
Abstract
Apoptosis plays a crucial role in development and tissue homeostasis. Some key survival pathways, such as DNA damage checkpoints and DNA repair, have been described to be inactivated during apoptosis. Here, we describe the processing of the human checkpoint protein Claspin during apoptosis. We observed cleavage of Claspin into multiple fragments in vivo. In vitro cleavage with caspases 3 and 7 of various fragments of the protein, revealed cut sites near the N- and C-termini of the protein. Using mass spectrometry, we identified a novel caspase cleavage site in Claspin at Asp25. Importantly, in addition to cleavage by caspases, we observed a proteasome-dependent degradation of Claspin under apoptotic conditions, resulting in a reduction of the levels of both full-length Claspin and its cleavage products. This degradation was not dependent upon the DSGxxS phosphodegron motif required for SCFβ-TrCP-mediated ubiquitination of Claspin. Finally, downregulation of Claspin protein levels by short interfering RNA resulted in an increase in apoptotic induction both in the presence and absence of DNA damage. We conclude that Claspin has antiapoptotic activity and is degraded by two different pathways during apoptosis. The resulting disappearance of Claspin from the cells further promotes apoptosis.
Similar content being viewed by others

Main
Living organisms require accurate transfer of genetic information from one cell to its daughter cells. DNA damage, caused by endogenous (e.g. free radicals) or exogenous factors (e.g. exposure to ultraviolet (UV) light), interferes with this faithful transfer of information. To prevent such dangerous consequences of DNA damage, eukaryotic cells pause the progression of the cell cycle, which allows them to repair the damage before proceeding into a further stage of cellular division. In higher eukaryotes, when the genotoxic damage is too severe to be repaired efficiently, apoptosis is triggered. Apoptotic signalling converges on the effector caspases (caspases 3, 6 and 7), which cause the destruction of a cell by limited proteolysis of a subset of intracellular proteins.1 Upon cleavage, these caspase targets become either more active, inactive or deregulated in general.
The inactivation of cell survival mechanisms, and particularly, routes that maintain genomic integrity, seems to be a common feature during apoptosis.2, 3, 4 The ATM (ataxia telangiectasia mutated)- and Rad3-related (ATR)–Chk1 is a key survival pathway activated by many types of DNA damage, including lesions caused by UV light, stalled replication forks and double-strand breaks.5 ATR is recruited at an early stage to the site of DNA damage by ATRIP.6 ATR activation also requires the 9-1-1 complex (Rad9, Rad1 and Hus1) that is recruited independently of ATR to DNA lesions.7 Once activated, ATR phosphorylates a number of downstream targets, including the effector kinase Chk1, that is responsible for sending the inhibitory signal to the cell cycle progression machinery.8, 9 A key player in mediating the ATR-dependent activation of Chk1 is Claspin.10, 11
In Xenopus and human cells, Claspin is phosphorylated in response to DNA damage in an ATR-dependent manner,12 promoting the association of Claspin with Chk1 and facilitating Chk1 activation and subsequent checkpoint arrest.10 Both Claspin and its homologue in yeast, Mrc1, are involved in the checkpoint that is triggered in the presence of stalled replication forks during S phase of the cell cycle.13, 14 Claspin also has a role as a positive regulator of cell proliferation.15
In this report, we describe that Claspin, but not other proteins involved in the ATR-Chk1 signalling pathway, is processed during apoptosis. This processing involves both cleavage by caspases 3 and 7 and degradation via the proteasome. Mimicking the degradation of Claspin in vivo by knocking down protein levels by RNA interference (RNAi) promotes apoptosis both in the presence and absence of DNA damage. Our studies indicate that Claspin is an important target for inactivation during apoptosis.
Results
The fate of the ATR-Chk1 pathway in apoptosis
The ATR-Chk1 pathway is triggered in response to genotoxic stress. To investigate whether this pathway is active during apoptosis, we induced apoptosis in HL60 cells with the topoisomerase II inhibitor etoposide16 and analysed whole cell extracts by immunoblotting with a Chk1-specific antibody. After 1 h of etoposide treatment, Chk1 was phosphorylated, as indicated by a mobility shift in SDS-PAGE (Figure 1b). However, 3 and 5 h after addition of etoposide to culture medium, when 55 and 63% of the cells, respectively, are apoptotic (Figure 1a, subG1) the mobility shift of Chk1 is no longer detectable (Figure 1b). To determine what might cause the inactivation of Chk1 during apoptosis, we investigated if any of the proteins involved in the ATR-Chk1 pathway9 is cleaved during apoptosis. ATR, ATRIP, Chk1, Rad1, Rad9 and Hus1 all maintain normal levels of expression throughout apoptosis (Figure 1b). In contrast, Claspin levels dramatically decreased 3 and 5 h after etoposide addition (Figure 1b). This coincided with the cleavage of PARP1 by caspases (Figure 1b). We also observed a very limited cleavage of Rad9 in apoptotic cells (Figure 1b), as described before.17

Inactivation of the Chk1 pathway during apoptosis. (a) HL60 cells were treated with 68 μM etoposide and collected after 0, 1, 3 and 5 h. DNA content was analysed by FACS. (b) HL60 cells were treated as above, lysed and analysed by Western blotting with the indicated antibodies. PARP1 is a positive control for apoptotic cleavage, and Ku70 was used as a loading control. *A nonspecific cross-reacting band.
Cleavage of Claspin during apoptosis
In order to characterise the Claspin fragments produced during apoptosis in HL60 cells, we raised antibodies against the N-terminus (amino acids 85–248, Nt) and C-terminus (amino acids 1006–1340, Ct) of Claspin. Western blotting analysis reveals that Claspin migrates as an ∼270 kDa band instead of the predicted 151 kDa, as described before (Figure 2a, top arrow).10 We also observe a protein band corresponding to what may be another isoform of Claspin that migrates with an apparent molecular weight of about 200 kDa (Figure 2a, bottom arrow). This isoform shows a mobility shift after 1 h of etoposide treatment, similar to that of the full-length protein (Figure 2a, bottom arrow). By 3 h of treatment with etoposide, both isoforms are barely detectable, and there is a gradual appearance of smaller molecular weight fragments. The Nt Claspin antibody detects three protein fragments that migrate with apparent molecular weights between 90 and 120 kDa (Figure 2a, arrowheads ii–iv). The Ct Claspin antibody identified smaller protein fragments with apparent molecular weights of 20, 35, 45 and 70 kDa (Figure 2a, right panel, arrowheads v–viii). Both antibodies detected an ∼235 kDa fragment (Figure 2, arrowhead i) that appears transiently 3 h after addition of etoposide, but which is barely detectable after 5 h, probably because of further cleavage into smaller fragments. Together, these data suggest that Claspin contains several cleavage sites.

