
Abstract
In mammals and Drosophila, apoptotic caspases are under positive control of the CED-4-like proteins Apaf-1 and ARK, respectively. In an EMS-mutagenesis screen, we isolated 33 ark mutants as recessive suppressors of hid-induced apoptosis. The ark mutants are loss-of-function alleles characterized by reduced developmental apoptosis. Using the phenotypic series of these alleles, we identified helical domain I in the nucleotide oligomerization domain as critical for ARK's apoptotic activity. Interestingly, the WD40 region may also have an unanticipated positive requirement for the apoptotic activity of ARK. Considering structural information, we discuss the roles of these domains for assembly and activity of the ARK apoptosome, and propose that the WD40 region is anti-apoptotic in the absence of apoptotic signals, and pro-apoptotic in the presence of such signals. Furthermore, a defined null allele reveals that ark is required for most, but not all apoptosis suggesting the existence of an ARK-independent apoptotic pathway.
Similar content being viewed by others

Main
Programmed cell death or apoptosis is a naturally occurring process of cell suicide that plays crucial roles in the development, homeostasis, and defense of metazoans.1 Genetic studies in the nematode Caenorhabditis elegans have implicated CED-4-like genes as essential for apoptosis.2 Subsequently, the CED-4-like genes Apaf-1 (apoptotic protease activating factor-1) in mammals and ARK (Apaf-1-related killer, also known as Dark, D-Apaf-1 and Hac-1) in Drosophila were discovered.3, 4, 5, 6 CED-4-like proteins are characterized by the presence of a N-terminal caspase activation and recruitment domain (CARD), and a nucleotide oligomerization domain (NOD, also referred to as CED-4 homology domain and nucleotide-binding domain). Based on structural analysis of inactive, ADP-bound WD40-depleted Apaf-1, the NOD was divided into several distinct domains: α/β domain, helical domain I (HD1), winged-helix domain (WHD), and helical domain II (HD2).7 Apaf-1 and ARK also contain in their C-terminal half a series of WD40 repeats which are not found in CED-4 (reviewed in Cain et al.8). WD40 repeats are short ∼40 amino-acid motifs, often terminating in a Trp-Asp (WD) dipeptide, and are thought to form a circularized β-propeller structure. WD40-repeat containing proteins coordinate multi-protein complex assemblies, where the repeating units serve as a rigid scaffold for protein interactions.9
Among the CED-4-like proteins, Apaf-1 is best characterized both biochemically and structurally. Apaf-1 functions to multimerize and activate Caspase-9, an initiator caspase that triggers apoptosis when activated.8 The CARD of Apaf-1 is required for homotypic interaction with the CARD of Caspase-9.10 However, inactive Apaf-1 is a globular monomer that is kept in an auto-inhibited state by complex formation between the CARD and the WD40 repeats,11 thus preventing CARD–CARD interactions. Hence, the WD40 repeats have an inhibitory function for Apaf-1 activity in the absence of apoptotic signals. Consistently, deletion of the WD40 repeats results in the activation of Apaf-1.12, 13 In response to apoptotic stimuli, cytochrome c is released from mitochondria and binds to the WD40 repeats, thus displacing the CARD from WD40 inhibition. This displacement allows binding of dATP/ATP to the NOD, which then promotes assembly of seven Apaf-1/cytochrome c protein complexes to form the apoptosome, a wheel-like particle with seven Y-shaped spokes and a hub.11 The CARD domains are arranged in the center of the apoptosome. The α/β domain, HD1, and WHD of the NOD form the hub which encircles the CARD ring, whereas HD2 links the hub to the WD40 region. The WD40 region form the spokes containing two β-propellers with seven and six WD40 repeats, respectively, which bind cytochrome c.11 In the final step, Caspase-9 is tethered to the hub of the apoptosome through CARD–CARD interactions with Apaf-1.10, 11 The active apoptosome in turn activates Caspase-3, an effector caspase.
Drosophila ARK has a similar domain architecture compared to Apaf-1 suggesting the possible conservation of this function in Drosophila. In addition, ARK possesses a C-terminal extension of ∼180 residues which is not found in Apaf-1.14 ARK has also been shown to physically interact with the initiator caspases DRONC and DREDD,4, 5, 15 although only DRONC is considered to be a caspase-9 homolog in Drosophila.16 The 3D structure of ARK has recently been demonstrated by electron cryo-microscopy at 18.8 Å resolution.14 The overall structure of the ARK apoptosome is similar to the Apaf-1 apoptosome. It is a ring-like particle with a central CARD ring, a hub comprised of the α/β domain, HD1, and WHD of the NOD, and spokes containing the WD40 region forming two β-propellers with eight (β8 propeller) and six (β6 propeller) WD40 repeats, respectively.14 Both apoptosomes require dATP for assembly. However, there are also a few notable differences. The ARK apoptosome is comprised of eight subunits, and two of these rings associate face-to-face to form a double apoptosome. In contrast, the Apaf-1 apoptosome is a single ring composed of seven subunits. The connection (termed C1) between the two rings in the ARK double apoptosome appears to be mediated by HD1 of the NOD.14 Interestingly, Cytochrome c is not required for assembly of and was not found in the ARK apoptosome, even when provided in excess,14 consistent with reports which showed that cytochrome c is not required for the induction of apoptosis in Drosophila.17, 18, 19 In contrast to Apaf-1, due to the presence of eight densely packed subunits in the ARK apoptosome, a connection (C2) is established between the β6 and β8 propellers in the WD40 region of adjacent subunits.14 This connection may stabilize the conformation of the WD40 region.
ark is required for cell death induced by the RHG genes reaper, hid and grim.4, 5 The primary pro-apoptotic function of the RHG proteins is to liberate caspases, mainly DRONC, from DIAP1 (Drosophila inhibitor of apoptosis protein 1) inhibition.20 It has been proposed that free DRONC is constitutively activated through binding to ARK.21 Thus, whereas in mammals activation of Caspase-9 by Apaf-1 and cytochrome c is the major control element for apoptosis, it emerges that Drosophila employs release of DRONC from DIAP1 inhibition as the main means of PCD regulation (reviewed by Cashio et al.22). The RHG proteins are fully dedicated to overcome DIAP1-mediated DRONC inhibition.
Here, we report the isolation and identification of 33 EMS-induced loss-of-function alleles of ark as recessive suppressors of hid-induced apoptosis. This is the first unbiased generation and analysis of ark/apaf-1 mutants in any organism. Interestingly, whereas nonsense mutations are randomly distributed throughout the ark gene, missense mutations cluster in the HD1 of the NOD and in the WD40 region. Surprisingly, missense mutations in the CARD were not recovered. Using the phenotypic series of these alleles and considering structural information, we discuss models about the roles of individual domains for assembly and activity of the ARK apoptosome, and conclude that the WD40 region may also have an unanticipated positive requirement for the apoptotic activity of ARK. Furthermore, a defined null allele suggests the existence of an ARK-independent cell death pathway in Drosophila. We also show that compensatory proliferation induced by hid, and inhibition of apoptosis by ark mutant clones accounts for the strong suppression of GMR-hid.
