
Abstract
Our recent study has demonstrated that cellular redox imbalance can directly initiate apoptosis in a mitotic competent PC-12 cell line without the involvement of reactive oxygen species (ROS). However, whether cell apoptosis induced by ROS is, in fact, mediated by a loss of redox balance caused by the oxidant is unresolved. The linkage between oxidant-mediated apoptosis and the induction of cellular redox was examined in PC-12 cells using the oxidant, tert-butylhydroperoxide (TBH). TBH caused cell apoptosis in 24 h that was preceded by an early increase (30 min) in oxidized glutathione (GSSG). Pretreatment with N-acetyl cysteine prevented TBH-induced GSSG increases and cell apoptosis. Altered Bax/BcL-2 expression and release of mitochondrial cytochrome c occurred post-redox imbalance and was kinetically linked to caspase-3 activation and poly ADP-ribose polymerase cleavage. Moreover, cell apoptosis was attenuated by inhibition of caspase-9, but not caspase-8, and blockade of mitochondrial ROS generation and permeability transition pore attenuated caspase 3 activation and cell apoptosis. Collectively, these results show that TBH-induced GSSG elevation is associated with the disruption of mitochondrial integrity, activation of caspase-3 and cell apoptosis. This redox induction of the apoptotic cascade was dissociated from cellular GSH efflux.
Similar content being viewed by others


Introduction
Oxidants have widely been shown to initiate the cellular apoptotic cascade by perturbing the balance between the cellular signals for survival and suicide.1 It is well appreciated that oxidants can perturb the intracellular redox balance. Glutathione (GSH) is the most abundant intracellular low molecular weight thiol, and is among the many detoxification processes that maintain cellular redox homeostasis.2 Previous studies have shown that a loss of cellular GSH through efflux from cells upon oxidant exposure was linked to apoptotic initiation.3,4,5,6 The issue that oxidant-mediated induction of a shift in cellular GSH/GSSG status in favor of GSSG can enhance cell apoptosis has previously been demonstrated in our studies with intestinal cells.7 Our data support the hypothesis that the cell apoptosis mediated by lipid hydroperoxides is associated with the induction of cellular redox imbalance caused by the hydroperoxide. More recently, we have demonstrated that chemical oxidation of cellular GSH to GSSG without involvement of reactive oxygen species (ROS), can directly initiate apoptosis in a mitotic competent cell (PC-12), apoptosis through activation of mitochondrial apoptotic signaling.8 Interestingly, these studies showed that cell apoptosis was mediated by increases in GSSG levels (relative to GSH) rather than changes in GSH levels per se. Collectively, these findings raise an interesting question as to whether, mechanistically, the hydroperoxide effect on cell apoptosis is, in fact, mediated by the increase in GSSG-to-GSH ratio caused by the oxidant.
The objective of the current study was designed to address this question using tert-butyl hydroperoxide (TBH) as a model hydroperoxide whose cellular catabolism by GSH peroxidase results in enhanced GSSG formation through oxidation of GSH and a decreased GSH-to-GSSG redox state. This contribution of TBH-induced redox imbalance to cell apoptosis was studied in pheochromocytoma (PC-12) cells, a model that has been previously been characterized by Greene and Tischler to study the cellular and molecular aspects of neuronal apoptosis when cells were induced to differentiate in culture with nerve growth factor.9 Given the ease to readily induce cell differentiation in culture, we have selected this cell model for our current and ongoing studies because of its potential utility for elucidating differential vulnerability of cell stages (undifferentiated or terminally differentiated) to test the effects of oxidant and redox stress and apoptotic signaling. In the current study, we have investigated a role for cellular redox as a functional mediator in hydroperoxide-induced cell apoptosis in the mitotic competent undifferentiated state. Studies in progress are investigating the apoptotic responses in the terminally differentiated state. Since mitochondria have been widely implicated in the apoptotic cascade mediated by oxidative stress,1,10 we have investigated the effect of TBH on key components of mitochondrial apoptotic signaling and assessed the contribution of TBH-induced mitochondrial ROS production to cell apoptosis.
Results
TBH induces apoptosis in PC-12 cells and the attenuation by NAC and GSH
In preliminary experiments, we exposed PC-12 cells to varying concentrations of TBH (0–1 mM) and determined the extent of cell apoptosis at 24 h. The results show that TBH dose-dependently induced cell apoptosis at hydroperoxide levels between 20 and 300 μM (20–50%, respectively) at 24 h. Higher concentrations of TBH (500 μM and 1 mM) were cytotoxic. Based on the results that 100 μM TBH induced 30–40% cell apoptosis with little evidence of cytotoxicity, this concentration was used in subsequent experiments to examine the relationship among TBH-induced redox imbalance, mitochondrial dysfunction and PC-12 apoptosis.
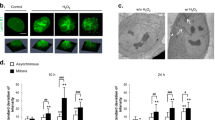
Figure 1 summarizes the results on the effect of 100 μM TBH and thiol agents on apoptosis of naïve PC12 cells. A 24 h culture of control cells typically exhibited ∼5% cell death that was characteristic of apoptosis, as evidenced by nuclear condensation, fragmentation of cell nucleus and formation of apoptotic bodies (indicated by arrows, Figure 1A), consistent with normal cell turnover. Cell apoptosis was significantly increased with TBH treatment (35%, Figure 1A,B), suggesting a link between oxidative challenge and enhanced cell apoptosis. To determine whether TBH-induced cell apoptosis was associated with altered cellular redox status, we pretreated cells with the GSH precursor, N-acetyl cysteine (NAC), concomitant with TBH exposure. Notably, the presence of NAC protected the cells against TBH-induced apoptosis and brought the level of apoptosis to near control values (Figure 1C). In addition, exogenous addition of GSH also resulted in a significant attenuation of cell apoptosis induced by TBH (Figure 1C). These results are consistent with the notion that cell apoptosis induced by TBH is mediated by an altered cellular redox caused by the hydroperoxide.