Cleavage of Claspin during apoptosis. (a) HL60 cells were treated with 68 μM etoposide and collected at 1 h intervals up to 5 h. The cells were analysed by Western blotting with the indicated antibodies. (b) HeLa cells were left untreated or treated with UV (600 J/m2). After 8 h, the cells were collected and analysed by Western blotting. (c) HeLa cells were treated with 30 ng/ml TNFα+10 μg/ml CHX for 0, 4 or 8 h, and analysed by Western blotting. (d) HL60 cells were left untreated or pretreated for 30 min with ZVAD-fmk (40 μM) before adding 68 μM etoposide. Cells were collected 0, 1, 3 and 5 h later, and analysed by Western blotting. The blots were reprobed with mouse anti-actin as a loading control and mouse anti-PARP1 as a positive control for apoptotic cleavage. The position of full-length Claspin (top arrow), a possible isoform of Claspin (bottom arrow), fragments produced during apoptosis (arrowheads and roman numerals), and nonspecific cross-reacting bands (asterisks), are indicated
Apoptotic cleavage of Claspin was also observed upon induction of apoptosis in HeLa cells by UV irradiation (Figure 2b) or by incubation with tumour necrosis factor alpha (TNFα) and cycloheximide (CHX) (Figure 2c). Therefore, apoptotic cleavage of Claspin is not specific to HL60 cells and is induced by both the intrinsic and extrinsic apoptotic pathways. In HeLa cells, we could only detect two cleavage products with the Nt Claspin antibody, corresponding to the ∼120 and ∼235 kDa bands (Figure 2b and c, arrowheads ii and i, respectively) detected in HL60 cells.
Claspin cleavage by caspases 3 and 7
To demonstrate that Claspin cleavage was caspase-dependent, we treated HL60 cells with the broad specificity caspase inhibitor, ZVAD-fmk, and found that it inhibited the cleavage of Claspin (Figure 2d).
To characterise the sites at which Claspin is cleaved by caspases, we introduced glutathione-S-transferase (GST) fusion proteins of four fragments that span the full-length of the Claspin open-reading frame in an in vitro caspase assay using recombinant caspases 3, 6 and 7. Caspase 3 cleaved both GST-Claspin(1–336) and GST-Claspin(1006–1340) (Figure 3a, bands a-d). Interestingly, caspase 7 appeared to cleave GST-Claspin(1006–1340) in a similar manner to caspase 3, although it was not able to cleave the N-terminal fragment GST-Claspin(1–336) (Figure 3b). We were unable to detect cleavage of any of the Claspin fragments by caspase 6 (data not shown).

Cleavage of Claspin by caspases 3 and 7. (a and b) Bacterially expressed fragments of GST-Claspin were purified and then treated in vitro with or without recombinant caspase 3 (a) or caspase 7 (b) for 30 min at 37°C. The cleavage products were analysed by SDS-PAGE and visualised by silver staining. Caspase cleavage products are indicated (arrows). *A nonspecific band originating from the recombinant caspase 7 preparation. (c) Scaled-up caspase digestion of Claspin fragments 1–336 (wt), 1006–1340 (wt) and 1006–1340 (D1072A) analysed by SDS-PAGE and Coomassie staining. The cleavage products indicated by arrowheads were analysed by MALDI-TOF spectrometry. (d) Schematic representation of GST-Claspin(1–336). The last three amino acids of GST (small letters) together with the first 56 amino acids of Claspin (capital letters) form a single tryptic peptide. The predicted m/z of the peptides that would be produced by caspase cleavage at Asp25 are shown along with the spectra of full-length GST-Claspin(1–336) and the caspase cleavage products a and b. For the sake of clarity, only the relevant parts of the spectra are shown. Cleavage at Asp25 produces 2955 and 2971 Da peptides from the sequence upstream of the cleavage site, and these were uniquely found in the mass spectrum of fragment b. The 3017 and 3145 Da peptides, derived from the sequence downstream of Asp25, were uniquely identified in the mass spectrum of fragment a. (e) HeLa cells were transfected with wild-type or D25A mutant Claspin(1–260)-GFP. Two days after transfection, apoptosis was induced with 300 J/m2 UV light or 30 ng/ml TNFα+10 mg/ml CHX. The cells were collected 8 h later and analysed by immunoblotting. The membrane was reprobed with anti-actin and anti-PARP1 antibodies as loading and apoptosis controls, respectively. (f) HeLa cells were transfected with wild-type or D25/1072A double mutant full-length HA-Claspin. Two days after transfection, apoptosis was induced with 300 J/m2 UV irradiation. Cells were collected 0, 4 and 8 h after treatment and analysed by immunoblotting. Nonspecific cross-reacting bands are indicated with an *
Identification of caspase cleavage sites
To identify the cleavage sites, the in vitro caspase reactions were scaled up and visualised by Coomassie staining (Figure 3c). Bands that appeared in the cleaved sample but not in the uncleaved sample were analysed by MALDI-TOF-MS. The mass spectrum of fragment b contained peaks corresponding to GST, whereas fragment a did not, indicating that fragment b is the N-terminal part of the protein. Fragment b is only slightly larger than GST (27 kDa), therefore the cleavage site must be close to the Nt of Claspin. The presence of 2955 and 2971 Da peptides in the mass spectrum of fragment b, and 3017 and 3145 Da peptides in the mass spectrum of fragment a, are consistent with caspase cleavage after Asp25 (Figure 3d).
