
Abstract
Manipulating the selectivity-determining step in post-C–C coupling is crucial for enhancing C2 product specificity during electrocatalytic CO2 reduction, complementing efforts to boost rate-determining step kinetics. Here we highlight the role of single-site noble metal dopants on Cu surfaces in influencing C–O bond dissociation in an oxygen-bound selectivity-determining intermediate, steering post-C–C coupling toward ethylene versus ethanol. Integrating theoretical and experimental analyses, we demonstrate that the oxygen binding strength of the Cu surface controls the favorability of C–O bond scission, thus tuning the selectivity ratio of ethylene-to-ethanol. The Rh-doped Cu catalyst with optimal oxygen binding energy achieves a Faradaic efficiency toward ethylene of 61.2% and an ethylene-to-ethanol Faradaic efficiency ratio of 4.51 at –0.66 V versus RHE (reversible hydrogen electrode). Integrating control of both rate-determining and selectivity-determining steps further raises ethylene Faradaic efficiency to 68.8% at 1.47 A cm−2 in a tandem electrode. Our insights guide the rational design of Cu-based catalysts for selective CO2 electroreduction to a single C2 product.

Similar content being viewed by others


Main
Copper-based catalysts drive the electrochemical CO2 reduction reaction (CO2RR) to produce fuels and chemicals containing multiple carbon centers (C2+)1,2. However, Cu catalysts typically give a wide distribution of products, making electrocatalytic CO2-to-C2+ conversion far from practical2. So far, tremendous effort has been devoted to improving the selectivity of overall C2+ products by enhancing the binding energy of adsorbed CO (*CO) or increasing local CO concentration2,3. These two interplaying parameters govern the kinetics of C–C coupling, which is regarded as the rate-determining step (RDS) for C2+ product formation2,3. Experimental observations suggest the pathways toward ethylene (C2H4) and ethanol (C2H5OH), the two major C2 products in CO2RR, may share several common intermediates until a bifurcation step at the post-C–C coupling stage4,5. The relative stability of a selectivity-determining intermediate (SDI) determines the bifurcation leading to C2H4 versus C2H5OH (refs. 6,7,8,9,10). Controlling the branching post-C–C coupling, which is a relatively underexplored strategy, can be as important as promoting C–C coupling kinetics in determining the selectivity toward a specific C2 product.
Compared with the progress made in understanding the mechanisms of C–C coupling steps, identifying the SDI remains elusive, partially because state-of-the-art operando technologies are unable to probe the key C2 intermediate directly. Density functional theory (DFT) studies dominate the research on the late stages of CO2RR and have proposed variable SDIs with tautomeric forms6,7,8,9. The SDI configurations can be classified based on the atom(s) coordinated to the Cu surface: carbon bound (for example, Cu–C–OH) or oxygen bound (for example, Cu–O–C)6,7,8,9. Previous studies developed various catalysts to enhance C2H5OH generation over C2H4 and applied different SDI structures to rationalize their selectivity-determining steps (SDSs)11,12,13,14,15,16,17. However, the key SDI branching C2 alcohol and hydrocarbon has not been validated by a generalized catalyst design strategy.
Regulating the C–O bond strength of the SDI is critical to direct the pathways to hydrocarbons versus oxygenates18,19,20,21,22. In particular, cleavage versus preservation of the C–O bond in the oxygen-bound SDI (for example, *CH2CHO) determines the selectivity toward C2H4 versus C2H5OH (refs. 9,10). Hydrogenation of *CH2CHO leads to either the formation of acetaldehyde (*CH3CHO, the precursor to C2H5OH) or the desorption of C2H4 with adsorbed oxygen (*O) remaining on the catalyst surface9. The relative stability of the *O species after C–O bond cleavage is an adequate descriptor to evaluate the potential bifurcation to C2H4 versus C2H5OH formation10. The oxygen-binding strength forms a linear scaling relationship useful for understanding the adsorption configurations and energetics of oxygen-bound intermediates, which in turn influence product selectivity10,21,22,23,24,25,26.
Here, we report a generalizable concept for tuning oxygen affinity27 to direct post-C–C coupling selectivity toward C2H4 or C2H5OH via the stabilization and activation of *O and *CH2CHO intermediates on single-site noble-metal-doped Cu catalysts (MCu; M = Rh, Ir, Ru, Pd, Ag, and Au). The oxygen-binding strength on the MCu surface dictates the cleavage or preservation of the C–O bond in the *CH2CHO intermediate. Specifically, the RhCu catalyst efficiently shifts the product distribution toward C2H4 at the expense of C2H5OH, as compared with bare Cu. DFT calculations reveal that the Rh dopant, with stronger oxygen affinity than Cu, stabilizes *CH2CHO adsorption and elongates/weakens C–O bond. In contrast, dopants with weaker oxygen affinity than Cu, such as Au, promote C2H5OH formation by preserving the C–O bond. Dopants that bind oxygen too strongly, such as Ru, are limited by slow *O removal from the dopant site. Our studies offer a universal reaction descriptor to rationalize catalyst design for selective CO2RR toward a single C2 product.