TBH-induced apoptosis in PC12 cells and the attenuation by exogenous NAC or GSH. PC-12 cells were treated for 24 h with 100 μM TBH and cell apoptosis was determined by DAPI staining as described in Materials and Methods. (A) Representative slides of control cells and TBH-treated cells stained with DAPI. Arrows point to cells undergoing apoptosis as evidenced by nuclear fragmentation and formation of apoptotic bodies. (B) Quantitative determination of TBH-induced apoptosis. (C) Effect of exogenous NAC (5 mM) or GSH (2 mM) on TBH-induced apoptosis. For B and C results are expressed as mean±S.E. for 10 and 12 separate experiments, respectively. *P<0.05 versus control, #P<0.05 versus TBH treatment
Kinetic changes of intracellular redox status induced by TBH and the effect of NAC
To determine the effect of TBH on intracellular redox, we measured the kinetics of TBH-induced changes in cellular GSH and GSSG at different times after treatment of PC12 cells with 100 μM TBH with or without NAC (5 mM). The results in Figure 2 show that the GSH/GSSG status in untreated cells remained constant throughout the 6-h incubation, in agreement with our previous observations.8 Interestingly, treatment of cells with TBH caused an increase in cellular GSH by 15 min that remained above baseline levels (Figure 2A), consistent with the stimulation of the GSH redox cycle activity during hydroperoxide challenge.11 The concomitant treatment of PC12 cells with TBH and NAC resulted in significant increases in cellular GSH levels at all time points (Figure 2A), indicating that NAC not only preserved the cellular GSH pool, but also caused GSH synthesis. Correspondingly, TBH caused an early and significant surge in GSSG at 30 min that returned to near baseline levels at 1 h (Figure 2B). These results are consistent with an initial rapid TBH-induced oxidation of GSH to GSSG and the subsequent reduction of GSSG through the action of GSSG reductase. Notably, the TBH-induced early increase in GSSG at 30 min was abrogated by NAC (Figure 2B). Importantly, the significant increase in GSSG resulted in a marked decrease in the GSH-to-GSSG ratio shortly after TBH challenge (at 30 min, Figure 2C). Accordingly, NAC eliminated the cellular redox imbalance caused by TBH and restored the GSH-to-GSSG ratio to control values (Figure 2C).

Kinetics of change in intracellular GSH and GSSG induced by TBH or TBH plus NAC. PC12 cells were treated with 100 μM TBH in the presence or absence of 5 mM NAC for 0–6 h. At designated times, samples were collected and derivatized for analyses of GSH and GSSG by HPLC as described in Materials and Methods. (A): GSH, (B): GSSG, (C): GSH-to-GSSG ratio at 30 min post treatment. Cellular concentrations of GSH and GSSG are expressed as nmol/mg protein and presented as mean±S.E. for three separate experiments. *P<0.05 versus corresponding control, # versus TBH treatment
TBH-induced GSH efflux is a late event associated with cell death
Because previous studies have implicated the efflux of GSH from cells as an initiator of apoptotic signaling,3,4,5,6 we sought to determine whether TBH induces GSH efflux from PC-12 cells. Cells were treated with 100 μM TBH, and extracellular GSH levels were determined in the media at different times after TBH exposure. We found that TBH treatment did not increase GSH in the media over a 6-h incubation period (Figure 3). A significant amount of GSH (1.2 nmol/1×106 cells, Figure 3) was detected at 24 h which coincided with substantial cell apoptosis (see Figure 1B), consistent with substantial loss of cellular GSH as a consequence of cell death. Extracellular levels of GSSG were low and remained unaltered at all times (data not shown).

Effect of TBH on GSH efflux. PC-12 cells were treated for 0–24 h with 100 μM TBH and the incubation media were collected and derivatized for analysis of extracellular GSH. Media GSH is expressed as nmol per 105 cells and presented as mean±S.E. for three separate preparations. *P<0.05 versus zero time
TBH-induced changes in mitochondrial proteins (cytochrome c, Bax, BcL-2)
Our previous studies have demonstrated that cellular redox imbalance can initiate mitochondrial apoptotic signaling.8 To determine whether the redox shift caused by TBH also induce mitochondria-mediated apoptotic signaling, we examined the central components of the mitochondrial pathway. The loss of mitochondrial cytochrome c and its release into the cytoplasm has been implicated as an essential event in initiating mitochondrial signaling of caspase-3 dependent cascade.12,13 Figure 4A illustrates the kinetics of loss of mitochondrial cytochrome c and its appearance in the cytoplasm. The results show that baseline cytochrome c levels are high in the mitochondria. TBH treatment resulted in a significant loss of mitochondrial cytochrome c at 6 h with a corresponding increase in cytoplasmic cytochrome c. Since the mitochondrial release of cytochrome c has been linked to the gate-keeper function of two heterodimeric mitochondrial proteins, Bax and BcL-2 which control mitochondrial cytochrome c release into the cytoplasm,13,14 we determine the changes in these proteins. The results in Figure 4B shows that the Bax protein was essentially absent in control PC-12 cells, in agreement with our previous observations.8 Notably, expression of Bax in the mitochondria occurred at 4–6 h post TBH treatment, while a significant increase in total cellular Bax protein levels did not occur until 6 h post- oxidant exposure (Figure 4B). In comparison, total cellular BcL-2 protein level was high in control cells and remained relatively unchanged for 4 h post-TBH treatment; thereafter the BcL-2 expression decreased markedly at 6 h (Figure 4C) that temporally correlated with the increase in both cellular and mitochondrial Bax. Accordingly, the ratio of Bax to BcL-2 was significantly increased between 4–6 h (Figure 4D), consistent with a shift from an anti-apoptotic mitochondrial signal to an apoptotic signal downstream of the induction of redox imbalance at 30 min post TBH treatment. Notably, the time course of loss of mitochondrial cytochrome c and its release into the cytoplasm corresponded to the kinetic shift in expression Bax relative to BcL-2 and the translocation of Bax from the cytoplasmic to the mitochondrial compartments (Figure 4A–C, respectively).