Results
Isolation and identification of ark mutants
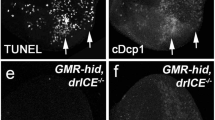
Ectopic expression of the cell death-inducing gene hid under control of the eye-specific GMR enhancer (GMR-hid) results in a strong eye ablation phenotype compared to wild-type (Figure 1a and b).23 In order to isolate mutants in ark, we took advantage of the GMR-hid eye-ablation phenotype and conducted an EMS-mutagenesis screen using the previously described GheF (G MR-h id e y- F LP) method.24 In this method, we screened for suppressors of GMR-hid in homozygous mutant eye clones obtained by ey-FLP/FRT-mediated recombination in otherwise heterozygous animals (for details see Xu et al.24). Among 32 000 F1 progeny, we isolated 33 mutants as suppressors of the GMR-hid eye phenotype (Figure 1c–i). Inter se complementation analysis showed that these mutations affect the same genetic function. We determined by recombination mapping, by complementation analysis with Df(2R)ED2751 (deleting ark) and with the hypomorphic arkCD4 allele,5 and by DNA sequencing analysis that these suppressors are allelic to ark. Furthermore, although these mutants are pupal-lethal, it is possible to obtain homozygous viable ark mutant flies using certain allelic combinations, for example arkL46/arkP46 These flies exhibit phenotypes similar to the ones published for the hypomorphic arkCD4 allele, including melanotic tumors and abnormal wings4, 5 (data not shown).

Phenotypic series of ark mutant alleles as recessive suppressors of GMR-hid. (a) Wild-type eye. (b) The unsuppressed GMR-hid ey-Flp (GheF) eye ablation phenotype in wild-type background. (c–i) Representative examples of the suppression of the GheF eye ablation phenotype in ark mutant mosaics. The exact genotype of each of these flies: GMR-hid ey-FLP (GheF); FRT42D arkx/FRT42D P[w+], where x denotes the allele indicated in each panel
The level of suppression of the GMR-hid eye phenotype by the ark mutants varies from weak to strong (Figure 1; Table 1). For example, arkS7, arkP56, and arkD3 restore the GMR-hid eye phenotype almost completely back to wild-type size, whereas arkJ7 has a moderately strong effect on GMR-hid. Other alleles such as arkM20 weakly rescue the GMR-hid eye phenotype (Figure 1; for a complete list see Table 1). These differences suggest that the ark alleles isolated in the GheF screen represent an allelic series, ranging from weak to strong alleles.
Molecular analysis of ark mutants
By DNA sequencing analysis, we identified missense and nonsense mutations in 21 of the EMS-induced ark alleles. Figure 2 indicates the relative positions of these mutations in the ARK protein. Table 1 lists the molecular lesions associated with the ark alleles and the strength of the suppression of GMR-hid.

Relative location of the molecular lesions associated with the ark alleles. Schematic outline of the ARK protein, depicting the CARD, NOD and WD40 repeats. The NOD is further divided into α/β-domain, helical domain 1 (HD1) and winged-helix domain (WHD).14 Helical domain II (HD2) comprises the arm linking the NOD with the WD40 region.14 The relative location of the missense mutations and the allele names are indicated by arrows above the outline, nonsense mutations are below. Asterisks denote alleles for which two mutations were identified. CTD – C-terminal domain. Not drawn to scale
From the location of these mutations we can draw some interesting conclusions. The nonsense mutations are relatively randomly distributed throughout the gene (Figure 2). In contrast, the missense mutations cluster in a stretch of 53 residues in the NOD, and in the first and tenth WD40 repeat (we refer to the WD40 repeats as defined by Kanuka et al.4). Interestingly, missense mutations were not recovered in the CARD of any of the 33 ark alleles. This is a surprising observation, as the CARD has been implicated for interaction with and activation of DRONC, and is believed to be critical for the apoptotic activity of ARK (see Discussion for possible interpretations). However, one nonsense mutation was recovered in the CARD of arkH16 which has a premature termination codon at position 42 (Figure 2; Table 1). This truncation likely disrupts the interaction with DRONC, and because the NOD and the WD40 region are also deleted, arkH16 is unlikely to produce a functional ARK protein, and thus is a defined null allele. As the null allele arkH16 is a strong suppressor of GMR-hid, and all ark alleles were recovered as suppressors of GMR-hid, we conclude that the ark alleles isolated in this study represent loss-of-function alleles. This is also confirmed by specific cell death assays, presented below.
Eight of the missense mutations are clustered in a small region of the NOD, ranging from residues 316 to 369 (Figure 2; Table 1). We refer to these mutants collectively as arkNOD alleles. One of the arkNOD mutations, Cys346 → Tyr, was independently recovered in three alleles (arkA24, arkG8, arkL46; see Materials and Methods), suggesting some degree of saturation in the mutagenesis. The clustering of these mutations implies that this part of the NOD plays a critical role for ARK function and activity (see Discussion).
Finally, we isolated missense and nonsense mutations in the WD40 repeats of ark (Figure 2; Table 1), collectively referred to as arkWD40. arkB2 and arkD3 carry missense mutations in the first WD40 repeat, whereas arkE4 and arkN5 affect the tenth WD40 repeat. These alleles as well as the ones which induce premature termination codons in the WD40 region (arkG29, arkG15, arkB5, arkJ7) were recovered as suppressors of the GMR-hid phenotype (Figure 1) suggesting that they are loss-of-function alleles. This is a surprising finding because based on analysis of Apaf-1 it is generally believed that the WD40 repeats are auto-inhibitory.12, 13 Deletion of the WD40 repeats is therefore expected to induce a gain-of-function character to the ARK protein. However, the recovery of arkWD40 mutants as suppressors of GMR-hid suggests that they also might have a positive role for the regulation of ARK activity.
In summary, we have isolated mutations in ark as suppressors of the GMR-hid eye phenotype. Our genetic and molecular analysis suggests that arkH16 is a complete loss-of-function allele, whereas a cluster of mutations in the NOD suggests an important role of this domain for control of ARK activity. However, most important is the surprising finding that arkWD40 mutations were recovered as suppressors of GMR-hid, that is, as loss-of-function alleles, implying a previously unknown function of the WD40 repeats for control of ARK activity (see Discussion).
ark mutants block most, but not all, cell death in the embryo
All of the ark mutants were recovered as suppressors of GMR-hid (Figure 1, Table 1). This suggests that they suppress the ectopic cell death caused by GMR-hid in eye imaginal discs. Indeed, TUNEL analysis of GMR-hid eye imaginal discs demonstrate that these mutants suppress cell death caused by GMR-hid (data not shown; see also Figure 5j). However, we wished to determine the role of ark for normal developmental cell death. We analyzed this role of ark for embryonic cell death. Owing to the large maternal contribution, we obtained ark mutant embryos by inducing germline clones in otherwise heterozygous females.25 ark mutant embryos were analyzed by Acridine Orange (AO) which labels specifically dying cells.26 As CED-4-like proteins act as scaffolding molecules for the activation of caspases8 we also tested whether caspase activation is affected in ark mutant embryos by performing antibody labelings with anti-cleaved caspase-3 antibody.