A similar analysis with GST-Claspin(1006–1340) identified a 2207 Da peak in the mass spectrum of fragment d (data not shown), consistent with cleavage at Asp1072, which has also been described recently.18 In order to search for new caspase sites, we performed the analysis with a mutated version of GST-Claspin(1006–1340) containing an Asp1072Ala substitution. The two major cleavage products of the wild-type protein, c and d, are absent from the mutant sample, confirming that they arise from cleavage at Asp1072 (Figure 3c). Both wild-type and mutant Claspin(1006–1340) also contained low-abundance cleavage products (Figure 3c, unlabelled arrowheads), which may indicate the presence of other caspase cleavage sites in the Ct of Claspin. However, we were unable to identify any such sites using MS.
Expression of caspase-resistant Claspin in vivo
To look at cleavage at Asp25 in vivo, we expressed amino acids 1–260 of Claspin fused to enhanced green fluorescent protein (EGFP) in HeLa cells. When apoptosis was induced by UV irradiation or with TNFα+CHX, the wild-type fusion protein was cleaved to produce a fragment that migrated just below the full-length band. When Asp25 was mutated to Ala, the fusion protein was no longer cleaved (Figure 3e). Interestingly, the overall protein levels of both wild-type and mutant proteins decreased significantly upon apoptosis induction (see below).
Next, we expressed HA-tagged full-length wild-type Claspin and an Asp25/1072Ala double mutant in HeLa cells and induced apoptosis with UV irradiation. Both the wild-type and the caspase-resistant version of the protein were degraded upon induction of apoptosis, and we could not detect any caspase cleavage fragments (Figure 3f). This result suggests the involvement of other pathways in the degradation of Claspin. If caspase cleavage at D1072 was the only effect of apoptosis on Claspin, as was previously reported,18 one would expect that the D25/1072A mutant would be stable under apoptotic conditions, but this is not the case.
Proteasomal degradation of Claspin during apoptosis
A closer look at the levels of Claspin and its cleavage products during apoptosis (Figure 2) reveals that the sum of the intensities of the smaller fragments does not account for the decrease in the levels of the full-length protein. The same effect is observed when a construct encoding the first 260 amino acids of Claspin fused to EGFP is expressed in Hela cells (Figure 3f). To see if Claspin might also be degraded by the ubiquitin–proteasome pathway, we treated HL60 cells with the proteasome inhibitor MG132 before inducing apoptosis with etoposide. The levels of Claspin 1 h after addition of etoposide were the same, with or without MG132 (Figure 4a, compare lanes 3 and 6). However, 3 h after etoposide addition, the levels of Claspin in cells treated with MG132 and etoposide are much higher than in cells treated with etoposide alone (Figure 4a, compare lanes 4 and 7). This difference in Claspin levels is not due to a change in the degree of induction of apoptosis, as the degree of PARP1 cleavage is similar in response to both treatments. The stabilisation of full-length Claspin is not due to an indirect inhibition of caspase activity by MG132, as the amount of caspase cleavage products was even higher in MG132-treated cells compared to cells not treated with the proteasome inhibitor (Figure 4b, compare bands indicated by arrowheads ii–iv in lanes 4 and 5 with lanes 7 and 8). The involvement of the proteasome in the degradation of Claspin was confirmed by repeating the experiment with the highly specific proteasome inhibitor, epoxomicin19 (Figure 4c, compare lanes 4 and 5 with lanes 8 and 9).

Proteasomal degradation of Claspin during apoptosis. (a and b) HL60 cells were pretreated for 2 h with MG132 (10 μM) before incubating for 0, 1, 3 or 5 h in the presence of etoposide (68 μM). (c) HL60 cells were pretreated for 2 h with epoxomicin (5 μM) before incubating for 0, 1, 3 or 5 h in the presence of etoposide (68 μM). (d) HL60 cells were incubated with etoposide (68 μM) for 2 h before the addition of 10 μM MG132. Cells were collected 2, 3 or 5 h after the addition of etoposide. (e) HL60 cells were pretreated with MG132 for 2 h, and with ZVAD-fmk for 30 min before adding etoposide. Cells were collected at the time points indicated. (f) Cells were incubated with or without 25 μM ALLM for 15 min before induction of Calpain activity by incubation with Ionomycin for 90 min. In all the experiments, the cells were lysed and analysed by Western blot. Actin or Ku70 were used as controls for equal loading, and PARP1 was used as a positive control for apoptotic cleavage. The position of full-length Claspin (top arrow), a possible isoform of Claspin (bottom arrow), fragments produced during apoptosis (arrowheads), and nonspecific cross-reacting bands (asterisks), are indicated
Proteasome inhibitors have a dual effect on apoptosis – initially they inhibit apoptosis, and after longer exposure, they promote apoptosis.20 To avoid any confounding nonspecific effects, we performed the proteasome inhibitor experiments in two different ways. In the experiments described above, we added the proteasome inhibitor 2 h before addition of etoposide. Alternatively, we also added MG132 2 h after etoposide, once the cells had already committed to apoptosis. Even when added after etoposide, MG132 inhibited the degradation of Claspin (Figure 4d, compare lanes 2 and 5), indicating that Claspin degradation by the proteasome occurs during apoptosis.
Finally, we treated cells with a combination of caspase and proteasome inhibitors. Under these conditions, the levels of Claspin were completely stabilised even up to 5 h after addition of etoposide (Figure 4e). The levels of Claspin were higher in cells treated for 5 h with a combination of etoposide, MG132 and ZVAD-fmk, compared to cells treated with etoposide and ZVAD-fmk only (compare Figures 4e and 2d). ZVAD-fmk also partially inhibits other apoptotic proteases such as Calpains. To study if Claspin can be cleaved by these calcium-dependent proteases, we treated HL60 cells with the calcium-specific ionophore, Ionomycin. As a positive control, we used PARP1 which is cleaved by Calpain to produce several fragments about 35, 45, 50 and 89 kDa in size, in contrast to the single 89 kDa band produced by caspase cleavage of PARP1.21 Treatment of HL60 cells for 90 min with Ionomycin caused almost complete cleavage of PARP1 into small molecular weight fragments, whereas the levels of Claspin remained practically the same (Figure 4f). Treatment of HL60 cells with Ionomycin together with Calpain inhibitor II (ALLM) inhibited the degradation of PARP1 and caused a striking increase in the levels of Claspin, that was also seen when Claspin was treated by ALLM alone (Figure 4f). Owing to the fact that Ionomycin does not greatly affect the levels of Claspin when compared with PARP1 degradation, and the fact that ALLM also inhibits proteasomal activity, we interpret these results to mean that Claspin stabilisation by ALLM is probably due to inhibition of proteasomal degradation of Claspin rather than inhibition of Calpain activity.