TBH-induced changes in mitochondrial proteins. PC12 cells were treated with 100 μM TBH and at various time points, samples were removed and mitochondrial and cytosolic fractions were prepared and subjected to Western immunoblot for analysis of mitochondrial and cytosolic cytochrome c levels. In parallel experiments, Bax and Bcl-2 expression were determined by Western analyses in total cell lysates. Mitochondrial Bax expression was also determined in the mitochondrial fractions. The data are one representative of three separate immunoblots for cytochrome c (A), and 4 blots for Bax and BcL-2 (B). For each immunoblot (except mitochondrial cytochrome c), the membranes were reprobed for β-actin and the results verified equal protein loading in each lane (data not shown); (C) ratio of Bax to BcL-2 normalized to β-actin, and presented as mean±S.E. of four separate preparations. *P<0.05 versus zero time
TBH-induced caspase-3 activation and PARP cleavage kinetically correlate with loss of mitochondrial integrity
Because the activation of caspase-3 has been associated with oxidant-induced apoptosis,1,15 we determined the kinetics of caspase-3 activation and its temporal relationship to the loss of mitochondrial integrity. Activation of caspase-3 was assessed in three ways: cleavage of the inactive precursor, procaspase-3 (CPP32); direct measure of caspase-3 activity; and caspase-3-mediated proteolytic cleavage of its target substrate, PARP. Figure 5A illustrates the results of activation of CPP32. Interestingly, the expression of CPP32 was essentially absent in control cells, in agreement with previous findings in PC-12 cells.8 After TBH treatment the proenzyme levels increased significantly at 30 min and remained elevated for 4 h, which is consistent with release of procaspase-3 from the mitochondria to the cytosol.16 At 6 h CPP32 levels decreased, indicating a cleavage of the pro-enzyme to the active enzyme. Direct measurement of caspase-3 activity confirmed a rise in enzyme activity beginning at 4 h and a significant increase at 6 h (Figure 5B). The time course of activation of caspase-3 directly correlated with the cleavage of PARP at 6 h from a native 116 kDa protein to an 85 kDa product (Figure 5C). Kinetically, the activation of caspase-3 was preceded by the induction of redox imbalance, and paralleled the upregulation of Bax and the loss of mitochondrial cytochrome c. (see Figures 2 and 4, respectively), thus correlating the loss of redox balance with mitochondrial initiation of PC-12 apoptosis. To test whether caspase-3 activation was the direct result of mitochondria initiated rather than death-receptor initiated signals amplified through the mitochondria, we treated cells with inhibitors of caspases-9 or -8, the respective initiator caspases of the mitochondria or death receptor pathways. The results in Figure 6 show that cell apoptosis was attenuated by inhibition of caspase-9, while treatment with caspase-8 inhibitor was without effect on apoptosis, indicating that the TBH-induced PC-12 apoptosis was mediated by direct mitochondria signals.

TBH-induced caspase-3 activation and PARP cleavage. PC12 cells were treated with 100 μM TBH, and at various times, samples were collected and total cell lysates prepared and processed for Western blot analyses of CPP32 expression (A). Caspase-3 activity (B) was determined in cell extracts using a fluorometric assay kit as described in Materials and Methods. Nuclear extracts were collected as described by Stefanis in Materials and Methods and analyzed for PARP (116 kDa) expression and its cleavage product (85 kDa) (C). The immunoblots of CPP32 and PARP are one representative of three separate experiments. Each immunoblot for CPP32 was reprobed for β-actin and the results verified equal protein loading in each lane (data not shown). Caspase-3 activities are expressed as relative fluorescence unit (RFU) per 106 cells and presented as mean±S.E. for four separate experiments. *P<0.05 versus zero time

Attenuation of TBH-induced PC-12 apoptosis by caspase-9, but not caspase-8 inhibitor. PC12 cells were treated with 100 μM TBH for 24 h in the absence or presence of the inhibitors of caspase-9 (LEHD-CHO) or caspase-8 (IETD-CHO). Cell apoptosis was determined by DAPI staining. Results are mean±S.E. for four separate experiments. *P<0.05 versus control. #P<0.05 versus minus inhibitor (-In) and caspase-8 inhibitor (Cas-8 In)
To verify that loss of mitochondrial permeability transition pore (MPT) function mediated caspase-3 activation, we pretreated PC-12 cells with cyclosporin A, an inhibitor of pore activity17 prior to TBH exposure. Because the adenine nucleotide translocase has been suggested to be a component of the MPT in some cell types or can form a non-specific pore,18 we also pretreated cells with atractyloside, an inhibitor of the adenine nucleotide translocase, and examined its effect on TBH-induced cell apoptosis. The results in Figure 7A shows that CPP32 cleavage was effectively prevented by treatment with cyclosporin A. Accordingly, TBH-induced cell apoptosis was significantly attenuated by cyclosporin A (Figure 7B), confirming the importance of the MPT in mediating caspase-3 activation and cell apoptosis.19 However, pretreatment with atractyloside resulted in an accelerated TBH-induced activation of CPP32 at 2 h (Figure 7A) as compared to caspase-3 activation at 6 h in the presence of TBH alone (see Figure 5A). Cell apoptosis was unaffected by atractyloside treatment (Figure 7B). These results are consistent with the notion that inhibition of the adenine nucleotide translocase promotes either non-specific pore formation18 or MPT opening.19

Effect of cyclosporin A and atractyloside on TBH-induced caspase-3 activation and cell apoptosis. PC-12 cells were treated with 100 μM TBH in the absence or presence of 1 μM cyclosporin A, or 10 μM atractyloside for either 0–6 h for determination of caspase-3 activation (A) or for 24 h for determination of cell apoptosis (B). For evaluation of caspase-3 activation, cell lysates were prepared and processed for Western analysis of CPP32 cleavage. The immunoblots of CPP32 are one representative of three separate experiments. The membranes of each immunoblot were reprobed for β-actin and the results verified equal protein loading in each lane (data not shown). For assessment of cell apoptosis, cells grown on glass cover slips were stained with DAPI and apoptotic cells were counted. Results on cell apoptosis are mean±S.E. for four separate experiments. *P<0.05 versus control, #P<0.05 versus TBH treatment
To test whether TBH-induced PC-12 cell apoptosis was mediated by enhanced mitochondrial ROS generation caused by TBH, we pretreated cells with known inhibitors of mitochondrial sites of ROS production, namely, rotenone, an inhibitor of Complex I (NADH dehydrogenase), and antimycin A, an inhibitor of Complex III (cytochrome bc1 complex). The results (Figure 8) show that rotenone as well as antimycin A treatment significantly attenuated TBH-induced apoptosis, indicating that blockade of electron flux at Complex I and Complex III can effectively prevent the apoptotic outcome. This suggests that TBH-induced production of mitochondrial ROS is a critical step in mitochondrial apoptotic signaling. Taken together, these results support our contention that TBH-induced, redox-mediated mitochondrial dysfunction is directly linked to the initiation of the apoptotic cascade which involves ROS generation, Bax upregulation and mitochondrial association, BcL-2 downregulation, mitochondrial cytochrome c release, permeability transition pore opening, and caspase-3 activation.