GMR-hid induces compensatory proliferation, and ark clones protect the newly formed cells from hid-induced apoptosis. (a) The suppressed eye of GMR-hidw− is almost entirely composed of ark mutant tissue, phenotypically marked by absence of red eye pigment (white). Genotype: ey-FLP; FRT42D arkG8/FRT42D P[w+]; GMR-hidw−. (b) Adult eye of an ey-FLP-induced mosaic of arkG8. The ark mutant tissue is marked by absence of red eye pigment (white); the twin spots (ts) and heterozygous cells are marked by red eye pigment. Genotype: ey-FLP; FRT42D arkG8/FRT42D P[w+]. (c) Wild-type eye-antennal disc labeled for BrdU as proliferation marker. MF, morphogenetic furrow; SMW, second mitotic wave. (d) Wild-type eye-antennal disc labeled with anti-Hid antibody. (e) GMR-hid eye-antennal disc labeled with anti-Hid antibody. The bracket indicates the expression domain of GMR-driven hid expression posterior to the morphogenetic furrow. (f–h) GMR-hid eye-antennal disc labeled with BrdU (f) and anti-cleaved caspase-3 antibody (caspase-3*) (g). (h) is the overlay of (f) and (g). Arrows in (f) indicate the morphogenetic furrow (MF), the second mitotic wave (SMW) and the zone of compensatory proliferation (ZCP). The arrows in (g) indicate hid-induced cell death overlapping with the SMW and slightly posterior to the ZCP. (i–k) GMR-hid eye-antennal disc containing ey-FLP-induced ark clones labeled for BrdU (i) and anti-cleaved caspase-3 antibody (caspase-3*) (j). (k) is the overlay of (i) and (j). White lines in (j) and (k) mark ark clones. Genotype: GheF; FRT42D arkG8/FRT42D P[w+]
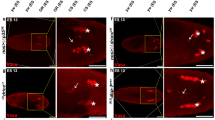
The null allele arkH16 allows determining the null phenotype of ark. Before the initiation of this project this experiment was not possible because only hypomorphic alleles of ark were available.4, 5, 6 To remove the strong maternal contribution, germline clones (see Materials and Methods) were analyzed for arkH16 and the ark alleles presented in Figure 3. In arkH16 mutant embryos, AO-positive cell death and caspase-3-like activity are significantly reduced, but not completely blocked (see arrow in Figure 3) suggesting that ark+ is required for most, but not all, embryonic cell death. An ARK-independent apoptotic pathway exists in the embryo.

ark mutant embryos are characterized by reduced rates of apoptosis. (a) Acridine orange labelings of wild-type (top left) and several ark alleles indicated in the panels. White arrows point to the dying cells in the head region of ark mutant embryos. All embryos in this panel and panel B were obtained from germline clone (see Materials and Methods). (b) Anti-cleaved caspase-3 labelings of wild-type (top left) and several ark alleles indicated in the panels
Eight additional ark alleles affecting the NOD and the WD40 repeats were selected for further analysis of embryonic cell death. Compared to wild-type embryos, AO-positive cell death and caspase-3-like activity is strongly reduced in all ark mutants (Figure 3), consistent with an essential role of ark for embryonic cell death. Interestingly, the mutants show a general decrease in the extent of cell death in the trunk region of the embryo, whereas some cell death in the head region remains (Figure 3a, arrows). arkNOD alleles (arkS7) are characterized by strong reduction of embryonic cell death (Figure 3), although it is not as strong as observed for arkH16 Similar results were obtained for arkC1 and arkG8 (data not shown). These findings underscore the importance of the NOD for ark activity and cell death.
arkWD40 alleles (arkE4, arkD3, and arkB5) cause significant reduction of embryonic cell death (Figure 3). Consistent with conclusions made earlier, this analysis suggests that these mutants are loss-of-function alleles. Even arkJ7, which has a premature termination codon at position 1282 deleting the unique C-terminal domain, displays reduced rates of embryonic cell death (Figure 3). This observation suggests that also the C-terminal domain is required for ARK's apoptotic activity.
Of particular interest is the analysis of arkP56. This allele contains a premature termination codon at position 499 in the linker between the NOD and the WD40 region (Figure 2; Table 1), thus completely truncating the WD40 region, but leaving the CARD and NOD intact. This mutant mimics a putative splice variant of ark which produces a smaller isoform of 531 residues.4 As observed for other arkWD40 mutants, AO-positive cell death and caspase-3-like activity are significantly reduced in arkP56 embryos (Figure 3). However, compared to the null mutant arkH16, arkP56 still retains some weak apoptotic activity. In conclusion, this analysis establishes that ARK requires the WD40 repeats for its apoptotic activity (see Discussion).
Mutants affecting the WD40 repeats contain extra cells
The reduction of the global cell death pattern of arkWD40 mutants suggests a positive requirement of the WD40 region for cell death. However, to further confirm this conclusion we wished to determine this requirement in a well-defined cellular model system for apoptosis. The midline glia (MG) are transient cells in the developing central nervous system, that are required for the separation and ensheathment of commissural axon tracts.27 During mid-embryogenesis, approximately 10 MG per segment have been formed. Subsequent to the establishment of commissure morphology, a subset of the MG cells undergo cell death, leaving about three ensheathing MG per segment by the end of embryogenesis at stage 17 (Figure 4a). Embryos of the hypomorphic arkCD4 allele have been shown to contain additional MG.28 Thus, MG apoptosis is an ideal model to address the role of the WD40 repeats at a cellular level. We tested arkE4 and arkJ7, containing a point mutation in the 10th WD40 repeat and deleting the C-terminal domain, respectively (Figure 2), and compared MG survival of these mutants to the null allele arkH16. arkH16 mutant embryos contain on average 10.7 MG per segment (n=7) (Figure 4b). This number is similar to dronc mutants and H99 mutants that delete the pro-apoptotic genes reaper, hid and grim.24 Similarly, the WD40 mutants arkE4 and arkJ7 contain on average 9.1 and 8.3 MG per segment (n=7) at stage 17 (Figure 4c and d). Thus, consistent with the AO- and anti-cleaved caspase-3- labelings (Figure 3), ark mutant embryos affecting the WD40 region block developmental cell death.