Distinct fates of Claspin in response to high and low levels of DNA damage
To understand whether the degradation of Claspin is the normal response of the cell to DNA damage or whether it is only apoptosis-specific, we compared the levels of Claspin in cells treated with high (68 μM) and low (0.5 μM) doses of etoposide. In contrast to the rapid degradation of Claspin in apoptotic cells, in cells exposed to low concentrations of etoposide, compatible with cell survival, the levels of Claspin steadily increased (Figure 5a). Similar results were obtained with cells treated with hydroxyurea (HU), which stalls replication forks. HU does not initially induce apoptosis and the levels of Claspin gradually accumulate. However, after prolonged exposure (5 h), apoptosis is induced, PARP1 is cleaved and Claspin levels decline (Figure 5a).

Downregulation of Claspin promotes apoptosis. (a) HL60 cells were treated with 68 or 0.5 μM etoposide or 10 mM HU for 0, 1, 3 and 5 h and analysed by immunoblotting. The membrane was reprobed with anti-tubulin and anti-PARP1 antibodies as loading and apoptosis controls, respectively. (b) Schematic representation of Claspin mutants lacking the caspase cleavage sites at D25 and D1072, and the phosphorylation-dependent β-TrCP binding motif at S30/34. (c) HeLa cells were transfected with wild-type and 2DS HA-Claspin. Two days after transfection, apoptosis was induced with 300 J/m2 UV light or 30 ng/ml TNFα+10 mg/ml CHX. Cells were collected 7 h after treatment and analysed by immunoblotting. Nonspecific cross-reacting bands are indicated with an *. (d) HeLa cells were transfected with wild-type, 2D or 2DS EGFP-Claspin, or empty pEGFP-C1 vector. One day after transfection, the cells were synchronised in G1 phase by treating with 2.5 mM thymidine for 20 h. Three hours after removal of the thymidine, apoptosis was induced with 150 J/m2 UV light. The cells were collected 36 h after treatment and analysed by immunoblotting. (e) U2OS cells were transfected twice with control (GFP, luciferase) or three distinct Claspin-specific siRNA oligos in two sets of experiments. Two days after the second transfection, the cells were collected and analysed by immunoblotting. The membrane was reprobed with anti-Ku70 or Ku80 antibody as a loading control. (f) U2OS cells were transfected twice with control or Claspin-specific siRNA oligos. Three days after the second transfection, the cells were collected and analysed by FACS. Cell cycle profiles from a single experiment are shown on the left. The population of subG1 cells is indicated with a line. A graphical representation of the percentage of apoptotic cells (means and standard errors of three independent experiments) is shown on the right
Proteasomal degradation of Claspin during apoptosis is not dependent upon the S30/34 phosphodegron
As pointed out above, the 2D HA-Claspin mutant (D25/1072A) is as unstable in apoptotic cells as wild-type (wt) Claspin (Figure 3f), which we attribute to the role of the proteasome in the degradation of Claspin during apoptosis. A number of recent reports have described the ubiquitination of Claspin by the ubiquitin ligase SCFβ-TrCP leading to its degradation in the transition from G2 to mitosis and in recovery from DNA damage-induced checkpoint arrest.22, 23, 24 The interaction with β-TrCP and subsequent ubiquitination is dependent upon phosphorylation of serines 30 and 34 (Ser30 and Ser34) in Claspin (Figure 5b). To determine if β-TrCP is also responsible for the degradation of Claspin during apoptosis, we mutated Ser30 and Ser34 to alanine, on top of the D25/1072A mutations (2DS mutant, Figure 5b). We expressed wild-type and 2DS HA-Claspin in HeLa cells and found that upon induction of apoptosis by either UV or TNFα+CHX, the levels of both proteins fell below detection levels (Figure 5c). The instability of the 2DS Claspin mutant suggests that its proteasomal degradation during apoptosis is via a different route from that described for Claspin so far. To confirm that this degradation was not merely an artefact of the HA-tagged construct, we repeated the experiment with Claspin cloned in frame with EGFP. The levels of both wild-type and 2DS EGFP–Claspin did not fall as dramatically after induction of apoptosis as with the HA-Claspin constructs, which we attribute to the very high levels of expression of the EGFP–Claspin fusion proteins in comparison to the HA-fusions (data not shown). However, as with the HA-tagged constructs, both wild-type and 2DS EGFP-Claspin levels decreased to a similar degree upon induction of apoptosis, supporting the idea that phosphorylation at Ser30 and Ser34 is not required for proteasomal degradation of Claspin during apoptosis (Supplementary Figure 1a). The S30/34A mutation affects the stability of Claspin during the progression of the cell cycle. In order to avoid any confounding effects introduced by cell cycle variation, we synchronised HeLa cells in G1 and then treated the cells with UV during S phase. As in asynchronous cells, wild-type, 2D and 2DS Claspin were all equally unstable upon induction of apoptosis with UV (Figure 5d). Similar results were obtained with etoposide treatment (data not shown).
The effect of overexpression of wild-type and 2D Claspin in apoptosis
Although both wild-type and 2D Claspin are unstable in apoptotic cells, we took advantage of the high level of expression of Claspin by the pEGFP-C1 vector to see if wild-type or caspase-resistant Claspin could offer some protection against apoptosis. A common feature of apoptosis is the production of double-strand breaks by the nuclease CAD. As Claspin is involved in the DNA damage response, we hypothesized that its protective role might relate to a response to this damage and therefore, we decided to study the behaviour of cells expressing EGFP–Claspin in the presence of two drugs that cause double-strand breaks, camptothecin and etoposide. Induction of apoptosis using camptothecin for 24 h caused similar levels of apoptosis (∼50%) in both control and Claspin-transfected cells (Supplementary Figure 1b). When we induced apoptosis under milder conditions, using 20 μM etoposide, there was little apoptosis after a 24 h incubation with the drug (data not shown), but by 40 h approximately 70% of the control cells were apoptotic (Supplementary Figure 1b). In contrast, only about 40% of the cells transfected with either wild-type or 2D Claspin were apoptotic. Both with camptothecin and with etoposide treatment, there was no difference in the amount of apoptosis in cells transfected with wild type compared to the caspase-resistant 2D mutant.