Effect of rotenone (Rot) and antimycin A (AA) on TBH-induced cell apoptosis. PC-12 cells were grown on cover slips and treated with 100 μM TBH in the absence or presence of 50 μM rotenone or 1 μM antimycin A for 24 h. Cells were stained with DAPI and apoptotic cells were counted. Results are mean±S.E. for four separate experiments. *P<0.05 versus control, #P<0.05 versus TBH treatment
Discussion
Oxidants, such as hydroperoxides, have been shown to activate the cellular apoptotic cascade.1,20 What is unknown, however, is whether the hydroperoxide effect on cell apoptosis is mediated by peroxide-induced alteration in cellular redox balance. In our current study, we provide evidence that the induction of apoptosis in a mitotic competent undifferentiated cell, PC-12, by tert-butyl hydroperoxide was mediated by an early loss of redox (GSH/GSSG) balance caused by the oxidant. Indeed, previous studies by Mirkovic et al 21 have shown that an early window of redox balance (30 min–4 h) was essential for protecting cells from radiation-induced apoptosis. Our results indicate the following sequelae of events that are consistent with mitochondrial signaling in apoptosis,10 namely, TBH-induced loss of redox balance, mitochondrial generation of ROS and loss of mitochondrial integrity, release of mitochondrial cytochrome c, activation of caspases-9 and -3. Several lines of evidence support this contention. First, the temporal dissociation of TBH-induced increase in GSSG at 30 min from the appearance of apoptotic death at 24 h support the notion that a rapid loss of GSH/GSSG homeostasis is a critical upstream event in PC-12 apoptosis. The results that exogenous GSH and NAC restored cellular redox balance and prevented cell apoptosis confirm this suggestion. Second, the kinetics of total cellular BcL-2 and Bax expression and Bax translocation to the mitochondria, loss of mitochondrial cytochrome c and release into the cytoplasm, and subsequent and caspase-3 activation and PARP cleavage revealed that all these events occurred at 6 h downstream of the induction of redox imbalance (30 min) and preceded apoptotic death at 24 h. This sequence of events clearly established the time line for TBH-induced cell apoptosis. The results that blockade of mitochondrial ROS formation and inhibition of MPT function attenuated cell apoptosis support the involvement of the mitochondria in apoptotic signaling. Furthermore, the apoptotic cascade is mediated through direct mitochondria initiated rather than death-receptor initiated signals amplified through the mitochondria as evidenced by the attenuation of TBH-induced cell apoptosis by inhibition of caspase-9, the initiator caspase of mitochondrial apoptotic signaling.
The involvement of mitochondrially derived ROS in apoptotic signaling in TBH-induced apoptosis is notable. Our results implicate both mitochondrial Complex I (NADH dehydrogenase) and Complex III (cytochrome bc1 complex and the Q cycle) as the major mitochondrial sites of ROS production. This is evidenced by the protection against apoptotic cell death by rotenone, a specific inhibitor of electron transfer at Complex I and by antimycin A, the specific inhibitor of Complex III (Figure 8). Ongoing studies in our laboratory are investigating the role of the mitochondrial form of superoxide dismutase (MnSOD) in protection against TBH-induced apoptosis. We have generated PC-12 clones that overexpress MnSOD, and preliminary data show that MnSOD overexpression afforded significant protection against TBH-induced cell apoptosis that directly correlated with decreased ROS generation and maintenance of cellular GSH/GSSG status (Pias and Aw, unpublished data). In parallel studies in CaCo-2 intestinal cells, we found that pretreatment of cells with rotenone or antimycin A both resulted in attenuation of lipid hydroperoxide-induced apoptosis (Wang and Aw, unpublished data). Notably, pretreatment of cells with TTFA, an inhibitor of succinate dehydrogenase (Complex II) was without effect on cell apoptosis (Wang and Aw, unpublished data). These findings are consistent with current paradigms that Complexes I and III, but not Complex II, are major intramitochondrial sources of superoxide formation.22,23 Taken together, these results support our contention that enhanced mitochondrial production of ROS induced by hydroperoxide challenge is an important trigger of the mitochondrial redox signaling events that subsequently leads to cell apoptosis, in agreement with previous studies.10
An interesting finding in this study is that the significant rise in GSSG occurred shortly after TBH challenge (30 min) was transient as evidenced by its return to baseline levels by 1 h (Figure 2B). Importantly, this initial GSSG rise is consistent with the hypothesis that significant oxidation of GSH is a primary inciting event of the apoptotic cascade. Once initiated, normalization of cellular GSSG levels, and recovery of redox balance did not prevent the progression of cell apoptosis to its ultimate biological endpoint at 24 h. Thus, it appears that an initial disruption of redox status is the critical step in peroxide-induced PC-12 apoptosis, and that sustained redox imbalance is not necessary or critical to affect the final apoptotic outcome. Our data suggest that there may exist an irreversible temporal checkpoint for redox recovery such that failure to compensate for this imbalance within a defined time window will cause apoptosis to continue to its final biological endpoint despite subsequent restoration of cellular redox homeostasis. Given the rapidity of recovery of redox status at 1 h, the threshold duration for redox signaling in PC-12 cells must occur within a time window of 30–60 min after TBH challenge. Interestingly, we observed a similar time line for redox-mediated signaling (30–60 min) in peroxide-induced apoptosis in intestinal cells.7 Previous studies by Mirkovic et al 21 have shown that the window of redox balance that was essential for protecting cells from radiation-induced apoptosis was between 30 min and 4 h. Taken together, our results and others show that redox initiation of apoptotic signaling is a kinetically rapid process, and the duration within which cells can be rescued from apoptotic death is dependent on the cell type and the apoptotic stimuli. Curiously, while significant GSSG increases were associated with TBH challenge, we found no loss of cellular GSH (Figure 2); rather, there appears to be a compensatory increase in the GSH pool. This is especially evident in the presence of NAC, a GSH precursor. The reason for the lack of a decrease in GSH with TBH exposure is somewhat unexpected, and may be related to the large pool size of GSH that could mask small and transient decreases. Moreover, our finding of a higher cellular GSH content during oxidative stress may be consistent with the stimulation of the GSH redox cycle activity for regeneration of GSH during hydroperoxide challenge.11 Collectively, our results suggest that it is a TBH-induced rise in cellular GSSG rather than a decrease in GSH that is the important mediator of cell apoptosis.