arkWD40 mutants contain additional midline glia (MG) cells and produce detectable proteins. (a) Stage 17 wild-type embryos contain on average 3 MG per segment. (b) Stage 17 arkH16 embryos contain on average 10.7 MG per segment (N=7). (c) Stage 17 arkE4 embryos contain on average 9.1 MG per segment (N=7). (d) Stage 17 arkJ7 embryos contain on average 8.3 MG per segment (N=7). (e) Immunoblot analysis of head extracts probed with anti-ARK antibody.29 Relative molecular weight (Mr) in kDa is indicated on the left. Note P35 does not denote an ark allele, but encodes a caspase inhibitor. An unspecific band indicated by * serves as loading control. The genotypes of the head extracts are as follows: lane 1, FRT42D P[y+] (wild-type); lane 2, GheF; FRT42D arkP56/FRT42D P[w+]; lane 3, GheF; FRT42D arkD3/FRT42D P[w+]; lane 4, GheF; FRT42D arkE4/FRT42D P[w+]; lane 5, Ghef; GMR-P35
Interestingly, MG survival in the hypomorphic arkJ7 allele is not identical in every segment. The segments in the trunk region contain more MG compared to segments in the head and posterior abdomen (see enlargement in Figure 4d). This observation is mirror-imaged by the AO and anti-cleaved caspase-3 labelings of arkJ7 embryos in which cell death is high in the head and posterior region of the embryo but largely blocked in the trunk (Figure 3). These analyses reveal that cells – depending on their location – require different minimum ark activity to undergo apoptosis. This differential requirement is also visible in stronger alleles where AO-positive cell death is usually strongly reduced in the trunk region of the mutant embryos, but only weakly affected in the head region (Figure 3).
arkWD40 alleles with missense mutations produce stable ARK proteins
The genetic analysis presented above establishes that the arkWD40 mutants are loss-of-function alleles. One possible explanation for this genetic behavior might be that arkWD40 alleles affect the stability of the mutant ARK proteins. Thus, to address this possibility we performed immunoblot analysis. It has previously been shown that ARK protein is undetectable under non-apoptotic conditions29 (Figure 4e, lane 1). However, the same study29 showed that under apoptotic conditions ARK immunoreactivity was increased. We confirmed this finding by analyzing ARK protein in head extracts from GMR-hid animals that co-express the caspase inhibitor P35 (GMR-P35) in otherwise ark+ background. ARK protein (∼170 kDa) is detectable in GMR-hid/GMR-P35 animals (Figure 4e, lane 5). As expression of GMR-P35 results in a similar rescue of the GMR-hid phenotype23 compared to ark mosaics, analyzing mutant ARK protein in head extracts from GMR-hid, ark mosaics provides a convenient assay. The anti-ARK antibody was raised against an epitope in the C-terminus,29 preventing us from analyzing truncation mutants for protein stability. However, we were able to assay for ARK protein stability of the missense mutations arkD3 and arkE4 and as negative control of arkP56 which contains a premature stop codon at residue 499 and thus has lost the epitope recognized by the ARK antibody.29 ARKD3 and ARKE4 proteins are detectable in head extracts of GMR-hid ark mosaics (Figure 4e, lanes 3 and 4) whereas ARKP56 protein is not, demonstrating specificity of the antibody (lane 2). Thus, this analysis suggests that the loss-of-function phenotypes of arkD3 and arkE4 are not caused by instability of the mutant proteins.
Compensatory proliferation and suppression of apoptosis accounts for rescue of GMR-hid by ark
Finally, we addressed the question how ark mutant clones suppress the GMR-hid phenotype. Obviously, through suppression of GMR-hid-induced cell death. However, we obtained evidence that suppression of cell death alone is not fully sufficient for the strong rescue of the GMR-hid eye phenotype by ark mutant clones. This evidence stems from a comparative clonal analysis of ark mosaics in the presence and absence of GMR-hid. First, we determined the genetic identity of the rescued tissue of GMR-hid by clonal analysis based on red/white pigment selection in the eye using a GMR-hidw− transgene. (Note that the GMR-hid transgene used in Figure 1 is marked with P[w+] which provides red eye pigment, and does not allow a clonal analysis based on red/white pigment selection). As shown in Figure 5a, the rescued eye tissue of GMR-hidw− is genetically ark−, which is marked by absence of red eye pigment (white). Thus, consistent with the expectation, the strongly suppressed GMR-hidw− eye phenotype is due to ark mutant tissue. The heterozygous and wild-type tissues are eliminated by GMR-hid-induced cell death and replaced by ark mutant tissue.
However, when this clonal analysis was done in the absence of GMR-hid, the size of the ark mutant clones (white area in Figure 5b) is equal or smaller than the wild-type twin spots and heterozygous cells (red eye pigment), and makes up at best 50% of the total eye size (Figure 5b). However, ey-FLP generates ark mutant clones and wild-type twin spots before GMR-driven hid expression. Thus, because the wild-type and heterozygous cells are eliminated by GMR-hid, and because the ark mutant clones make up at the most 50% of the eye tissue by the time GMR expresses hid, the rescued adult GMR-hid eye would be expected to have approximately 50% of the wild-type size. However, in reality, we observe a rescue of up to 95% of the wild-type size in ark mosaics (Figure 1). Thus, it appears that inhibition of GMR-hid-induced cell death does not fully account for the strong rescue of GMR-hid in ark mosaics. Other mechanisms contribute to the suppressed eye phenotype.
We considered ectopic cell proliferation as a contributing factor to the suppression of GMR-hid in ark clones. We labeled third-instar eye imaginal discs with BrdU as proliferation marker. In wild-type eye imaginal discs, posterior to the morphogenetic furrow (MF), a wave of proliferating cells, referred to as second mitotic wave (SMW), is detectable (Figure 5c). The GMR enhancer induces hid expression posterior to the MF as visualized by α-Hid antibody labelings (see bracket in Figure 5e). Consistent with the expression of hid posterior to the MF in GMR-hid discs, the second mitotic wave overlaps with a zone of caspase-3-positive cells indicating that these cells undergo apoptosis (Figure 5f–h). Unexpectedly, GMR-hid eye imaginal discs contain an additional zone of proliferating cells posterior to the second mitotic wave (Figure 5f; labeled ZCP, see below) which was not found in wild-type discs (Figure 5c). Thus, GMR-hid triggers ectopic cell proliferation. Ectopic cell proliferation in dying tissue referred to as compensatory proliferation is not unprecedented. Recently, it has been demonstrated that dying cells can induce compensatory proliferation in response to X-ray treatment or in diap1 mutant clones.30, 31, 32 However, in these studies compensatory proliferation could only be identified by expression of the caspase inhibitor P35 to prolong the life of the dying cell. In our experiment, P35 was not present suggesting that compensatory proliferation can be detected even without inhibition of cell death by P35. Furthermore, Ryoo et al. (2004)30 and Huh et al. (2004)31 implicated wingless (wg) as the signaling mechanism for compensatory proliferation. We do not detect wg signaling in GMR-hid eye discs (data not shown). Thus, a different mechanism appears to induce compensatory proliferation in GMR-hid eye discs. However, if a zone of compensatory proliferation (ZCP) in GMR-hid eye discs exists, why is the resulting adult eye still small (Figure 1a)? By anti-cleaved caspase-3 labeling we detect a zone of dying cells immediately posterior to the ZCP, presumably eliminating the newly formed cells (Figure 5g and h). Thus, the small eye phenotype of GMR-hid is caused by cell death in the SMW as well as in the ZCP.