Knockdown of Claspin levels induces apoptosis
The results above suggest that degradation of Claspin might be required for the progression of apoptosis. To test this hypothesis, we downregulated Claspin protein levels by RNAi, in order to mimic the total levels of the protein in apoptotic cells. Transfection of U2OS cells with three independent Claspin-specific short interfering RNAs (siRNAs) caused a significant downregulation of Claspin levels when compared to cells transfected with two different control siRNAs (Figure 5e), comparable to the downregulation observed in apoptotic cells. Cell cycle analysis of cells transfected with Claspin-specific siRNA showed a two- to four-fold increase in the subG1 population of cells when compared with siRNA control cells. Only 8–10% of cells transfected with control siRNAs (GFP and luciferase) were apoptotic, compared to 20–30%, of the cells transfected with Claspin siRNAs (Figure 5f). Moreover, we also studied the effect of the lack of Claspin in the presence of double-strand breaks in the DNA, in an attempt to mimic initial stages of apoptosis. When low levels of DNA damage were induced with etoposide or ionising radiation, the percentage of apoptotic cells in the Claspin downregulated population increased marginly, ranging between 23 and 37% for etoposide and 22 and 45% for ionising radiation, depending on the siRNA used. This was significantly higher than the percentage of apoptotic cells in the control siRNA-transfected populations, which ranged between 9 and 14% (Supplementary Figure 1c).
Discussion
In this report, we describe the inactivation of the ATR-Chk1 DNA damage checkpoint pathway by cleavage and degradation of Claspin during apoptosis. While we were performing this work, a report was published regarding the cleavage of Claspin by caspase 7 during apoptosis at a single site – Asp1072.18 Our data complement the previous study and suggest that the fate of Claspin during apoptosis is more complex. Using antibodies against the Nt and Ct of Claspin, we detected multiple fragments produced during apoptosis, indicating that Claspin is cleaved at more than one site. It is possible that we were able to detect a greater number of cleavage products partly because of the different antibodies used in the two studies.
Using an in vitro caspase assay and mass spectrometry, we confirmed Claspin cleavage at Asp1072 and identified a novel cleavage site near the Nt of the protein, at Asp25. Cleavage at Asp25 in vivo was confirmed by expression of wild-type and mutant N-terminal fragments of Claspin in HeLa cells. On their own, these two cleavage sites do not explain all the Claspin fragments observed in vivo. The results of the in vitro caspase assay suggested that the Ct of Claspin might contain other sites that are cleaved less efficiently than D1072; however, we were unable to identify these sites. It is also possible that the fragments produced in vivo are due to activity of other apoptotic proteases, such as Calpains. However, cells treated with a calcium ionophore to activate Calpains, the levels of Claspin remained relatively stable when compared with a known Calpain substrate, PARP1. Alternatively, the multiplicity of fragments could originate from the expression of more that one isoform of Claspin in normal cells (200 and 270 kDa bands in Figure 2).
In addition to cleavage of Claspin by caspases, we show that Claspin is degraded by the proteasome during apoptosis. Firstly, full-length Claspin in which both caspase cleavage sites had been mutated was not resistant to degradation in apoptotic cells (Figure 3f). And secondly, both the full-length protein and caspase cleavage fragments of Claspin are stabilised in cells exposed to different proteasome inhibitors (Figure 4a and b). During the preparation of this manuscript, a report was published in which the proteasomal degradation of Claspin in normal cells was described;25 however, the degradation of Claspin by the proteasome during apoptosis is a novel observation. The proteasome plays a complex role in cell death, having both negative and positive effects on apoptosis.20, 26 To our knowledge, there have only been a handful of reports of proteins degraded during apoptosis by a combination of caspase cleavage and proteasomal degradation.27, 28, 29 These reports describe the degradation of caspase-generated fragments of these proteins by the proteasome, presumably via the N-end rule pathway, which degrades proteins with unfavourable N-terminal residues.30 However, the fact that a caspase-resistant version of Claspin(1–260)-GFP is downregulated in apoptotic cells despite mutation of Asp25, suggests that proteasomal degradation of Claspin is not only due to the N-end rule pathway (compare GFP-Claspin(1–260)-D25A in apoptotic and non-apoptotic cells Figure 3e). Furthermore, Claspin degradation by the proteasome does not appear to be dependent on caspase cleavage, as the level of the full-length protein decreases even in the presence of caspase inhibitors (Figure 2d and Clarke et al.18). Similarly, the proteasome inhibitor did not prevent cleavage by caspases (Figure 4b). Therefore, in the case of Claspin these two pathways seem to be acting in parallel to eliminate the protein from the cell and are not dependent on one another.
The fact that the GFP-Claspin(1–260)-D25A fragment is unstable in apoptotic cells (Figure 3e) would suggest that a proteasomal degradation signal is present within the first 260 amino acids of Claspin; however, the nature of this signal remains elusive. Three papers published during the review of this manuscript have identified amino acids 29–34 of Claspin as a phosphorylation-dependent proteasomal degradation motif required for binding of the ubiquitin ligase SCFβ-TrCP to Claspin.22, 23, 24 However, mutation of Ser30 and Ser34, which has been shown to abolish SCFβ-TrCP-mediated degradation of Claspin22, 23, 24 did not increase the stability of a caspase-resistant Claspin construct in apoptotic cells. These data indicate that the stability of Claspin during apoptosis is regulated by a ubiquitin ligase and/or a degradation signal different from that responsible for its degradation during normal cell cycle progression and recovery from checkpoint arrest. Zhang et al.31 have identified a deubiquitination enzyme, USP28, that causes the stabilisation of a number of checkpoint proteins, including Claspin, upon DNA damage. The deubiquitination activity of USP28 would explain the stabilisation of Claspin that we observed in cells treated with low levels of etoposide and HU. However, it would not be sufficient to explain the proteasomal degradation of Claspin during apoptosis induced with high levels of etoposide, as even in the absence of deubiquitination activity, a ubiquitin ligase would still be required to ubiquitinate Claspin and target it for proteasomal degradation. Therefore, the regulation of Claspin by the ubiquitin proteasome pathway appears to be rather complex, including at least one deubiquitinating enzyme, USP28, and two ubiquitin ligases: SCFβ-TrCP and an as yet unidentified ubiquitin ligase that is active during apoptosis.