Results
Catalyst design principles from DFT
We applied DFT calculations as a proof-of-concept to evaluate our proposed strategy that guides post-C–C coupling steps toward a single C2 product. The SDI of the *CH2CHO species proposed previously9 originates from four proton-coupled electron transfer steps of the *COCOH intermediate in a sequence following C–C coupling. The C2H4 pathway proceeds via C–O bond scission, releasing C2H4 and leaving an adsorbed *O (Fig. 1a). The *O adsorbate is protonated to *OH and then to H2O. Alternatively, hydrogenation of the α-carbon in *CH2CHO leads to *CH3CHO and consequently to C2H5OH, where the C–O bond is preserved (Fig. 1a). We postulate that doping single-site metals into the Cu surface with distinct oxygen binding affinity can modulate the favorability of C–O bond dissociation in the SDI. To test this hypothesis, we computationally screened several single-site noble metal dopants (Rh, Ir, Ru, Pd, Ag, and Au) that were chosen because of their thermodynamic stability in a Cu matrix28.

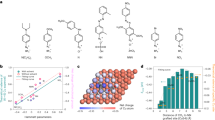
a, Schematic illustration of branching pathways to C2H4 or C2H5OH from the *CH2CHO intermediate as the SDI. The black, red, and white spheres represent C, O, and H atoms, respectively. b, Adsorption energies of *O and *CH2CHO intermediates on MCu catalysts relative to pristine Cu. c, Reaction free energy for first hydrogenation steps of the *CH2CHO intermediate to C2H5OH or C2H4 over pristine Cu and oxophilic MCu catalysts. d, Free energy diagrams under –0.65 V versus RHE for hydrogenation of the *CH2CHO intermediate to C2H4 and *OH on pristine Cu and oxophilic MCu catalysts. The numbers are kinetic barriers in eV.
We investigated the relative adsorption stability of *O, *CH2CHO, and other related intermediates along the post-C–C coupling reaction pathways (Fig. 1b and Supplementary Fig. 1). Pd, Ag, and Au dopants, which have weaker oxygen affinity than Cu, induce only a slight change in adsorption energies and configurations of oxygen-bound intermediates. The *O adsorbate prefers to sit on a hollow site composed of four Cu atoms (Cu4 sites) compared with the site composed of one dopant and three Cu atoms (that is, Cu3Ag, Cu3Au, and Cu3Pd sites) at low doping concentration (Supplementary Fig. 2). This observation is intuitive, as the Cu–O bond is stronger than the Ag–O, Au–O, and Pd–O bonds. The dopants with stronger oxygen affinity than Cu (that is, Rh, Ir, and Ru) create sites that directly participate in the stabilization of the *O species (Supplementary Fig. 2c). By defining oxygen affinity as the adsorption energy of *O on the Cu3M sites, the oxygen affinity of sites created with the dopant metals are ordered as Ru > Rh > Ir > Cu > Pd > Ag > Au. Note that an analysis of oxygen-binding energies and Bader charges as a function of adsorption distance from the dopant confirmed that the dopant is local to the doping center within the Cu3M ensemble and that the bulk Cu electronic structure far away from the dopant is not affected at the employed low doping concentration (Supplementary Fig. 3).
The stronger interaction between *CH2CHO and the catalyst surfaces with enhanced oxygen affinity elongates the C–O bond length in the *CH2CHO intermediate, weakening the C–O bond strength10. The C–O bond lengths for oxophilic MCu (M = Rh, Ir, and Ru) surfaces are ~1.36 Å, which are longer than those for pristine Cu (1.32 Å) and oxophobic MCu (M = Pd, Ag, and Au) surfaces (~1.32 Å; Supplementary Table 1). As such, we hypothesize that the oxophilic surfaces direct the reaction path bifurcation to C2H4 by promoting C–O bond scission.
We subsequently assessed the elementary steps of *CH2CHO hydrogenation on pristine Cu and oxophilic MCu surfaces. It is thermodynamically favorable for the C–O bond of *CH2CHO to be cleaved to produce C2H4 compared with the formation of *CH3CHO on oxophilic MCu surfaces, especially on the RuCu catalyst (Fig. 1c). Reaction energies of the following *O protonation step in the C2H4 pathway are also influenced by improved surface oxygen affinity (Supplementary Fig. 4). The kinetic barriers for the C2H4 pathway are shown in Fig. 1d, Supplementary Fig. 5, and Supplementary Table 2. The activation energy barrier for C–O bond scission decreases as surface oxygen affinity increases. However, the *O hydrogenation step will become energetically prohibitive if the oxygen affinity is too high because removing *O becomes rate-limiting on highly oxophilic surfaces (that is, as seen on the RuCu site). The Rh dopant has optimal oxygen affinity: it balances the activation energy barriers of C–O bond scission and *O protonation. Thus, we expect RhCu to show the highest selectivity toward C2H4 among the MCu catalysts.
