299–1308 (2006).
Cuenda, A. & Cohen, P. Stress-activated protein kinase-2/p38 and a rapamycin-sensitive pathway are required for C2C12 myogenesis. J Biol.Chem. 274, 4341–4346 (1999).
Zetser, A., Gredinger, E. & Bengal, E. p38 mitogen-activated protein kinase pathway promotes skeletal muscle differentiation. Participation of the Mef2c transcription factor. J Biol.Chem. 274, 5193–5200 (1999).
Guttridge, D. C. Signaling pathways weigh in on decisions to make or break skeletal muscle. Current opinion in clinical nutrition and metabolic care 7, 443–450 (2004).
Fearon, K. C., Glass, D. J. & Guttridge, D. C. Cancer cachexia: mediators, signaling and metabolic pathways. Cell metabolism 16, 153–166, doi: 10.1016/j.cmet.2012.06.011 (2012).
Banerjee, A. & Guttridge, D. C. Mechanisms for maintaining muscle. Current opinion in supportive and palliative care 6, 451–456, doi: 10.1097/SPC.0b013e328359b681 (2012).
Hettmer, S. & Wagers, A. J. Muscling in: Uncovering the origins of rhabdomyosarcoma. Nature medicine 16, 171–173, doi: 10.1038/nm0210-171 (2010).
Tapscott, S. J., Thayer, M. J. & Weintraub, H. Deficiency in rhabdomyosarcomas of a factor required for MyoD activity and myogenesis. Science 259, 1450–1453 (1993).
Khanjyan, M. V., Yang, J., Kayali, R., Caldwell, T. & Bertoni, C. A high-content, high-throughput siRNA screen identifies cyclin D2 as a potent regulator of muscle progenitor cell fusion and a target to enhance muscle regeneration. Human molecular genetics 22, 3283–3295, doi: 10.1093/hmg/ddt184 (2013).
Polesskaya, A. et al. Genome-wide exploration of miRNA function in mammalian muscle cell differentiation. PloS one 8, e71927, doi: 10.1371/journal.pone.0071927 (2013).
Xu, C. et al. A zebrafish embryo culture system defines factors that promote vertebrate myogenesis across species. Cell 155, 909–921, doi: 10.1016/j.cell.2013.10.023 (2013).
Hollenberg, S. M., Cheng, P. F. & Weintraub, H. Use of a conditional MyoD transcription factor in studies of MyoD trans-activation and muscle differentation. Proc.Natl.Acad.Sci.USA 90, 8028–8032 (1993).
Chen, J., Bardes, E. E., Aronow, B. J. & Jegga, A. G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 37, W305–311, doi: 10.1093/nar/gkp427 (2009).
Falcone, G. et al. v-Src inhibits myogenic differentiation by interfering with the regulatory network of muscle-specific transcriptional activators at multiple levels. Oncogene 22, 8302–8315, doi: 10.1038/sj.onc.1206915 (2003).
Lim, M. J. et al. Suppression of c-Src activity stimulates muscle differentiation via p38 MAPK activation. Arch Biochem Biophys 465, 197–208, doi: 10.1016/j.abb.2007.06.004 (2007).
Nishiyama, T., Kii, I. & Kudo, A. Inactivation of Rho/ROCK signaling is crucial for the nuclear accumulation of FKHR and myoblast fusion. J Biol Chem 279, 47311–47319, doi: 10.1074/jbc.M403546200 (2004).
Shu, L. & Houghton, P. J. The mTORC2 complex regulates terminal differentiation of C2C12 myoblasts. Molecular and cellular biology 29, 4691–4700, doi: 10.1128/MCB.00764-09 (2009).
Ge, Y. & Chen, J. Mammalian target of rapamycin (mTOR) signaling network in skeletal myogenesis. J Biol Chem 287, 43928–43935, doi: 10.1074/jbc.R112.406942 (2012).
Erbay, E. & Chen, J. The mammalian target of rapamycin regulates C2C12 myogenesis via a kinase-independent mechanism. J Biol Chem 276, 36079–36082, doi: 10.1074/jbc.C100406200 (2001).
Shu, L., Zhang, X. & Houghton, P. J. Myogenic differentiation is dependent on both the kinase function and the N-terminal sequence of mammalian target of rapamycin. J Biol Chem 277, 16726–16732, doi: 10.1074/jbc.M112285200 (2002).
Dowling, R. J. et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 328, 1172–1176, doi: 10.1126/science.1187532 (2010).
Hsu, P. P. et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332, 1317–1322, doi: 10.1126/science.1199498 (2011).
Guertin, D. A. & Sabatini, D. M. Defining the role of mTOR in cancer. Cancer cell 12, 9–22, doi: 10.1016/j.ccr.2007.05.008 (2007).
Hay, N. & Sonenberg, N. Upstream and downstream of mTOR. Genes & development 18, 1926–1945, doi: 10.1101/gad.1212704 (2004).
Weintraub, H. et al. The myoD gene family: Nodal point during specification of the muscle cell lineage. Science 251, 761–766 (1991).
Calautti, E. et al. Fyn tyrosine kinase is a downstream mediator of Rho/PRK2 function in keratinocyte cell-cell adhesion. The Journal of cell biology 156, 137–148, doi: 10.1083/jcb.200105140 (2002).