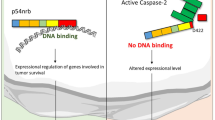
Caspase cleavage sometimes produces fragments that have dominant-negative activity because of the uncoupling of functional domains.2, 32 The same has been suggested for the C-terminal fragment released by caspase 7 cleavage of Claspin described by Clarke et al.,18 which maintains its DNA binding activity, but is physically separated from the N-terminal Chk1 activating domain. However, our results show that not only is Claspin cleaved at two or more sites generating many different fragments, but that it is also simultaneously degraded by the proteasome pathway during apoptosis. The final result of the activity of these two pathways is that very little full-length or cleaved Claspin remains in the cell during apoptosis (Figures 2 and 4). Overexpression of wild-type and 2D Claspin protected cells against etoposide-, but not camptothecin-induced DNA damage. This could be due to the different mechanisms affected by these two drugs: etoposide inhibits Topoisomerase II, whereas camptothecin inhibits Topoisomerase I. Alternatively, it could be due to the strength of the apoptotic stimuli, as etoposide required more time to induce a similar level of apoptosis in HeLa cells, and therefore overexpression of Claspin could have performed a protective role in the initial stages, before commitment to apoptosis and the degradation of Claspin. It is interesting that both with camptothecin and etoposide treatment, there was no difference in the levels of apoptosis between cells transfected with the wild-type and the 2D mutant Claspin. We have seen that both proteins are degraded by the proteasome during apoptosis; however, the wild-type construct would also produce caspase-cleavage products, and our results suggest that these do not cause any additional cytotoxicity. Therefore, it is the absence of full-length Claspin that is particularly cytotoxic to the cells.
To mimic the situation in apoptotic cells, we knocked down the protein levels of Claspin by RNAi and observed a two- to fourfold increase in apoptosis in U2OS cells. Lin et al.15 has shown that overexpression of Claspin positively regulates cell growth, which provides a rationale for the inactivation of Claspin during apoptosis: not only there is a need to eliminate Claspin in order to inactivate the DNA damage checkpoint, but there is also a need to inactivate its growth-promoting function. The mere fact that the cell recruits two different pathways to process Claspin during apoptosis suggests that it is an important target for inactivation.
We have shown that the fate of Claspin in apoptosis is strikingly different to its fate in response to low levels of DNA damage. Apoptosis-inducing DNA damage leads to degradation of Claspin, whereas in the presence of low levels of DNA damage, compatible with cell survival, Claspin is stabilised. We propose a model in which, upon DNA damage, Claspin is initially stable and able to activate Chk1. However, when the DNA damage is too severe to be repaired, the cells initiate apoptosis and Claspin is degraded by caspases and the proteasome (Figure 6). The degradation of Claspin abolishes the activation of Chk1 and also further promotes apoptosis. The involvement of two pathways in the degradation of Claspin could be a mechanism to speed up the inactivation of Claspin such that the proposed dominant-negative activity of caspase cleavage fragments18 would be more potent if the amount of full-length Claspin is decreased, or alternatively it could be a ‘fail-safe’ mechanism to ensure its inactivation during apoptosis even if one of the degradation pathways is incapacitated for some reason.

The inactivation of the DNA damage checkpoint pathways in apoptosis. In response to low levels of DNA damage (left), the PI3-like kinases ATM and ATR are activated, and with the help of the adaptor proteins BRCA1 and Claspin, they phosphorylate Chk2 and Chk1, and signal to the cell cycle progression machinery to stop. During apoptosis (right), the DNA damage checkpoint pathways are inactivated. Signalling to Chk2 is inhibited by caspase cleavage of ATM2 and BRCA1,37 whereas signalling to Chk1 is inhibited by caspase cleavage and proteasomal degradation of Claspin
Materials and Methods
Reagents and antibodies
All reagents were obtained from Sigma unless otherwise stated. To generate antibodies against the Nt of Claspin, the sequence encoding amino acids 85–248 (numbering according to AF297866) was cloned in frame with an N-terminal 6xHis tag in the pET-28 vector (Novagen). For antibodies against the Ct of Claspin, we used a construct encoding amino acids 1006–1340 of Claspin fused to GST, which was a gift from Dr. Junjie Chen (Mayo Clinic, MN, USA). The N- and C-terminal fragments were expressed in bacteria and purified according to the manufacturer's instructions with either Ni-NTA resin (Qiagen) or glutathione-sepharose beads (Amersham), respectively. Polyclonal sera were raised against these polypeptides in rabbits, and then affinity purified. Mouse anti-PARP1 antibody was from Serotech. Mouse anti-HA antibody was purified from the culture medium of 12CA5 hybridoma cells. HRP-conjugated secondary antibodies were obtained from Jackson ImmunoResearch Laboratories.
Induction of apoptosis
HL60 cells were maintained at a density of 200 000–600 000 cells/ml. Apoptosis was induced by the addition of 68 μM etoposide to the culture medium. For immunoblotting, cells lysed in urea/SDS buffer (6 M urea, 1% SDS, 50 mM Tris, pH 8 and 150 mM NaCl) and sonicated on ice for 10 s at 80% amplitude using a UP100H sonicator (Dr. Hielscher GmbH). The samples were analysed by SDS-PAGE followed by immunoblotting. For fluorescence-activated cell sorting (FACS) analysis, the cells were fixed with 70% ethanol, then stained with 20 μg/ml propidium iodide in phosphate-buffered saline containing 1% tween and 40 μg/ml ribonuclease A. Cell cycle profiles were analysed using an EPICS XL-MCL (Beckman Coulter) flow cytometer. The caspase inhibitor ZVAD-fmk (Alexis Biochemicals), and the proteasome inhibitors MG132 and epoxomicin, were added to cells at a final concentration of 40, 10 and 5 μM, respectively.
Apoptosis was induced in HeLa cells by addition of 30 ng/ml TNFα (Immunotools) and 10 μg/ml CHX to the medium. Alternatively, the cells were irradiated with 300–600 J/m2 of UV light.