It is worth noting that the *CH2CHO intermediate does not solely adsorb through *O on any of the MCu surfaces. However, regardless of whether the initial state merely binds through *O, the product state on all of the doped surfaces has the *O atom from the C–O scission step binding on the Cu3M ensemble as shown in Supplementary Fig. 5. As such, stronger oxygen binding is an effective descriptor because it quantifies the stability of the products that have undergone C–O bond scission. Finally, we note that the inclusion of implicit solvation energy corrections only causes marginal variations to adsorption energies of oxygen-bound intermediates and energy barriers of C–O bond scission (Supplementary Fig. 6).
Synthesis and characterization of MCu catalysts
Guided by DFT computations, we sought to synthesize a series of MCu catalysts by cation exchange. For example, Rh-doped CuO pre-catalyst (RhCuO) is prepared by first exchanging Cu2+ with Rh3+ on the Cu(OH)2 matrix29 followed by a calcining treatment (Methods; Supplementary Fig. 7). The RhCuO is reduced in situ to RhCu catalyst at −0.35 V (versus RHE, thereafter) in a flow cell. In situ Raman spectra show that CuO is electrochemically reduced to metallic Cu as evidenced by the vanishing of characteristic Raman bands associated to copper oxide with reduction time (Fig. 2a). Ex situ X-ray diffraction (XRD) characterization confirms the bulk metallic Cu phase in the RhCu catalyst (Supplementary Fig. 8). Additionally, no characteristic peaks attributed to Rh species are observed in XRD patterns for the RhCuO pre-catalyst and RhCu catalyst, indicating a low Rh content and/or the absence of Rh particles. The mass content of Rh for RhCu catalyst is 1.47 wt% determined by inductively coupled plasma optical emission spectrometry (ICP-OES; Supplementary Table 3). High-resolution X-ray photoelectron spectroscopy (XPS) of Cu 2p displays that the Cu valence state is reduced from +2 to 0/+1 during the pre-reduction step (Fig. 2b). High-resolution Rh 3d XPS spectrum reveals that Rh mainly presents a trivalent state in RhCuO pre-catalyst while a metallic state in RhCu catalyst30 (Fig. 2c). Note that a small fraction of Cu+ (Supplementary Fig. 9) and Rh3+ species are detected on the RhCu surface, arising from the inevitable oxidation during the sample transfer in XPS analysis31. The oxide scale is also detected by ex situ Raman spectra (Supplementary Fig. 10). The Rh doping and in situ pre-reduction cause little variations in morphology and size of catalyst particles as shown in the scanning electron microscopy (SEM) characterizations (Supplementary Figs. 11 and 12). Transmission electron microscopy (TEM) images show that RhCu catalyst has a particle size of 15–20 nm (Fig. 2d). The atomic structure of RhCu catalyst was further examined by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). Isolated Rh sites (bright dots in blue circles) clearly indicate the monoatomic dispersion of Rh and the absence of Rh clusters or nanoparticles (Fig. 2e and Supplementary Fig. 13). The STEM energy-dispersive X-ray spectroscopy (STEM-EDS) mapping confirms the uniform distribution of Rh single sites on the Cu matrix (Fig. 2f).

a, In situ Raman spectrum of the RhCuO pre-catalyst at –0.35 V under CO2RR, the pre-reduction process of RhCuO turning into RhCu. b,c, Cu 2p XPS spectrum (b) and Rh 3d XPS spectrum (c) of the RhCuO and RhCu catalyst. Sat. represents satellite peak. d, TEM image of the RhCu catalyst. e, HAADF-STEM image of the RhCu catalyst. The blue circles highlight Rh atoms. f, STEM-EDS mapping of the RhCu catalyst, showing atomic dispersion of Rh sites on the Cu matrix. g,h, Ex situ XANES spectra (g) and EXAFS spectra in R-space with the k-range for Fourier transforms from 2 to 10 Å–1 (h) at the Rh K-edge of the RhCuO and RhCu catalyst. The spectra of Rh foil and Rh2O3 are shown as references. i, In situ Cu K-edge XANES spectra of the RhCu catalyst under CO2RR. The spectra of Cu, Cu2O, and CuO are shown as references.