The attenuation of TBH-induced apoptosis by exogenous NAC directly correlated with the abrogation of the GSSG surge and restoration of the GSH/GSSG redox status, consistent with a role for redox modulation of apoptosis. Mechanistically, NAC could function in one of two ways. First, a direct non-enzymatic interaction of TBH with NAC could spare the oxidation of GSH, thereby preserving the cellular GSH pool. In addition, NAC could serve as a cysteine precursor for de novo GSH synthesis. The results that NAC treatment caused a net increase in the total intracellular GSH pool (GSH+GSSG, Figure 2), indicates that the major role of NAC was in the synthesis of new GSH, although we cannot rule out a direct role of NAC as a reductant in TBH elimination. The specific mechanism(s) by which TBH-induced redox shifts mediate PC-12 cell apoptosis is unresolved. One mechanism may involve target-specific thiol oxidation of the redox-sensitive MPT to control pore opening and closure. GSH has been shown to interact with MPT to change the threshold potential for pore opening, and this event is closely associated with the function of Bax and BcL-2 which controls the translocation of apoptotic releasing factors like cytochrome c from the mitochondrial matrix to the cytosol.24,25 Studies by Marchetti et al 26 have demonstrated that oxidation of thiol groups in the MPT pore determines pore function. Functional divalent thiol-reactive agents were found to induce apoptosis that was associated with formation of disulfide crosslinks that blocked the gatekeeping function of BcL-2.27 The current findings of significant increases in GSSG, the protective effects of NAC and cyclosporin A (Figures 1, 2 and 7), are all consistent with a role for MPT in mitochondrial apoptotic signaling that could involve thiol oxidation of the pore protein. Our finding that caspase-3 activation was accelerated by atractyloside (Figure 7) supports previous results that atractyloside enhances mitochondrial membrane permeabilization through either increased opening of the MPT19 or induction of non-specific pore formation by the adenine nucleotide translocase.18 The loss of cellular GSH from cells through direct efflux has been proposed as a key mechanism in the initiation of the apoptotic cascade.3,4,5,6 However, our results did not support a role for GSH efflux in apoptotic signaling. Notably, we did not see significant efflux of cellular GSH until 24 h post TBH treatment (Figure 3) that coincided with significant cell apoptosis. This suggests that the loss of cellular GSH was a consequence of cell death and therefore not a contributing factor to the initiation of PC-12 apoptosis induced by TBH. Another mechanism that TBH-induced redox shift can modulate apoptosis may be tied to its role in transcription factor activation and binding to enhancer–promoter elements in DNA. Recent studies have demonstrated that hydrogen peroxide increased the binding of the nuclear factor kappaB (NF-κB) to DNA in HeLa cell extracts.28 Thus, a highly oxidized environment such as occurs with increased ROS production and GSSG formation could favor gene transcription activity. In our study, the kinetics of TBH-induced redox shift (30 min) and the induction of mitochondrial signaling and activation of caspase-3 (6 h) suggest that a transcription-dependent mode of apoptotic induction is likely. A potential for redox modulation of transcriptional activity of apoptotic genes is an avenue of research that warrants further investigation. Ongoing efforts in our laboratory are devoted to this area of investigation. Regardless of mechanism, our study provides evidence that redox perturbation induced by TBH leads to loss of mitochondrial integrity, activation of caspase-3 and enhanced apoptosis.
Our data are consistent with redox imbalance as a mediator of the TBH effect. However, it is noteworthy that there are clear kinetic differences in apoptotic signaling by TBH (which involves both redox shifts and ROS) and by the thiol agent, diamide, that induces redox signaling per se independent of ROS.8 Notably, both agents mediate PC-12 apoptosis by initiating mitochondrial apoptotic signaling, but diamide-induced mitochondrial changes were significantly more rapid than those induced by TBH. Specifically, diamide caused apoptotic initiation, i.e., Bax upregulation and loss of mitochondrial cytochrome c at 30 min, and caspase-3 activation at 1 h as compared to TBH-induced changes at 6 h despite the fact that both agents caused a similar rapid increase in GSSG (within 15–30 min). The reason for this difference is unclear. One possible explanation for the kinetic difference may be related to a broader time window of redox imbalance for redox signaling mediated by diamide. Indeed, diamide caused significant increases in GSSG beginning at 5 min after treatment that continued to increase to 30 min,8 while TBH treatment caused GSSG increases between 20–30 min which is a narrower defined time window available for redox signaling. An alternate explanation is that, apart from causing redox changes, ROS may directly activate other signaling mechanisms in TBH-treated cells. Similar findings in our studies on ROS- or redox-mediated NFκBκB signaling in posthypoxic endothelial cells support this latter suggestion.29 Thus, despite the many similarities, the differences do underscore a fundamental distinction between signaling mechanisms that are mediated directly by ROS and those that are mediated through redox changes induced by ROS. Notwithstanding, our results are consistent with TBH-induced redox imbalance being a major contributor to the initiation of mitochondrial redox signaling and apoptotic cell death in undifferentiated PC-12 cells. The collective findings from our current and previous study underscore the potential importance of redox status in oxidant-mediated apoptosis in mitotic competent undifferentiated cells. Whether similar apoptotic responses and regulation occurs in terminally differentiated cells in response to oxidant challenge is unknown and warrants further investigation.