GMR-hid eye discs containing ark clones have a similar proliferation pattern as GMR-hid discs without ark clones (Figure 5i). However, as expected, caspase-3-like activity is significantly reduced in ark clones both in the SMW and ZCP (Figure 5j and k; white lines indicate clonal boundaries). In conclusion, this result implies that the suppression of cell death in the SMW, but in particular in the ZCP, contributes to the expansion of ark mutant tissue, presumably resulting in the strong suppression of GMR-hid by ark clones in the adult eye.
Discussion
We have isolated 33 mutants of ark, the Apaf-1 homologue in Drosophila, as suppressors of GMR-hid-induced apoptosis. The strong suppression of GMR-hid by the ark mutants is the result of compensatory proliferation and inhibition of apoptosis. Interestingly, in a heterozygous condition these alleles do not suppress the GMR-hid eye-ablation phenotype (data not shown), suggesting that a 50% reduction in the gene dose of ark is not sufficient to dominantly modify GMR-hid. This conclusion implies that ark is produced in excess over its genetic requirement. Overexpression of ark (GMR-Gal4 UAS-ark) also does not cause a detectable phenotype (data not shown). Thus, the cell can afford to produce excessive amounts of ARK without damaging consequences. This conclusion suggests that the activation of ARK is under strict genetic control.
The molecular and genetic characterization of the ark mutants is consistent with an essential role of ark for developmental apoptosis in Drosophila. Before the initiation of this project, hypomorphic ark alleles were reported which were caused by P-element insertions.4, 5, 6 Although these P-alleles provided important information about the role of ark for developmental cell death, the determination of the null phenotype was not possible. The phenotypic series of the ark alleles presented in this study not only allowed determining the null phenotype of ark, but also dissecting genetically the role of the individual domains of ARK.
ARK is required for most, but not all, embryonic cell death
It has previously been shown that ARK interacts physically with and activates the apical caspase DRONC, presumably through formation of an apoptosome-like complex.4, 5, 15, 18 This interaction requires the CARD. The arkH16 allele carries a premature termination codon at position 42 in the CARD, which likely disrupts the interaction with DRONC, and is a putative null allele. Although arkH16 mutant embryos display significantly reduced cell death and caspase activity, cell death was not completely blocked. Using AO and cleaved caspase-3 antibody labelings as two independent assays, we consistently detected a few cells that underwent cell death in arkH16 mutants. This observation suggests that ark is not required for all embryonic cell death. We reported recently a similar finding for null mutants of dronc, the putative caspase-9 homolog.24 These observations suggest the existence of an ARK/DRONC- (or apoptosome-) independent cell death pathway in Drosophila. This conclusion is in agreement with studies in mammalian cells, where Bcl-2 regulated caspase activation can occur independently of apoptosome formation.33 Caspase activity is still noticeable in cells lacking apaf-1, and absence of Apaf-1 does not have any effect on apoptotic cell death in thymocytes. Furthermore, whereas most apaf-1−/− mice die perinatally, 5% of apaf-1−/− animals survived to adulthood,34 suggesting an alternative cell death pathway in mammals.
Despite the importance of the CARD of ARK for interaction with DRONC, no point mutations in the CARD in any of the 33 ark alleles were identified (Figure 2). This is even more astounding given the isolation of eight mutants in a short stretch of only 53 residues in the NOD, one of which was independently isolated three times indicating some degree of saturation. Does the lack of these mutants imply that the CARD is not important for ARK function? Of course, we cannot exclude the possibility that if we screen for more ark alleles that we would also recover point mutations in the CARD. However, in analogy to Apaf-1 (Acehan et al.11), it is believed that the CARD of ARK interacts with DRONC and with the WD40 region in a mutually exclusive way. Point mutations in the CARD that weaken or abolish the interaction with the WD40 region, may be dominantly active and may cause organismal lethality even in a heterozygous condition because ARK could now engage in apoptosome formation independently of an apoptotic signal. Therefore, such CARD mutations might not be recoverable as modifiers of GMR-hid. However, it should still be possible to recover point mutations that specifically affect the interaction with DRONC. It is also possible that the CARD is more flexible than other parts of ARK such that genetic changes may be tolerated and the interaction with DRONC remains intact. In any case, it is puzzling that no point mutations were recovered in the CARD.
Mutations in the NOD reveal a critical domain for ARK function
The NOD belongs to the AAA+ (ATPases associated with various cellular activities) family of ATPases,7, 35 and mediates oligomerization of Apaf-1, for which it requires ATP/dATP.8 Based on structural analysis of inactive, ADP-bound WD40-depleted Apaf-1, the NOD was divided into several distinct domains: α/β domain, helical domain I (HD1), winged-helix domain (WHD), and helical domain II (HD2).7 The α/β and WHD domains keep ADP-bound WD40-depleted Apaf-1 in an auto-inhibited state.7
Although the structure of ARK has not been determined at the atomic level, a recent electron cryo-microscopy study reveals a similar domain architecture of the NOD of ARK.14 Eight of the missense mutations, all causing a loss-of-function phenotype, are clustered in a small region in the NOD, spanning 53 amino acids. Interestingly, seven of eight missense mutations were identified in HD1. The eighth mutation was found in the first helix of the WHD, immediately adjacent to HD1. No mutation was identified in the α/β domain, consistent with its auto-inhibitory role. None of the affected residues in HD1 has been implicated in ADP binding or domain interactions based on the structure of inactive ADP-bound WD40-depleted Apaf-1 (Riedl et al.7) suggesting that nucleotide binding may not be affected by these mutations.
Structural information may provide an explanation for the loss-of-function phenotype of the arkHD1 mutants. The connection (C1) between the two rings of the double apoptosome of ARK appears to be mediated by HD1 of each subunit.14 Thus, the arkHD1 mutations may affect the C1 connection between the rings of the double apoptosome. However, it is unclear whether the ARK double apoptosome exists in vivo. Yu et al.14 did not exclude the possibility that formation of the double apoptosome may only occur in vitro due to unphysiologically high concentrations of ARK (0.5–1.0 mg/ml) and dATP (10 mM) in the assembly preparation. However, the clustering of arkHD1 mutations supports the possibility that the double apoptosome exists in vivo.