Stark, D. A., Karvas, R. M., Siegel, A. L. & Cornelison, D. D. Eph/ephrin interactions modulate muscle satellite cell motility and patterning. Development 138, 5279–5289, doi: 10.1242/dev.068411 (2011).
Urness, L. D., Sorensen, L. K. & Li, D. Y. Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nature genetics 26, 328–331, doi: 10.1038/81634 (2000).
Amit, S. et al. Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes & development 16, 1066–1076, doi: 10.1101/gad.230302 (2002).
Ge, Y., Yoon, M. S. & Chen, J. Raptor and Rheb negatively regulate skeletal myogenesis through suppression of insulin receptor substrate 1 (IRS1). J Biol Chem 286, 35675–35682, doi: 10.1074/jbc.M111.262881 (2011).
Greene, M. W., Sakaue, H., Wang, L., Alessi, D. R. & Roth, R. A. Modulation of insulin-stimulated degradation of human insulin receptor substrate-1 by Serine 312 phosphorylation. J Biol Chem 278, 8199–8211, doi: 10.1074/jbc.M209153200 (2003).
Bentzinger, C. F. et al. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell metabolism 8, 411–424, doi: 10.1016/j.cmet.2008.10.002 (2008).
Risson, V. et al. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. The Journal of cell biology 187, 859–874, doi: 10.1083/jcb.200903131 (2009).
Ge, Y., Waldemer, R. J., Nalluri, R., Nuzzi, P. D. & Chen, J. RNAi screen reveals potentially novel roles of cytokines in myoblast differentiation. PloS one 8, e68068, doi: 10.1371/journal.pone.0068068 (2013).
Halevy, O. et al. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science 267, 1018–1021 (1995).
Novitch, B. G., Mulligan, G. J., Jacks, T. & Lassar, A. B. Skeletal muscle cells lacking the retinoblastoma protein display defects in muscle gene expression and accumulate in S and G2 phases of the cell cycle. J.Cell Biol. 135, 441–456 (1996).
Liu, N. et al. Requirement of MEF2A, C and D for skeletal muscle regeneration. Proceedings of the National Academy of Sciences of the United States of America 111, 4109–4114, doi: 10.1073/pnas.1401732111 (2014).
Widau, R. C. et al. p19Arf represses platelet-derived growth factor receptor beta by transcriptional and posttranscriptional mechanisms. Molecular and cellular biology 32, 4270–4282, doi: 10.1128/MCB.06424-11 (2012).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359, doi: 10.1038/nmeth.1923 (2012).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome biology 14, R36, doi: 10.1186/gb-2013-14-4-r36 (2013).
Trapnell, C. et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nature biotechnology 31, 46–53, doi: 10.1038/nbt.2450 (2013).
Silva, R. L. et al. Arf-dependent regulation of Pdgf signaling in perivascular cells in the developing mouse eye. EMBO J. 24, 2803–2814 (2005).
Cline, M. S. et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc 2, 2366–2382, doi: 10.1038/nprot.2007.324 (2007).
Keshava Prasad, T. S. et al. Human Protein Reference Database–2009 update. Nucleic Acids Res 37, D767–772, doi: 10.1093/nar/gkn892 (2009).
Chatr-Aryamontri, A. et al. The BioGRID interaction database: 2013 update. Nucleic Acids Res 41, D816–823, doi: 10.1093/nar/gks1158 (2013).
Birmingham, A. et al. Statistical methods for analysis of high-throughput RNA interference screens. Nat Methods 6, 569–575, doi: 10.1038/nmeth.1351 (2009).
Acknowledgements
The authors gratefully acknowledge Drs. N. Liu and E. Olson (UT Southwestern) for providing primary mouse myoblasts and advice on their cultivation; helpful discussions with J. Amatruda (UT Southwestern), G. Greene (U Chicago) and past and current members of the Skapek laboratory including D. Dighe, N. Iqbal, C. Sung, R. Widau; support from the UT Southwestern Medical Center Simmons Cancer Center Animal Resources Center (Supported by the NIH National Cancer Institute under award number 5P30CA142543); and grant support to SXS from the Cancer Prevention and Research Institute of Texas (RP120685-P2), St. Baldrick’s Foundation, Ted Mullin Fund for Sarcoma Research, Children’s Cancer Fund Dallas and Wipe Out Kids’ Cancer; and to YZ from the Hyundai Hope on Wheels Foundation.
Author information
Authors and Affiliations
Contributions
R.A.W., J.L., L.X., S.H., G.M., C.T. and Y.Z. each contributed to the collection and analysis of data from the screening optimization, screen conduct, secondary validation and mechanistic studies and gene expression analysis in normal myoblasts; J.A. and C.G. contributed to, in particular, to high throughput screen design, conduct and data analysis; SXS conceived of overall project, guided experimental design, data acquision, data analysis; all authors contributed to manuscript writing and in the deicsion to submit the manuscript for publication.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wilson, R., Liu, J., Xu, L. et al. Negative regulation of initial steps in skeletal myogenesis by mTOR and other kinases. Sci Rep 6, 20376 (2016). https://doi.org/10.1038/srep20376
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep20376
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