Materials and Methods
Materials
The following chemicals were obtained from Sigma Chemicals (St. Louis, MO, USA): agarose, N-acetyl cysteine (NAC), atractyloside (ATR), antimycin A (AA) tert-butyl hydroperoxide (TBH), cyclosporin A (CSA), 4′6-diamidino-2-phenylindole (DAPI), 2,4-dinitroflurobenzene (FDNB), protease inhibitors (aprotinin, leupeptin, pepstatin, phenylmethylsulfonylfluoride [PMSF]), glutathione (GSH and GSSG), iodoacetic acid, rotenone. Fetal calf serum and horse serum were obtained from Atlanta Biologicals (Norcross, GA, USA). Monoclonal antibodies against Bax, BcL-2, and CPP32 were acquired from Transduction labs (Lexington, KT, USA); the monoclonal antibody against β-actin was purchased from Oncogene (Cambridge, MA, USA). Cytochrome c and PARP polyclonal primary antibodies were obtained from Santa Cruz Biologicals (Santa Cruz, CA, USA). The fluorescent caspase-3 assay kit was obtained from Pharmingen (San Diego, CA, USA). Inhibitors of caspase-9 (LEHD-CHO) and caspase-8 (IETD-CHO) were from Calbiochem (San Diego, CA, USA). Dulbecco's Modified Eagle's Medium (DMEM) was obtained from GIBCO (Grand Isle, NY, USA). Nitrocellulose membranes, Bio-Rad protein dye assay kit, and the SDS-electrophoresis units were acquired from BIORAD Corporation (Hercules, CA, USA). The enhanced chemiluminescence system for Western immunoblot, the hyperfilm, and secondary IgG anti-mouse and anti-rabbit antibodies were purchased from Amersham (Arlington Heights, IL, USA). Fluorescent mounting media was obtained from DAKO Corporation (Carpinteria, CA, USA). Twelve mm circle number 1 glass cover slips for DAPI staining were procured from Fisher (Pittsburgh, PA, USA).
Cell culture and incubations
Pheochromocytoma cells (PC-12) were a generous gift from Dr. Nikki Holbrook (National Institute on Aging, Baltimore, MD, USA). PC-12 cells were cultured in DMEM containing 10% heat inactivated fetal bovine serum, 5% heat inactivated horse serum, penicillin (100 units/ml), streptomycin (100 μg/ml), amphotericin B (25 μg/ml), gentamycin (1 ml/l), and 2 mM glutamine in a 5% CO2 humidified environment/95% air at 37°C. The culture medium was changed every 2 days. For each experiment, PC12 cells were plated in standard DMEM culture media at a specified density the day before the experiment was performed. On the day of the experiment, the culture media were replaced with fresh DMEM prior to addition of TBH or other reagents. Whenever present, reagents were added to cell cultures at the following final concentrations: TBH, 100 μM; NAC, 5 mM; GSH, 2 mM; rotenone, 50 μM; cyclosporin A, 1 μM; atractyloside, 10 μM and antimycin A, 1 μM. Caspases-9 and -8 inhibitors were each prepared as 20 mM stock solutions in DMSO, and were added to cell cultures at final concentrations of 10 μM. In studies of GSH efflux, the culture media were replaced with fresh serum-free DMEM prior to TBH addition. At designated times of 30 min, 1, 2, 4, 6, and 24 h, the media were collected and treated with 5% TCA. The acid supernatants were derivatized for quantification of GSH and GSSG as described below.
Detection of apoptosis by DAPI
DAPI staining was performed according to the method of Wang et al.28 Briefly, 2×105 PC12 cells were grown on 12 mm circular glass cover slips in 24-well plates. Cells were treated with 100 μM TBH for 24 h. Cells were washed with PBS and fixed with cold 4% paraformaldehyde for 15 min. After washing with PBS, cells were fixed with cold 70% ethanol at −20°C for at least 1 h, and stained with 1 μg/ml DAPI for 30 min in the dark. The slides were washed three times with PBS, and mounted using DAKO fluorescent mounting fluid. Cells were viewed and counted using a fluorescent Olympus B×50 microscope with the 20× objective. At least six fields of total and apoptotic cells were counted on each slide. A total of 200 cells were counted.
Measurements of GSH and GSSG
Cellular glutathione (GSH and GSSG) was determined by the high-performance liquid chromatography (HPLC) method of Reed et al.30 Cells (2×106) were cultured in T-25 culture flasks and exposed to 100 μM TBH in 10 mls of standard DMEM. In some experiments, cells were concomitantly treated with the thiol-modifying agent, NAC (5 mM) and TBH at time zero. At time points ranging from 0 to 6 h cells were harvested by scraping in ice-cold 5% trichloroacetic acid (TCA). Cell suspensions were then centrifuged to remove the TCA-insoluble proteins. The acid supernatant was derivatized with 6 mM iodoacetic acid and 1% 2,4-dinitrophenyl fluorobenzene to yield the S-carboxymethyl and 2,4-dinitrophenyl derivatives of GSH and GSSG. Separation of GSH and GSSG derivatives was performed on a 250 mm×4.6 mm Alltech Lichrosorb NH2 10 micron column. Cellular GSH and GSSG contents were quantified by comparison to standards derivatized in the same manner.
Preparation of cell lysates for Western analyses of CPP32, Bax, BcL-2 and cytochrome c
Lysate preparation: CPP32, Bax and BcL-2
PC12 cells (2×106) were plated per T-25 dish, and treated with 100 μM TBH for specified times from 0–6 h. Thereafter, cells were ruptured with 300–600 μL of lysis buffer containing 300 mM NaCl, 50 mM Tris-HCl, 0.5% Triton X-100, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mM PMSF, and 1.8 mg/ml iodoacetamide for 30 min at 4°C. Cells were then scraped and cell extracts stored at −20°C until later use in Western analyses.
Lysate preparation: Bax and cytochrome c
PC12 cells were plated at a density of 2×106 cells and were harvested at indicated times (0–6 h) after TBH treatment. Briefly, culture media were removed and cells were washed two times with PBS. Cells were harvested in 1ml PBS by scraping, transferred to eppendorf tubes, and collected by centrifugation at 800 g for 10 min. The cell pellets were suspended in 0.5 ml of cold lysis buffer, incubated for 3 min on ice, and homogenized with 10 up and down strokes using a Glas-Col homogenizer. The suspensions were centrifuged at 750 g for 15 min at 4°C to collect the mitochondrial pellets. The pellets were stored at −80°C until further use.
Western blot analyses
Typically, 20 μg protein was resolved on 10% acrylamide gels (100 V, 90 min) and blotted onto nitrocellulose membranes. The membranes were individually probed with anti-CPP32, Bax or BcL-2 (1 : 500–1 : 1000). The secondary antibody used corresponded to the primary antibody (goat, mouse, etc) and was conjugated to horseradish peroxidase (1 : 1000). To detect cytochrome c, 20 μg protein was resolved on a 12% SDS–page gel at 50 V and 4°C for 6 h. Protein was transferred onto nitrocellulose membranes overnight at 20 V and 4°C. Membranes were probed with primary and secondary cytochrome c antibodies (1 : 500, 1 : 5000). Detection of chemiluminescence was performed with an ECL Western blotting detection reagent according to the manufacturer s recommendation. After exposure to one antibody, each membrane was stripped (6.25 mM Tris pH 6.7, 2%SDS, and 100 mM mercaptoethanol) and probed again for β-actin to verify equal protein loading in each lane.