X-ray absorption spectroscopy (XAS) measurements provide information on the electronic states and coordination structures of Rh dopant and Cu matrix. The normalized X-ray absorption near edge structure (XANES) spectra at the Rh K-edge indicate that the chemical states of RhCuO pre-catalyst and RhCu catalyst resemble those of Rh2O3 and Rh foil, respectively (Fig. 2g). The Rh K-edge extended X-ray absorption fine structure (EXAFS) spectra show that RhCuO pre-catalyst has only first shell Rh–O scattering path (Fig. 2h). The RhCu catalyst exhibits one dominated scattering at ~2.15 Å attributed to the Rh–Cu bond32 with a shoulder at ~1.51 Å ascribed to Rh–O bond29, mainly arising from the oxidation during ex situ measurements. The absence of Rh–Rh scattering (~2.50 Å)32 corroborates the atomic dispersion of Rh atoms in RhCu catalyst, which is also verified by the EXAFS fitting analysis (Supplementary Fig. 14 and Supplementary Table 4). In situ XAS measurements at Cu K-edge support that Cu maintains metallic nature during CO2RR (Fig. 2i and Supplementary Figs. 15–17). Control samples, including those with different Rh doping contents and other single-site dopants (Ir, Ru, Pd, Ag, and Au), were also prepared and characterized similarly (Supplementary Figs. 8, 9, 11, 12, and 18–26).
Electrocatalytic CO2RR performance of MCu catalysts
We evaluated the CO2RR performances of various MCu catalysts in a flow cell with 1 M KOH electrolyte. All MCu catalysts show similar total Faradaic efficiency (FE) of C2+ products (~74 ± 3%) at −0.65 ± 0.01 V (Fig. 3a). Two major CO2RR products are C2H4 and C2H5OH accompanied by minor products of acetate (CH3COO−) and n-propanol (C3H7OH; Supplementary Fig. 27). However, the distribution of C2H4 and C2H5OH varies by the dopant. The FE ratio of C2H4 to C2H5OH roughly exhibits a volcano-shaped dependence on the surface oxygen affinity of MCu. Using the performance of pristine Cu as a benchmark, noble metal dopants such as Rh, Ir, and Ru, with stronger oxygen affinity than Cu, improve the selectivity of C2H4 over C2H5OH, leading to an enhanced C2H4/C2H5OH FE ratio. In this oxophilic MCu group (M = Rh, Ir, and Ru), the FE of C2H4 increases in the order of Cu < RuCu < IrCu < RhCu. The maximum C2H4/C2H5OH FE ratio is achieved on RhCu, doubling to 4.51 compared with that of pristine Cu (2.33). The FE of C2H4 and C2H4/C2H5OH FE ratio drop with further increases in surface oxygen affinity in the RuCu catalyst. Too strong oxygen binding on the Ru site impedes the surface’s ability to release adsorbed *O atoms according to the DFT computations (Fig. 1d). By contrast, Pd, Ag, and Au dopants with weaker oxygen affinity than Cu slightly shift the selectivity from C2H4 to C2H5OH, resulting in lower C2H4/C2H5OH FE ratios than that of pristine Cu. In this oxophobic MCu group (M = Pd, Ag, and Au), the selectivity toward C2H5OH increases as the oxygen affinity of MCu decreases, following the order of Cu < PdCu < AgCu < AuCu. The AuCu catalyst exhibits the highest C2H5OH FE of 24.7% and the lowest C2H4/C2H5OH FE ratio of 1.75.

a, Comparison of FEs of C2H4, C2H5OH, and C2+ products, and C2H4/C2H5OH FE ratios among the MCu catalysts at –0.65 ± 0.01 V. b–d, Comparison of FEs of C2H4 and C2H5OH (b), C2H4/C2H5OH FE ratios (c), and jC2H4 (d) between pristine Cu and RhCu catalyst at different potentials. Data are presented as mean ± s.d. The error bars represent s.d. from measurements of three independent electrodes.
The CO2RR performances at different cathodic potentials follow a similar trend as Fig. 3a (Supplementary Figs. 28–30). As a representative, the catalytic performance of the RhCu at a broad potential window is shown in Fig. 3b–d. The RhCu catalyst exhibits superior C2H4 selectivity at the expense of C2H5OH formation at potentials more negative than −0.48 V (Fig. 3b), leading to a promoted C2H4/C2H5OH FE ratio of ~4.50 across the potential range of −0.48 to −0.70 V (Fig. 3c). RhCu reaches a peak C2H4 FE of 61.2% with a partial current density (jC2H4) of 653 mA cm−2 at −0.66 V (Fig. 3b,d). As a comparison, pristine Cu yields C2H4 FE of 47.7% and jC2H4 of 507 mA cm−2 at the same overpotential. Identical electrolysis conditions were maintained when comparing catalytic performances for different samples. We confirmed that neither CO2 transport, nor liquid electrolyte flow conditions posed limitations on performance in our flow cell setup (Supplementary Figs. 31 and 32). However, it is important to note that we have not accounted for the potential impact of variability in internal mass transport within the pores of the gas diffusion electrode (GDE). The modeling and characterization of these internal mass transport processes still present a substantial challenge33. We also investigated the possible influence of electrochemical surface area (ECSA), which was determined by the double-layer capacitance method (Supplementary Fig. 33 and Supplementary Table 5). The ECSAs for MCu catalysts are nearly invariant, indicating that surface roughness is not the main factor contributing to performance changes. The ECSA-normalized partial current densities of C2H4 and C2H5OH still follow the same order as geometric ones (Supplementary Fig. 34). The long-term stability of RhCu catalyst was evaluated at −0.66 V, where the best C2H4 selectivity was achieved. The RhCu catalyst maintained C2H4 FE for at least 35 h operated under a total current density (jtotal) of ~1,000 mA cm−2 (Supplementary Fig. 35). Post-reaction characterization shows that the Cu surface retained Rh dopants after the stability test (Supplementary Fig. 36 and Supplementary Table 4).