In vitro caspase assay
Fragments of Claspin cloned as fusions to GST33 were a kind gift from Dr. Junjie Chen (Mayo Clinic, MN, USA). Once expressed in bacteria, the GST fusion proteins were purified using glutathione sepharose beads, and equal amounts of protein were incubated in a final volume of 30 μl in the presence or absence of recombinant caspase 3, 6 or 7, purified as described previously.34 The reactions were carried out in caspase assay buffer35 for 30 min at 37°C and then stopped by addition of Laemmli sample buffer and boiling. The fragments were analysed on 11% SDS-PAGE gels and then detected by silver staining.36
Mass spectrometry
The caspase cleavage assay was scaled up and analysed on a Coomassie stained SDS-PAGE gel. Heat-inactivated caspase 3 was included in the negative control samples. The uncut protein as well as bands that appeared in the cleaved sample but not in the negative control were processed by in gel trypsin digestion using a Montage In-Gel Digest kit (Millipore). Mass spectra were obtained using an Autoflex MALDI-TOF mass spectrometer (Bruker). Peptide mass fingerprints of Claspin and GST were identified by searching the Swissprot and NCBI protein databases with the Mascot software (Matrix Science, UK). To identify the exact cleavage site, the peptide masses obtained from each band in the gel were compared with virtual digests performed with MS-digest (http://prospector.ucsf.edu/ucsfhtml4.0/msdigest.htm).
Calpain assay
Calpain activity was induced in HL60 cells by addition of Ionomycin to the culture medium at a final concentration of 10 μM. When indicated, 25 μM of the Calpain inhibitor ALLM was added to the culture medium 15 min before Ionomycin addition. Cells were collected 90 min after Ionomycin addition and analysed by immunoblotting.
Expression of recombinant Claspin in mammalian cells
The pcDNA3/HA-Claspin plasmid was a kind gift from Dr. PM Reaper (Vertex Pharmaceuticals, UK). Claspin(1–260)-GFP was created by cloning the region coding for amino acids 1–260 of Claspin into the SalI and BamHI sites in the pEGFP-N1 expression vector (Invitrogen). Mutation of Asp25, Asp1072, Ser30 and Ser34 to Ala was carried out by site-directed mutagenesis, and confirmed by sequencing. BamHI–XhoI fragments of pcDNA3-Claspin wild type, 2D (D25/1072A) and 2DS (D25/1072A and S30/34A) were cloned into BglII–SalI-digested pEGFP-C1 for expression as EGFP fusion proteins.
HeLa cells were transfected using the calcium phosphate method. Two days after transfection apoptosis was induced with TNFα+CHX or UV as described above.
SiRNA
Three siRNA duplexes targeted at distinct sequences in Claspin (#3: CCUUGCUUAGAGCUGAGUCdTdT, #7: GGAAAGAAAGGCAGCCAGAdTdT and #8: GCACAUACAUGAUAAAGAAdTdT) and control siRNA duplexes targeted at GFP (CACUUGUCACUACUUUCUCdTdT) and luciferase (CGUACGCGGAAUACUUCGAdTdT) were obtained from Dharmacon Research.
Two rounds of transfection of U2OS cells with the siRNA oligos using oligofectamine (Invitrogen) were performed on consecutive days. To determine the degree of Claspin downregulation by siRNA, cells were collected 2 days after the second transfection, lysed and analysed by immunoblotting as above.
Abbreviations
- ATM:
-
ataxia telangiectasia mutated
- ATR:
-
ATM- and Rad3-related
- RNAi:
-
RNA interference
- siRNA:
-
short interfering RNA
- TNFα:
-
tumour necrosis factor alpha
- GST:
-
glutathione-S-transferase
- EGFP:
-
enhanced green fluorescent protein
- CHX:
-
cycloheximide
- Nt:
-
N-terminus
- Ct:
-
C-terminus
- FACS:
-
fluorescence-activated cell sorting
- HU:
-
hydroxyurea
References
Fischer U, Janicke RU, Schulze-Osthoff K . Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ 2003; 10: 76–100.
Smith GC, d'Adda di Fagagna F, Lakin ND, Jackson SP . Cleavage and inactivation of ATM during apoptosis. Mol Cell Biol 1999; 19: 6076–6084.
Freire R, d’Adda Di Fagagna F, Wu L, Pedrazzi G, Stagljar I, Hickson ID et al. Cleavage of the Bloom's syndrome gene product during apoptosis by caspase-3 results in an impaired interaction with topoisomerase IIIalpha. Nucleic Acids Res 2001; 29: 3172–3180.
Casciola-Rosen LA, Anhalt GJ, Rosen A . DNA-dependent protein kinase is one of a subset of autoantigens specifically cleaved early during apoptosis. J Exp Med 1995; 182: 1625–1634.
Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J et al. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol 2006; 8: 37–45.
Zou L, Elledge SJ . Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003; 300: 1542–1548.
Bao S, Lu T, Wang X, Zheng H, Wang LE, Wei Q et al. Disruption of the Rad9/Rad1/Hus1 (9-1-1) complex leads to checkpoint signaling and replication defects. Oncogene 2004; 23: 5586–5593.
Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 2000; 14: 1448–1459.
Melo J, Toczyski D . A unified view of the DNA-damage checkpoint. Curr Opin Cell Biol 2002; 14: 237–245.
Kumagai A, Dunphy WG . Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol Cell 2000; 6: 839–849.
Yoo HY, Jeong SY, Dunphy WG . Site-specific phosphorylation of a checkpoint mediator protein controls its responses to different DNA structures. Genes Dev 2006; 20: 772–783.
Kumagai A, Kim SM, Dunphy WG . Claspin and the activated form of ATR-ATRIP collaborate in the activation of Chk1. J Biol Chem 2004; 279: 49599–49608.
Osborn AJ, Elledge SJ . Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev 2003; 17: 1755–1767.
Lee J, Kumagai A, Dunphy WG . Claspin, a Chk1-regulatory protein, monitors DNA replication on chromatin independently of RPA, ATR, and Rad17. Mol Cell 2003; 11: 329–340.
Lin SY, Li K, Stewart GS, Elledge SJ . Human Claspin works with BRCA1 to both positively and negatively regulate cell proliferation. Proc Natl Acad Sci USA 2004; 101: 6484–6489.