Western analyses for PARP cleavage
PC12 cells were plated at a density of 2×106 cells were harvested at indicated times (0–6 h) after TBH treatment according to the method of Stefanis et al.31 Media were collected and centrifuged for 5 min at 1000 r.p.m. to collect detached cells. The pellet was combined with cells in the flask. Cells were scraped and collected into buffer (25 mM HEPES, pH 7.5, 5 mM EDTA, 2 mM EDTA, 1% Triton X-100, 10 μg/ml each pepstatin and leupeptin, and 1 mM PMSF) and sonicated. The nuclear pellets were separated from supernatants by centrifugation. The pellets were stored at −20°C for Western analyses of PARP and its degradation product. Twenty micrograms of the protein was resolved on an 8% acrylamide gel and blotted onto nitrocellulose membranes. The membranes were probed with 1 : 1000 dilution of PARP (rabbit anti-mouse polyclonal antibody) and secondary IgG antibody 1 : 1000 and then treated with ECL and exposed to film as described above.
Fluorometric determination of caspase-3 activity
PC12 cells were seeded at a density of 1×106 and exposed to 100 μM TBH for time points ranging from 0–6 h. Cell lysates were prepared at designated times according to the protocol of the caspase-3 assay kit (PharMingen International). Briefly, cells were washed once with PBS and lysed with 0.5 mls of lysis buffer (10 mM Tris-HCl, [pH 7.5], 10 mM NaH2PO4/NaHPO4, [pH 7.5], 130 mM NaCl, 1% Triton X-100, 10 mM NaPPi, sterile filtered). Cell lysates were centrifuged at 14 000 r.p.m. at 4°C for 10 min to collect the supernatants for determination of caspase-3 activity. The assay mixture contained 2 mls of HEPES, 100 μl of cell lysate, and 10 μl of the non-fluorescent caspase-3 substrate (DEVD-AMC [N-acetyl-Aspartate-Gluatmate-Valine-Aspartate]-AMC [7-amino-4-methylcoumarin], 1 μg/1 μl). Incubations were carried out for 2 h at 37°C. Activated caspase-3 will cleave DEVD-AMC to its strongly fluorescent product, AMC. The formation of AMC was quantified using an AMINCO Bowman Series 2 luminescence spectrophotometer (Thermo Spectronic, Rochester, NY, USA), at excitation and emission wavelengths of 380 nm and 445 nm, respectively. Results are expressed as relative fluorescence unit (RFU) per 105 cells.
Protein assay
Protein was measured using Bio-Rad Protein Assay kit (Bio-Rad Laboratories, Hercules, CA, USA) according to the manufacturer's protocol.
Statistical analysis
Results are expressed as mean±S.E. Analysis of variance and Tukey's test were used to determine significance of differences. P values of <0.05 were considered as statistically significant.
Abbreviations
- PC12:
-
pheochromocytoma cells
- TBH:
-
tert-butyl hydroperoxide
- NAC:
-
N-acetyl cysteine
- ROS:
-
reactive oxygen species
- GSH:
-
reduced glutathione
- GSSG:
-
oxidized glutathione
- CPP32:
-
pro-caspase 3
- Bax/BcL-2:
-
pro-and anti-apoptotic members of BcL-2 family, respectively
- PARP:
-
poly ADP ribose polymerase
- MnSOD:
-
manganese superoxide dismutase
- MPT:
-
mitochondrial permeability transition pore
- NF-κB:
-
nuclear factor kappaB
- DAPI:
-
4′,6-diamidino-2-phenylindole
- CSA:
-
cyclosporin A
- ATR:
-
atractyloside
- FDNB:
-
2,4-dinitrofluorobenzene
- PMSF:
-
phenylmethylsulfonuylfluoride
- DMEM:
-
Dulbecco's Modified Eagle's medium
- HPLC:
-
high-performance liquid chromatography
- PBS:
-
phosphate buffered saline
- TCA:
-
trichloroacetic acid
- RFU:
-
relative fluorescence unit
- DEVD-AMC:
-
[N-acetyl-Aspartate-Glutamate-Valine-Aspartate]-AMC [7-amino-4-methylcoumarin]
- IETD-CHO:
-
[Acetyl-Alanine-Alanine-Valine-Alanine-Leucine-Leucine-Proline-Alanine-Valine-Leucine-Leucine-Alanine-Leucine-Leucine-Alanine-Proline-Isoleucine-Glutamate-Threonine-Aspartate-CHO]
- LEHD-CHO:
-
[Acetyl-Alanine- Alanine-Valine-Alanine-Leucine-Leucine-Proline-Alanine-Valine-Leucine-Leucine-Alanine-Leucine-Leucine-Alanine-Proline-Leucine-Histidine-Aspartate-CHO]
References
Chandra J, Samali A, Orrenius S . 2000 Triggering and modulation of apoptosis by oxidative stress Free. Rad. Biol. Med. 29: 323–333
Kaplowitz N, Aw TY, Ookhtens M . 1985 The regulation of hepatic glutathione Ann. Rev. Pharmacol. Toxicol. 25: 715–744
Van den Dobbelsteen DJ, Nobel CSI, Schlegel J, Cotgreave IA, Orrenius S, Slater AFG . 1996 Rapid and specific efflux of reduced glutathione during apoptosis induced by anti-Fas/APO-1 antibody J. Biol. Chem. 271: 15420–15427
Ghibelli L, Fanelli C, Rotilio G, Lafavia E, Coppola S, Colussi C, Civitareale P, Ciriole MR . 1998 Rescue of cells from apoptosis by inhibition of active GSH extrusion FASEB J. 12: 479–486
Li J, Miyakawa H, Liu J, Mori K, Takano T, Marumo F, Sato C . 1994 Characterization of glutathione efflux from HEP G2 Cells Res. Comm. Mol. Path. Pharm. 85: 261–270
Sagara J, Makino N, Bannai S . 1996 Glutathione efflux from cultured astrocytes J. Neurochem. 66: 1876–1881
Wang TG, Gotoh Y, Jennings MH, Rhoads CA, Aw TY . 2000 Cellular redox imbalance induced by lipid hydroperoxide promotes apoptosis in human colonic CaCo-2 cells FASEB J. 14: 1567–1576
Pias EK, Aw TY . 2002 Apoptosis in mitotic competent undifferentiated cells is induced by cellular redox imbalance independent of reactive oxygen species production FASEB J 16: in press
Greene LA, Tischler AS . 1976 Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor Proc. Natl. Acad. Sci. USA 73: 2424–2428