Validating the doping impact on oxygen binding
When surface oxygen affinity is enhanced by Rh doping, the improved adsorption of the oxygen-bound formyloxyl (*OCHO) intermediate could benefit formate (HCOO−) generation on the RhCu surface more than on pristine Cu, especially at low overpotentials19,34 (Supplementary Fig. 37). A dramatic enhancement in HCOO− formation over CO was observed if Rh content was further increased due to stabilized *OCHO intermediate toward HCOO− formation (Supplementary Figs. 38 and 39). However, heavy Rh doping reduces C−C coupling rates by limiting *CO coverage because of the even stronger competition from the *OCHO intermediate34,35. To eliminate the interference of HCOO− formation, CO electroreduction reaction (CORR) was conducted (Supplementary Fig. 40). The RhCu catalyst consistently demonstrated superior C2H4 FEs and C2H4/C2H5OH FE ratios compared with the pristine Cu throughout the investigated potential range under saturated *CO coverage conditions (Supplementary Fig. 41).
In situ Raman measurements were conducted to gain an insight into intermediate adsorption features over the pristine Cu and RhCu catalyst using a modified flow cell36. Two pronounced peaks at ~1,462 and ~1,530 cm−1 exclusively emerge on the RhCu catalyst at a low overpotential of −0.45 V (Supplementary Fig. 42). Considering that the improved yield of HCOO− originates from oxophilic sites at the low overpotential region, we are inclined to assign them to the symmetric O–CH–O stretch of bidentate *OCHO and the asymmetric O–C–O stretch of carboxylate (*OCO−), respectively37,38,39. These two peaks diminish at −0.65 V, in conjunction with the selectivity shift from HCOO− to C2+ products over the RhCu catalyst. Thus, oxophilic sites are likely occupied by intermediates related to C2 products, such as *CH2CHO and *O species, at relatively high overpotentials.
However, detecting the C2-relevant SDI and other intermediates from in situ Raman measurements is extremely challenging due to their high reactivity and low surface coverages40,41. Thus, we performed the ethylene oxide reduction reaction (EORR) as a model reaction to simulate the late stages of CO2RR to C2H4 (refs. 9,23,24). Prior theoretical and experimental studies suggested that after epoxy ring opening, ethylene oxide is reduced to C2H4 through C−O bond scission, and the adsorbed *O atom is released via protonation9,23,24, which is analogous to the elementary steps of *CH2CHO reduction to C2H4 (Supplementary Fig. 43a). We observed that EORR only produces C2H4 without any C2H5OH detected on MCu catalysts (Supplementary Fig. 44), in agreement with previous experiments23,24,42. The RhCu catalyst shows the most positive potential for C2H4 formation among all MCu samples (Supplementary Table 6). The reactivity of EORR to C2H4 over MCu catalysts follows the same order as the CO2-to-C2H4 conversion: RhCu > IrCu > RuCu > Cu > PdCu > AgCu > AuCu (Supplementary Fig. 43). A volcano-shaped correlation of C2H4 selectivity versus surface oxygen affinity also exists for EORR. The similarity in the volcano-like trends observed for both CO2RR and EORR lends credence to the hypothesis that modulating the surface oxygen affinity through single-site doping serves as a viable approach to manipulate the comparative stability of crucial SDI and *O species at the post-C–C coupling stages. Consequently, this strategy directs the reaction pathway toward a specific C2 product in the SDS.