Yu et al.14 also pointed out that access of DRONC to the double apoptosome may be blocked due to steric hindrance, and the double apoptosome may actually be catalytically inactive. Therefore, the arkHD1 mutations may ‘freeze’ the double apoptosome in an inactive state, not allowing DRONC access. However, alternative models exist. As it is unknown how ARK promotes DRONC activation, it is also possible that formation of the ARK double apoptosome – at least transiently – is required for DRONC activation, and the arkHD1 mutations actually block double apoptosome formation. In any case, our genetic analysis identifies HD1 as being critical for the apoptotic activity of ARK.
Role of the WD40 repeats for control of ARK activity
In addition to the CARD and the NOD, Apaf-1 and ARK also contain a series of WD40 repeats in the C-terminal half of the proteins (see Figure 2). The WD40 region of ARK contains two β-propellers with eight (β8) and six (β6) WD40 repeats, respectively.14 In Apaf-1 and possibly in ARK, the WD40 region is an auto-inhibitory domain which complexes the CARD and blocks access of Caspase-9 to Apaf-1.11 Deletion of the WD40 region generates constitutively active Apaf-1 in cell culture.12, 13 Cytochrome c binding to the WD40 region releases the inhibition and activates Apaf-1. However, in Drosophila, both biochemical and structural data have not established a similar role of cytochrome c for control of ARK. It is unknown whether a different co-factor exists, as postulated,14 or whether the WD40 region serves another function.
We have isolated arkWD40 alleles as suppressors of GMR-hid. In general, the arkWD40 mutants are characterized by reduced rates of cell death in both embryos and imaginal discs, suggesting that they are loss-of-function mutants. This is in contrast to the postulated inhibitory role of the WD40 repeats.4, 5, 11, 12, 13 However, it should be noted that the WD40 repeats exert their inhibitory function on monomeric, inactive Apaf-1. The arkWD40 mutants presented in this study affect the apoptotic activity of ARK, thus of activated ARK in the presence of apoptotic signals. This suggests that the WD40 repeats may also play a positive role for full activity of ARK.
One trivial reason for why the arkWD40 mutants behave as loss-of-function mutants is that they may encode unstable proteins. However, we showed by Western-blot analysis that ARK proteins carrying missense mutations in the first and tenth WD40 repeat are stable (Figure 4e). Furthermore, although cell death is reduced in arkWD40 mutants, there is still a considerable amount of dying cells in homozygous embryos compared to the null mutant (Figure 3) suggesting that these mutants still retain some apoptotic activity and thus produce appreciate protein amounts. Together, these observations suggest that at least the arkWD40 missense mutants do not affect the stability of the mutant ARK protein.
Another possibility to explain the loss-of-function phenotype of the nonsense arkWD40 alleles is mRNA instability mediated by nonsense-mediated mRNA decay (NMD). NMD is a cellular surveillance mechanism that identifies mRNAs containing premature termination codons and targets them for degradation.36 Thus, NMD might account for the observed phenotypes of arkWD40 nonsense mutants. However, NMD is not complete. Even with NMD model substrates the amount of mutant mRNA is reduced to only 25–30% of the wild-type mRNA level.37 As noted earlier, ark is produced in excess over its genetic requirement, especially in embryos which are loaded with maternal ark mRNA lasting until late larval stages. Thus, if the WD40 region of ARK has a similar inhibitory function as for Apaf-1, then the production of 25–30% of ARKWD40 truncation mutant proteins should still be sufficient to produce a strong gain-of-function phenotype, that is, unconstrained apoptosis. However, both in embryos and in imaginal discs we observe a strong loss-of-function phenotype of arkWD40 alleles. Thus, although we cannot exclude NMD as cause of the loss-of-function phenotype of the arkWD40 mutants, these considerations suggest that the WD40 region also has a pro-apoptotic requirement for ARK activity. This conclusion is also supported by the missense mutations in the WD40 region.
In cell culture, expression of WD40-depleted ARK is sufficient to induce apoptosis.4, 5 One of our mutants, arkP56 truncates ARK at residue 499, thus deleting the WD40 repeats entirely but leaving the CARD and the NOD intact. However, in contrast to the cell culture data, this mutant does not induce apoptosis, but instead blocks cell death in homozygous embryos (Figure 3). However, compared to the ark null phenotype, arkP56 still retains some apoptotic activity. This residual apoptotic activity might explain the discrepancy between the cell culture data and the arkWD40 data. In the cell culture experiments, WD40-depleted ARK was expressed under unphysiologically high levels such that a weak apoptotic protein might still cause an apoptotic phenotype.
What is the pro-apoptotic function of the WD40 repeats?
Our genetic analysis suggests that the WD40 region is required for the apoptotic activity of ARK. There is no obvious function or enzymatic activity of the WD40 repeats, other than protein-protein interactions with other molecules.9 As cytochrome c does not bind to the WD40 region of ARK (Yu et al.14), an additional role may exist for the WD40 repeats. The WD40 region may bind a different co-factor that activates ARK in vivo. Both missense and nonsense mutations in the WD40 may abolish this interaction and render ARK inactive. Such a co-factor has been postulated,14 but its identity is currently unknown.
An alternative possibility is that the WD40 repeats may have a structural requirement for stabilizing the ARK apoptosome. As the ARK apoptosome has to accommodate eight subunits instead of seven as for Apaf-1, this densely packed particle may require additional stabilization. It has been noted by Yu et al. 14 that a connection (termed C2) is formed between β8 and β6 propellers of adjacent subunits. Thus, the C2 connection may provide structural support for the integrity of the ARK apoptosome, and would be lost in nonsense mutants explaining the loss-of-function phenotype. This possibility may also explain the loss-of-function phenotype of the missense mutations affecting the first and tenth WD40 repeat (Figure 2). Although the resolution of the ARK apoptosome is not sufficient to identify individual WD40 repeats,14 it is possible that either of them or both participate in the C2 connection. A C2 connection has not been observed in the Apaf-1 apoptosome,11, 14 suggesting that it is a unique structural requirement for the ARK apoptosome.
A structural requirement might also be ascribed to the unique C-terminus of ARK (residues 1260–1440; see Figure 2). ARKJ7, which is truncated at residue 1262, is a moderate loss-of-function allele suggesting that the C-terminal domain also has a requirement for full apoptotic activity of ARK. Yu et al. 14 proposed that the C-terminal domain interacts with the α/β domain of the NOD. This interaction may also be required for stabilization of the apoptosome, and its disruption may result in partial loss-of-function.
Additionally, the arkWD40 mutants may impair the recruitment of effector caspases such as DrICE into the apoptosome. Consistent with this notion, it has been demonstrated that WD40-depleted Apaf-1 is able to recruit caspase-9 into the apoptosome, but not caspase-3.38 Interestingly, the WD40 region contains four putative effector caspase cleavage sites (DESD650; DEQD750; DVFD820; DAVD1292). Although it is not known whether these sites are cleavage targets in vivo, their presence underscores a possible role of the WD40 region for recruitment of effector caspases.