Cai J, Jones DP . 1998 Superoxide in apoptosis: Mitochondrial generation triggered by cytochrome c loss J. Biol. Chem. 273: 11401–11404
Chance B, Sies H, Boveris A . 1979 Hydroperoxide metabolism in mammalian organs Physiol. Rev. 59: 5227–5605
Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng T, Jones DP, Wang X . 1997 Prevention of apoptosis by BCL-2: release of cytochrome c from mitochondria blocked Science 75: 1139–1142
Rosse T, Olivier R, Monney L, Rager M, Conus S, Fellay I, Jansen B, Borner C . 1998 Bcl-2 prolongs cell survival after Bax-induced release of cytochrome c Nature 391: 496–499
Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD . 1997 The release of cytochrome c from the mitochondria: a primary site for Bcl-2 regulation of apoptosis Science 275: 1132–1136
Cohen GM . 1997 Caspases: the executioners of apoptosis Biochem. J. 326: 1–16
Kluck RM, Martin SJ, Hoffman BM, Zhou JS, Green DR, Newmeyer DD . 1997 Cytochrome c activation of CPP32-like proteolysis plays a critical role in Xenopus cell-free system EMBO J. 16: 4639–4649
Walter DH, Haendeler J, Galle J, Zeiher AM, Dimmeler S . 1998 Cyclosporin A inhibits apoptosis of human endothelial cells by preventing release of cytochrome c from mitochondria Circulation 98: 1153–1157
Vieira HL, Haouzi D, El Hamel C, Jacotot E, Belzacq AS, Brenner C, Kroemer G . 2000 Permeabilization of the mitochondrial inner membrane during apoptosis: impact of the adenine nucleotide translocator Cell Death Differ. 7: 1146–1154
Xu M, Wang Y, Kirai K, Ayub A, Ashraf M . 2001 Calcium preconditioning inhibits mitochondrial permeability transition and apoptosis Am. J. Physiol. 280: H899–H908
Abe K, Saito H . 1998 Characterization of TBH toxicity in cultured rat cortical neurons and astrocytes Pharmacol.Toxicol. 83: 40–46
Mirkovic N, Voehringer DW, Story MD, McConkey DJ, McDonnell TJ, Meyn RE . 1997 Resistance to radiation induced apoptosis in BcL-2 expressing cells is reversed by depleting cellular thiols Oncogene 15: 1461–1470
Turrens JF, Boveris A . 1980 Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria Biochem. J. 191: 421–427
Boveris A, Cadenas E, Stoppani AOM . 1976 Role of ubiquinone in the mitochondrial generation of hydrogen peroxide Biochem. J. 156: 435–444
Halestrap AP, Woodfield KY, Connern CP . 1997 Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase J. Biol. Chem. 272: 3346–3354
Constantini P, Chernyak BV, Petronili V, Bernardi P . 1996 Modulation of the mitochondrial permeability transition pore by pyridine nucleotides and dithiol oxidation at two separate sites J. Biol. Chem. 271: 6746–6751
Marchetti P, Decaudin D, Macho A, Zamzami N, Hirsch T, Susin SA, Kroemer G . 1997 Redox regulation of apoptosis: impact of thiol oxidation status on mitochondrial function Eur. J. Immunol. 27: 289–296
Zamzami N, Marzo I, Susin SA, Brenner C, Larochette N, Marchetti P, Reed J, Kofler R, Kroemer G . 1998 The thiol crosslinking agent diamide overcomes the apoptosis-inhibitor effect of Bcl-2 by enforcing mitochondrial permeability transition Oncogene 16: 1055–1063
Wang X, Martindale JL, Liu Y, Holbrook NJ . 1998 The cellular response to oxidative stress: influences of mitogen activated protein kinase signalling pathways on cell survival Biochem. J. 333: 291–300
Kokura S, Rhoads CA, Wolf RE, Yoshikawa T, Granger DN, Aw TY . 2001 NFκB signaling in posthypoxic endothelial cells: Relevance to E-selectin expression and neutrophil adhesion J. Vasc. Res. 38: 47–58
Reed DJ, Babson JR, Beatty PW, Brodie AE, Ellis WW, Potter DW . 1980 High-performance liquid chromatography analysis of nanomole levels of glutathione, glutathione disulfide, and related thiols and disulfides Anal Biochem. 106: 55–62
Stefanis L, Troy CM, Qi H, Shelanski ML, Greene LA . 1998 Caspase-2 processing and death of trophic factor deprived PC12 cells and sympathetic neurons occur independently of caspase-3 like activity J. Neurosci. 18: 9204–9215
Acknowledgements
This study was supported by a grant from the National Institutes of Health, DK44510.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by B Osborne
Rights and permissions
About this article
Cite this article
Pias, E., Aw, T. Early redox imbalance mediates hydroperoxide-induced apoptosis in mitotic competent undifferentiated PC-12 cells. Cell Death Differ 9, 1007–1016 (2002). https://doi.org/10.1038/sj.cdd.4401064
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401064
Keywords
This article is cited by
-
Apoptotic Pathways and Alzheimer’s Disease: Probing Therapeutic Potential
Neurochemical Research (2021)
-
Shikonin Protects PC12 Cells Against β-amyloid Peptide-Induced Cell Injury Through Antioxidant and Antiapoptotic Activities
Scientific Reports (2018)
-
Ethyl acetate extract of Wedelia chinensis inhibits tert-butyl hydroperoxide-induced damage in PC12 cells and D-galactose-induced neuronal cell loss in mice
BMC Complementary and Alternative Medicine (2014)
-
Glutathione depletion regulates both extrinsic and intrinsic apoptotic signaling cascades independent from multidrug resistance protein 1
Apoptosis (2014)
-
Nitrogen Compounds Prevent H9c2 Myoblast Oxidative Stress-Induced Mitochondrial Dysfunction and Cell Death
Cardiovascular Toxicology (2010)