Correlating theoretical descriptor and experimental reactivity
Incorporating various oxygen-affinity sites, such as noble metal atoms, into the Cu surface can steer the selectivity between C2H4 and C2H5OH via stabilizing and activating the key SDI in the post-C–C coupling stages. Specifically, introducing stronger oxygen-binding sites (for example, Rh, Ir, and Ru) to Cu surfaces facilitates C–O bond scission of the SDI (for example, *CH2CHO), favoring the C2H4 formation pathway. An optimal oxygen adsorption energy is required to reach the maximum selectivity toward C2H4 (Fig. 4a), which is derived from balancing the kinetic barriers of C–O bond cleavage in *CH2CHO and the subsequent hydrogenation of the *O adsorbate (Fig. 4b). The RhCu catalyst stands out as the most reactive one for C2H4 generation. While the RuCu catalyst exhibits the strongest oxygen adsorption energy, it is important to note that the binding of *O is exceptionally strong, exceeding that of Cu by 0.6 eV. This high affinity for oxygen can lead to catalyst poisoning, as the removal of oxygen atoms could become sluggish (as depicted in Fig. 4b). The Ir-doped surface is expected to perform similarly to RhCu but with slightly lower C2H4 selectivity because it has a slightly higher barrier for the C–O bond cleavage.

a, Relationship between experimentally measured C2H4/C2H5OH FE ratio versus DFT-calculated relative adsorption energy of the *O intermediate. The dashed line shows a volcano-shaped relationship to guide the eye. Data are presented as mean ± s.d. Error bars represent s.d. from measurements of three independent electrodes. b, Two-dimensional plot for ECSA-normalized experimental C2H4 activity versus DFT-calculated kinetic barriers of C–O bond scission and subsequent *O protonation along the C2H4 pathway.
Contrastingly, although PdCu, AgCu, and AuCu catalysts with weaker oxygen affinity induce lower C2H4 selectivity and C2H4/C2H5OH FE ratios than pristine Cu, the extent of the decrease is modest. This is consistent with our DFT predictions, where changes in adsorption energies of SDI are marginal because SDIs prefer to adsorb on Cu atoms neighboring the oxophobic dopant under the scenario of low doping content (Fig. 1b and Supplementary Fig. 2). However, we infer that a superior selectivity of C2H5OH over C2H4 can be achieved by elevating the doping level to an amount that forces intermediates to adsorb onto some of the oxophobic dopants for preserving the C–O bond. For instance, the 9–24at% Ag-modified Cu catalysts were reported to reach comparable or even higher FE of C2H5OH than C2H4, although different SDIs and mechanism were adopted in that work17.
Our combined experimental and computational approaches unravel the intricate puzzle of significant SDIs. Although numerous SDIs likely coexist, our findings indicate that the oxygen-bound SDI, such as *CH2CHO or its derivatives, could be prominent among them (see Supplementary Notes1 and 2 for more discussions). Therefore, the surface oxygen affinity might serve as a more universal descriptor for predicting selectivity between C2H4 and C2H5OH on different catalysts. Our DFT calculations support the hypothesis that the surface oxygen affinity can be modified by introducing single-site dopants. There is a clear correlation between oxygen adsorption energy and observed C2H4/C2H5OH production ratio as shown in Fig. 4a. Figure 4b maps the DFT prediction in Fig. 4a to the experimentally observed activity, demonstrating that the computationally predicted ‘volcano-shaped’ relationship with respect to oxygen binding strength is indeed observed in the experiments.
Collective promotion of RDS and SDS
Benefiting from the high activity and selectivity of C2H4, the RhCu catalyst presents a cathodic (half-cell) energy efficiency (EEhalf) of 37.2% for C2H4 in a flow cell at –0.66 V (Supplementary Fig. 45). To minimize the ohmic resistance and improve the full-cell energy efficiency (EEfull), the RhCu catalyst was incorporated into a zero-gap membrane electrode assembly (MEA) electrolyzer (Supplementary Fig. 46). The MEA cell delivers a C2H4 FE of 58.2% and jC2H4 of 590 mA cm−2 at the cell voltage of 3.8 V, where EEfull of C2H4 is 17.6% (Supplementary Fig. 47). Our previous research demonstrated that segmented tandem electrodes promoted the kinetics of C–C coupling by driving cascade CO2 → CO → C2+ conversion with maximized CO utilization43. We intend to promote the activity of both RDS (that is, C–C coupling step) and SDS (that is, post-C–C coupling step) by integrating the RhCu and CO-generating ZnO catalysts onto the co-planar segmented tandem electrode (ZnO/RhCu cs-GDE). The ZnO nanoparticles, serving as an extra CO supplier (Supplementary Fig. 48), are placed at the inlet segment of the catalyst layer to prolong CO residence time along the length of the electrode (Fig. 5a). In this way, cs-GDE efficiently intensifies *CO surface coverage of the RhCu surface to enhance the reaction kinetics of the C–C coupling step and in turn maximize the activity and selectivity toward C2+ products43. The ZnO/RhCu cs-GDE reaches C2+ FE of 85.4% and C2H4 FE of 68.8% at 3.7 V (Fig. 5b), corresponding to a C2H4 EEfull of 21.4% (Supplementary Fig. 49). The partial current densities of C2H4 and C2+ products are boosted to 1,012 and 1,256 mA cm−2, respectively (Fig. 5b).