Alexandre S, Rast C, Nguyen-Ba G, Poirier GG, Vasseur P . PARP degradation in apoptotic Syrian hamster embryo (SHE) cells compared to HL60 cell line. Biochimie 2000; 82: 1115–1122.
Lee MW, Hirai I, Wang HG . Caspase-3-mediated cleavage of Rad9 during apoptosis. Oncogene 2003; 22: 6340–6346.
Clarke CA, Bennett LN, Clarke PR . Cleavage of claspin by caspase-7 during apoptosis inhibits the Chk1 pathway. J Biol Chem 2005; 280: 35337–35345.
Groll M, Huber R . Inhibitors of the eukaryotic 20S proteasome core particle: a structural approach. Biochim Biophys Acta 2004; 1695: 33–44.
Butts BD, Hudson HR, Linseman DA, Le SS, Ryan KR, Bouchard RJ et al. Proteasome inhibition elicits a biphasic effect on neuronal apoptosis via differential regulation of pro-survival and pro-apoptotic transcription factors. Mol Cell Neurosci 2005; 30: 279–289.
Shah GM, Shah RG, Poirier GG . Different cleavage pattern for poly(ADP-ribose) polymerase during necrosis and apoptosis in HL-60 cells. Biochem Biophys Res Commun 1996; 229: 838–844.
Mamely I, van Vugt MA, Smits VA, Semple JI, Lemmens B, Perrakis A et al. Polo-like kinase-1 controls proteasome-dependent degradation of claspin during checkpoint recovery. Curr Biol 2006; 16: 1950–1955.
Peschiaroli A, Dorrello NV, Guardavaccaro D, Venere M, Halazonetis T, Sherman NE et al. SCFbetaTrCP-mediated degradation of claspin regulates recovery from the DNA replication checkpoint response. Mol Cell 2006; 23: 319–329.
Mailand N, Bekker-Jensen S, Bartek J, Lukas J . Destruction of Claspin by SCFbetaTrCP restrains Chk1 activation and facilitates recovery from genotoxic stress. Mol Cell 2006; 23: 307–318.
Chini CC, Wood J, Chen J . Chk1 is required to maintain claspin stability. Oncogene 2006; 25: 4165–4171.
Suh J, Lee YA, Gwag BJ . Induction and attenuation of neuronal apoptosis by proteasome inhibitors in murine cortical cell cultures. J Neurochem 2005; 95: 684–694.
Demontis S, Rigo C, Piccinin S, Mizzau M, Sonego M, Fabris M et al. Twist is substrate for caspase cleavage and proteasome-mediated degradation. Cell Death Differ 2006; 13: 335–345.
Mollerup J, Berchtold MW . The co-chaperone p23 is degraded by caspases and the proteasome during apoptosis. FEBS Lett 2005; 579: 4187–4192.
Rathore N, Matta H, Chaudhary PM . An evolutionary conserved pathway of nuclear factor-kappaB activation involving caspase-mediated cleavage and N-end rule pathway-mediated degradation of IkappaBalpha. J Biol Chem 2004; 279: 39358–39365.
Varshavsky A . The N-end rule: functions, mysteries, uses. Proc Natl Acad Sci USA 1996; 93: 12142–12149.
Zhang D, Zaugg K, Mak TW, Elledge SJ . A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell 2006; 126: 529–542.
Bischof O, Galande S, Farzaneh F, Kohwi-Shigematsu T, Campisi J . Selective cleavage of BLM, the bloom syndrome protein, during apoptotic cell death. J Biol Chem 2001; 276: 12068–12075.
Chini CC, Chen J . Human claspin is required for replication checkpoint control. J Biol Chem 2003; 278: 30057–30062.
Stennicke HR, Salvesen GS . Caspases: preparation and characterization. Methods 1999; 17: 313–319.
Perera D, Freire R . Human spindle checkpoint kinase Bub1 is cleaved during apoptosis. Cell Death Differ 2005; 12: 827–830.
Morrissey JH . Silver stain for proteins in polyacrylamide gels: a modified procedure with enhanced uniform sensitivity. Anal Biochem 1981; 117: 307–310.
Zhan Q, Jin S, Ng B, Plisket J, Shangary S, Rathi A et al. Caspase-3 mediated cleavage of BRCA1 during UV-induced apoptosis. Oncogene 2002; 21: 5335–5345.
Acknowledgements
This work was supported by grants obtained by RF from the EU (HPRN-CT-2002-00240) and from the Spanish Ministry of Science and Technology (SAF2004-07856 and GEN2003-20243-C08-07). JIS was supported by a postdoctoral research training contract from the EU (HPRN-CT-2002-00240). VAJS is supported by the Dutch Cancer Society (2005-3412) and the Association for International Cancer Research (05-005). JRF is supported by a grant from Caja Canarias and La Laguna University (541C8). RF is currently supported by a FIS (Fondo Investigaciones Sanitarias) contract. We thank J Chen (Mayo Clinic, MN, USA) for the GST-Claspin fragments and WG Dunphy (California Institute of Technology, USA) for the full-length Claspin cDNA construct. We also thank Antonio Perez for assistance with cloning and Eva Zemlickova for preliminary experiments.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Edited by B Zhivotovsky
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Supplementary information
Rights and permissions
About this article
Cite this article
Semple, J., Smits, V., Fernaud, JR. et al. Cleavage and degradation of Claspin during apoptosis by caspases and the proteasome. Cell Death Differ 14, 1433–1442 (2007). https://doi.org/10.1038/sj.cdd.4402134
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4402134
Keywords
This article is cited by
-
Increased sensitivity to apoptosis upon endoplasmic reticulum stress-induced activation of the unfolded protein response in chemotherapy-resistant malignant pleural mesothelioma
British Journal of Cancer (2018)
-
PERK inhibits DNA replication during the Unfolded Protein Response via Claspin and Chk1
Oncogene (2017)
-
Antiangiogenic and antihepatocellular carcinoma activities of the Juniperus chinensis extract
BMC Complementary and Alternative Medicine (2016)
-
USP29 controls the stability of checkpoint adaptor Claspin by deubiquitination
Oncogene (2015)
-
The Small Chaperone Protein p23 and Its Cleaved Product p19 in Cellular Stress
Journal of Molecular Neuroscience (2012)