Final Conclusions
We have isolated and characterized mutant ark alleles in Drosophila. A putative null mutant of ark blocks most, but not all developmental cell death suggesting the existence of an ARK- or apoptosome-independent pathway in Drosophila. Furthermore, the arkNOD mutants identify helical domain I as critical for ARK function, presumably by controlling the C1 connection between the two rings of the ARK double apoptosome. Finally, we show genetically that the WD40 repeats have a positive requirement for full apoptotic activity of ARK. Thus, it is conceivable that the WD40 repeats have two separate functions for the control of ARK activity. In the absence of apoptotic signals, they inhibit the activation of ARK through complex formation with the CARD as shown for Apaf-1. However, in the presence of apoptotic signals they also contribute to the activation of ARK. Further studies are required to characterize the pro-apoptotic role of the WD40 repeats for full activity of ARK. Our mutants will be a useful tool to further our knowledge on the role of ARK for cell death.
Materials and Methods
Isolation of ark alleles
For GheF screening, 20 independent mutagenesis experiments (labeled A-T) were performed. For each of them, 100 isogenized FRT42D P[y+] (Bloomington stock no. 2118) males were starved for 12 h and then incubated with 25 mM EMS in 5% sucrose solution for 24 h. After 3 h of recovery, the males in each experiment were mated to 200 GheF; FRT42D P[w+] females and incubated at 25°C. 32,000 F1 progeny was screened for suppression of GMR-hid. Each suppressor was named with the letter of the mutagenesis followed by a number in the order of isolation. Thus, arkA24, arkG8, and arkL46, which carry the same mutation (Table 1), were obtained in independent mutagenesis events.
Fly stocks and genetics
All ark alleles, except arkCD4 (Rodriguez et al.5), were recovered in this study. Mosaic eye clones (Figure 5) were obtained from GheF; FRT42D arkG8/FRT42D P[w+] larvae and labeled for caspase activity and proliferation (BrdU). For germline clone (GLC) analysis, ark alleles were recombined onto FRTG13 chromosomes. GLCs were induced by the DFS-FRT method as described.25 To visualize midline glia cells, males of the genotype P[sli-1.0-lacZ]; FRTG13arkx/CyO A405 were crossed to GLC arkx females, and embryos obtained from this cross analyzed by β-Gal immunohistochemistry. n denotes the number of embryos analyzed. The GMR-hidw− transgenic line was isolated by mobilizing GMR-hid, using Δ2–3 transposase. This transgene has lost the w+ marker, but maintains the hid ORF.
Immunohistochemistry and immunoblotting
Acridine Orange labelings, BrdU incorporation, and anti-cleaved caspase-3 (Cell Signaling Technology) and β-Gal immunohistochemistry were done as described.26, 39, 40 For immunoblotting, extracts of heads from flies of GheF; FRT42D arkP56/E4/D3/FRT42D and GheF;GMR-P35 genotype were prepared and analyzed using anti-ARK antibody (1:3000) as described.29 Wild-type control in all experiments was the FRT42D P[y+] stock used for the mutagenesis.
Abbreviations
- aa:
-
amino acid
- AAA+:
-
ATPases associated with various cellular activities
- AO:
-
acridine orange
- Apaf-1:
-
apoptotic protease activating factor-1
- ARK:
-
Apaf-1 related killer
- BrdU:
-
bromodeoxyuridine
- CARD:
-
caspase activation and recruitment domain
- CED-4:
-
cell death deficient-4
- CTD:
-
C-terminal domain
- DFS:
-
dominant female sterile
- DIAP1:
-
Drosophila inhibitor of apoptosis protein 1
- DREDD:
-
death-related ced-3/Nedd2-like protein
- DrICE:
-
Drosophila ICE
- DRONC:
-
Drosophila Nedd-2 like Caspase
- EMS:
-
ethyl-methyl sulfonate
- ey :
-
eyeless
- FLP:
-
Flippase
- FRT:
-
Flippase recombination target
- GheF:
-
GMR-hid ey-Flp
- GLC:
-
germline clones
- GMR:
-
glass multimer reporter
- HD1:
-
helix domain 1
- HD2:
-
helix domain 2
- hid :
-
head involution defective
- IAP:
-
inhibitor of apoptosis proteins
- kDa:
-
kilo Dalton
- MG:
-
midline glia
- MF:
-
morphogenetic furrow
- NMD:
-
non-sense mRNA-mediated decay
- NOD:
-
nucleotide oligomerization domain
- RHG:
-
Reaper Hid Grim
- SMW:
-
second mitotic wave
- TUNEL:
-
terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling
- wg:
-
wingless
- WHD:
-
winged-helix domain
- ZCP:
-
zone of proliferating cells
References
Baehrecke EH . How death shapes life during development. Nat Rev Mol Cell Biol 2002; 3: 779–787.
Yuan J, Horvitz HR . The Caenorhabditis elegans cell death gene ced-4 encodes a novel protein and is expressed during the period of extensive programmed cell death. Development 1992; 116: 309–320.
Zou H, Henzel WJ, Liu X, Lutschg A, Wang X . Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 1997; 90: 405–413.
Kanuka H, Sawamoto K, Inohara N, Matsuno K, Okano H, Miura M . Control of the cell death pathway by Dapaf-1, a Drosophila Apaf-1/CED-4-related caspase activator. Mol Cell 1999; 4: 757–769.
Rodriguez A, Oliver H, Zou H, Chen P, Wang X, Abrams JM . Dark is a Drosophila homologue of Apaf-1/CED-4 and functions in an evolutionarily conserved death pathway. Nat Cell Biol 1999; 1: 272–279.
Zhou L, Song Z, Tittel J, Steller H . HAC-1, a Drosophila homolog of APAF-1 and CED-4 functions in developmental and radiation-induced apoptosis. Mol Cell 1999; 4: 745–755.
Riedl SJ, Li W, Chao Y, Schwarzenbacher R, Shi Y . Structure of the apoptotic protease-activating factor 1 bound to ADP. Nature 2005; 434: 926–933.
Cain K, Bratton SB, Cohen GM . The Apaf-1 apoptosome: a large caspase-activating complex. Biochimie 2002; 84: 203–214.
Li D, Roberts R . WD-repeat proteins: structure characteristics, biological function, and their involvement in human diseases. Cell Mol Life Sci 2001; 58: 2085–2097.
Qin H, Srinivasula SM, Wu G, Fernandes-Alnemri T, Alnemri ES, Shi Y . Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature 1999; 399: 549–557.
Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW . Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell 2002; 9: 423–432.
Hu Y, Ding L, Spencer DM, Nunez G . WD-40 repeat region regulates Apaf-1 self-association and procaspase-9 activation. J Biol Chem 1998; 273: 33489–33494.
Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES . Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol Cell 1998; 1: 949–957.