a, Schematic illustration of the tandem electrode structure. b, FEs and partial current densities of C2H4, C2H5OH, and C2+ products at different applied cell voltages. Data are presented as mean ± s.d. The error bars represent s.d. from three independent measurements.
Discussion
With synergistic theoretical and experimental efforts, we demonstrate that single-site doping is an efficient strategy to modulate Cu surface oxygen affinity. Our research reveals that the oxygen-binding energy of the catalyst surface serves as a generalized descriptor, enabling the prediction of whether the post-C–C coupling pathway leans toward producing C2H4 or C2H5OH. The oxygen affinity of the dopant on the Cu surface has a decisive influence on the feasibility of C–O bond dissociation in the SDI, as evidenced by the volcano-type relationship between the C2H4/C2H5OH FE ratio and the oxygen adsorption energy. Strong oxygen-binding sites, such as Rh, promote C2H4 formation by facilitating C–O bond scission. By contrast, weak oxygen affinity dopants, such as Au, favor C2H5OH generation by preserving the C–O bond. Oxygen-affinity engineering can be extended to enhance the production of other high-value C1 liquid fuels that involve oxygen-bound intermediates as SDIs, such as formate and methanol.
Methods
Computational methods
All calculations were performed with the Vienna Ab Initio Simulation Package (VASP 5.4.4)44. The exchange-correlation energy was evaluated by the Perdew–Burke–Ernzerhof (PBE) functional45 with frozen core electrons treated with the projector-augmented-wave (PAW) theory46. Valence electrons treated self-consistently are Cu(3d104s1), Ag(4d105s1), Au(5d106s1), Pd(4d95s1), Ir(5d86s1), Rh(4d85s1), Ru(4d75s1), C(2s22p2), and O(2s22p4). First-order Methfessel–Paxton smearing was applied with a 0.2 eV width47. The plane-wave basis was truncated at 450 eV. Brilluoin zones were treated by a Monkhorst–Pack k-points mesh48 of 8 × 8 × 8 for bulk Cu and 2 × 2 × 1 for slab models. A four-layer model of Cu(100) with a 2 × 2 unit cell was used for both pristine and doped surfaces. The bottom two layers were frozen to mimic the bulk structure. A dipole correction was applied for all slab models. The van der Waals interaction correction was applied with the DFT-D3 method49. Transition states were found with the climbing image nudged elastic band method (cNEB)50,51. The potential-dependent reaction free energies and barriers are calculated with a formalism developed prviously52,53,54,55, where a water molecule is used as a proton shuttle in the O–H bond-formation reaction steps (see Supplementary Fig. 5 and Supplementary Methods 1 for more details). Gibbs free energies are calculated as:
where the contributions to the free energy are the total energy of the system determined by DFT optimization (EDFT), the zero-point energy (ZPVE), the enthalpic temperature correction (∫0TCvdT), and the entropy correction (TS).
Chemicals and materials
Copper (ii) nitrate trihydrate (Cu(NO3)2·3H2O), rhodium (iii) chloride (RhCl3), iridium(iii) chloride (IrCl3), ruthenium (iii) chloride trihydrate (RuCl3·3H2O), palladium (ii) chloride (PdCl2), silver nitrate (AgNO3), gold (iii) chloride trihydrate (HAuCl4·3H2O), sodium hydroxide (NaOH), potassium hydroxide (KOH), phosphate buffer saline (PBS), and gaseous ethylene oxide (≥99.5%) were purchased from Sigma Aldrich and used as received.
Catalyst synthesis
Cu(OH)2 precursor was first synthesized by a precipitation method: 0.5 M Cu(NO3)2·3H2O and 0.5 M NaOH were added dropwise into the deionized water under rapid stirring. The pH of the precipitating solution was kept at approximately 9. After aging for 8 h with stirring at room temperature and N2 protection, the blue slurry was washed and centrifuged with water until the pH was close to 7. The precipitates were dried overnight in vacuum to obtain Cu(OH)2 matrix. For a typical synthesis of the RhCu catalyst, a calculated amount of RhCl3 solution (1 mg ml−1) was injected into Cu(OH)2 solution (10 mg ml−1) by a syringe pump at a rate of 10 μl s−1. The mixture solution was stirred vigorously for 5 h. After the cation exchange process, the sample was washed, dried, and annealed at 330 °C for 3 h to obtain RhCuO product. RhCu catalyst was then formed in situ by reducing RhCuO pre-catalyst on the GDE at −0.35 V for 10 min in a flow cell system supplied with CO2 gas and 1 M KOH as electrolyte. ICP-OES was used to determine the mass loading of Rh. The preparation of other MCu samples with similar M atomic contents (Supplementary Table 7) was identical to that of the RhCu catalyst except for using the corresponding metal salts instead of RhCl3. The pristine Cu was obtained by direct calcination of Cu(OH)2 followed by electroreduction.