Yu X, Wang L, Acehan D, Wang X, Akey CW . Three-dimensional structure of a double apoptosome formed by the Drosophila Apaf-1 related killer. J Mol Biol 2006; 355: 577–589.
Quinn LM, Dorstyn L, Mills K, Colussi PA, Chen P, Coombe M et al. An essential role for the caspase dronc in developmentally programmed cell death in Drosophila. J Biol Chem 2000; 275: 40416–40424.
Dorstyn L, Colussi PA, Quinn LM, Richardson H, Kumar S . DRONC, an ecdysone-inducible Drosophila caspase. Proc Natl Acad Sci USA 1999; 96: 4307–4312.
Zimmermann KC, Ricci JE, Droin NM, Green DR . The role of ARK in stress-induced apoptosis in Drosophila cells. J Cell Biol 2002; 156: 1077–1087.
Dorstyn L, Read S, Cakouros D, Huh JR, Hay BA, Kumar S . The role of cytochrome c in caspase activation in Drosophila melanogaster cells. J Cell Biol 2002; 156: 1089–1098.
Dorstyn L, Mills K, Lazebnik Y, Kumar S . The two cytochrome c species, DC3 and DC4, are not required for caspase activation and apoptosis in Drosophila cells. J Cell Biol 2004; 167: 405–410.
Meier P, Silke J, Leevers SJ, Evan GI . The Drosophila caspase DRONC is regulated by DIAP1. EMBO J 2000; 19: 598–611.
Muro I, Hay BA, Clem RJ . The Drosophila DIAP1 protein is required to prevent accumulation of a continuously generated, processed form of the apical caspase DRONC. J Biol Chem 2002; 277: 49644–49650.
Cashio P, Lee TV, Bergmann A . Genetic control of programmed cell death in Drosophila melanogaster. Semin Cell Dev Biol 2005; 16: 225–235.
Grether ME, Abrams JM, Agapite J, White K, Steller H . The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev 1995; 9: 1694–1708.
Xu D, Li Y, Arcaro M, Lackey M, Bergmann A . The CARD-carrying caspase Dronc is essential for most, but not all, developmental cell death in Drosophila. Development 2005; 132: 2125–2134.
Chou TB, Noll E, Perrimon N . Autosomal P[ovoD1] dominant female-sterile insertions in Drosophila and their use in generating germ-line chimeras. Development 1993; 119: 1359–1369.
Abrams JM, White K, Fessler LI, Steller H . Programmed cell death during Drosophila embryogenesis. Development 1993; 117: 29–43.
Klambt C, Jacobs JR, Goodman CS . The midline of the Drosophila central nervous system: a model for the genetic analysis of cell fate, cell migration, and growth cone guidance. Cell 1991; 64: 801–815.
Rodriguez A, Chen P, Oliver H, Abrams JM . Unrestrained caspase-dependent cell death caused by loss of Diap1 function requires the Drosophila Apaf-1 homolog, Dark. EMBO J 2002; 21: 2189–2197.
Sang TK, Li C, Liu W, Rodriguez A, Abrams JM, Zipursky SL et al. Inactivation of Drosophila Apaf-1 related killer suppresses formation of polyglutamine aggregates and blocks polyglutamine pathogenesis. Hum Mol Genet 2005; 14: 357–372.
Ryoo HD, Gorenc T, Steller H . Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev Cell 2004; 7: 491–501.
Huh JR, Guo M, Hay BA . Compensatory proliferation induced by cell death in the Drosophila wing disc requires activity of the apical cell death caspase Dronc in a nonapoptotic role. Curr Biol 2004; 14: 1262–1266.
Perez-Garijo A, Martin FA, Morata G . Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development 2004; 131: 5591–5598.
Marsden VS, O'Connor L, O'Reilly LA, Silke J, Metcalf D, Ekert PG et al. Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature 2002; 419: 634–637.
Honarpour N, Du C, Richardson JA, Hammer RE, Wang X, Herz J . Adult Apaf-1-deficient mice exhibit male infertility. Dev Biol 2000; 218: 248–258.
Lupas AN, Martin J . AAA proteins. Curr Opin Struct Biol 2002; 12: 746–753.
Conti E, Izaurralde E . Nonsense-mediated mRNA decay: molecular insights and mechanistic variations across species. Curr Opin Cell Biol 2005; 17: 316–325.
Gatfield D, Unterholzner L, Ciccarelli FD, Bork P, Izaurralde E . Nonsense-mediated mRNA decay in Drosophila: at the intersection of the yeast and mammalian pathways. EMBO J 2003; 22: 3960–3970.
Hu Y, Benedict MA, Ding L, Nunez G . Role of cytochrome c and dATP/ATP hydrolysis in Apaf-1-mediated caspase-9 activation and apoptosis. EMBO J 1999; 18: 3586–3595.
Patel NH . Imaging neuronal subsets and other cell types in whole-mount Drosophila embryos and larvae using antibody probes. Methods Cell Biol 1994; 44: 445–487.
McCall K, Peterson JS . Detection of apoptosis in Drosophila. Methods Mol Biol 2004; 282: 191–205.
Acknowledgements
We apologize to all our colleagues whose work could not be cited due to space constraints. We thank George Jackson for the anti-ARK antibody; John Abrams for the arkCD4 allele and sharing information before publication; Hyung-Don Ryoo and Hermann Steller for antibodies; Bruce Hay and the Bloomington stock center for fly stocks; Eli Arama for pointing out that arkL46/arkP46 mutants produce viable adults; the MD Anderson DNA Analysis Core Facility for sequencing of ark alleles (supported by Core Grant No. CA16672 from the NCI). AB is a fellow of the MD Anderson Research Trust. This work was supported by grants from the NIH (GM068016) and The Robert A. Welch Foundation (G-1496) to AB.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by E Baehrecke
Rights and permissions
About this article
Cite this article
Srivastava, M., Scherr, H., Lackey, M. et al. ARK, the Apaf-1 related killer in Drosophila, requires diverse domains for its apoptotic activity. Cell Death Differ 14, 92–102 (2007). https://doi.org/10.1038/sj.cdd.4401931
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401931
Keywords
This article is cited by
-
Non-apoptotic enteroblast-specific role of the initiator caspase Dronc for development and homeostasis of the Drosophila intestine
Scientific Reports (2021)
-
Accurate elimination of superfluous attachment cells is critical for the construction of functional multicellular proprioceptors in Drosophila
Cell Death & Differentiation (2019)
-
Thin is required for cell death in the Drosophila abdominal muscles by targeting DIAP1
Cell Death & Disease (2018)
-
Drosophila caspase activity is required independently of apoptosis to produce active TNF/Eiger during nociceptive sensitization
Cell Death & Disease (2017)
-
Genetic characterization of two gain-of-function alleles of the effector caspase DrICE in Drosophila
Cell Death & Differentiation (2016)