Material characterizations
High-resolution SEM was performed on the FEI Apreo SEM. TEM, HAADF-STEM, and EDS elemental mapping were conducted using the JEOL ARM200F microscope, equipped with a cold field emission gun and operated at 200 kV voltage. XPS data were collected using an ESCALAB Xi+ spectrometer equipped with a monochromatic Al Kα radiation source operating at 200 W. XRD was carried out on the Malvern Panalytical Aeris research edition powder diffractometer using Cu Kα radiation. XAS measurements were performed on the Beamline 7-BM and 8-ID of the National Synchrotron Light Source II at Brookhaven National Laboratory. The Athena and Artemis software in Demeter package was employed for data processing and analysis56. The theoretical EXAFS signal was fitted to the experimental EXAFS data in R-space by Fourier transforming both the theoretical and experimental data.
Electrochemical measurements
The CO2RR performances of all samples were tested in a customized flow cell with 1 M KOH as electrolyte under ambient conditions. For preparing the cathode GDE, the catalyst was dispersed in isopropanol with 5 wt% Nafion to form a homogeneous ink. The catalyst ink was then spray-coated onto the gas diffusion layer (GDL, Sigracet 39BB). The mass loadings of all catalysts were controlled at ~0.35 mg cm−2. Nickel form was used as the anode. An anion exchange membrane (FAA-3-PK-130, Fuel Cell Store) was used to separate cathodic and anodic compartments. The electrolyte was fed by syringe pumps (IPS Series, Inovenso) at 2 ml min−1 and 5 ml min−1 to the cathode and anode chambers, respectively. CO2 gas was supplied to the cathode at 50 sccm via a mass flow controller (Alicat Scientific). A potentiostat (Gamry Interface 1010E) controlled a constant voltage to the flow cell and recorded the corresponding current. A programmable d.c. power supply (B&K Precision XLN3640) was employed when the current was higher than 1 A. The cathode potential was measured relative to the Ag/AgCl (3 M KCl) reference electrode. All potentials were converted to the RHE scale using: ERHE = EAg/AgCl + 0.209 V + 0.0591 × pH. An iR compensation was determined by potentiostatic electrochemical impedance spectroscopy.
During the electrochemical reaction, an on-line gas chromatograph (GC, SRI Instruments MultipleGas#5) equipped with both a thermal conductivity detector and a flame ionization detector was used to monitor the gas products. To calibrate the outlet gas flow rate of CO2, Ar, as an internal standard, was fed at 10 sccm and mixed with the outlet gas stream from the cathode before sampling to the GC loop. The FEs for gas products were calculated using the following equation:
where z is the number of electrons transferred for producing a target product; F is the Faraday constant; x is the molar fraction of a target product determined by GC; V is the molar flow rate of gas; and jtotal is the total current density.
Meanwhile, the liquid products after electrolysis were collected and quantified via 1H NMR (Bruker NEO 400 MHz spectrometer). The electrolyte (500 μl) was mixed with 100 μl internal standard of 5 mM 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt in D2O. The s.d. values were calculated based on the measurements of three independent electrodes.
The half-cell and full-cell energy efficiencies of C2H4 were calculated as follows:
where \({E}_{{\rm{C}}2{\rm{H}}4}^{0}\) is the equilibrium potential for CO2RR to C2H4, which is 0.08 V (ref. 2); Ecathode is the applied cathode potential; Ecell is the applied cell voltage; FEC2H4 is the FE of C2H4.
EORR was carried out in a flow cell with 1 M PBS as electrolyte. A neutral pH was chosen to avoid the hydrolysis and polarization of ethylene oxide under acidic and basic conditions57. The Nafion membrane was placed between the MCu GDE cathode and an IrO2 GDE anode. The catholyte was degassed by Ar and then bubbled with ethylene oxide to reach a concentration of 20 mM. During EORR, 2 vol% ethylene oxide balanced by Ar constantly flowed through the cathode. The electrochemical measurements and product quantification were identical to those of CO2RR.
The MEA electrolyzer-based CO2RR procedure is schematized in Supplementary Fig. 46. The GDE cathode and an IrO2/Ti mesh anode were separated by a Sustainion anion-exchange membrane (Dioxide Materials). Humidified CO2 at a rate of 50 sccm was supplied to the cathode. KOH (0.5 M) was pumped into the anode chamber at 15 ml min−1. A potentiostat (Gamry Interface 5000E) was applied to monitor current densities in a two-electrode system at different cell voltages without iR correction. A cold trap was placed downstream of the effluent gas at the cathode to separate gas and liquid products. Due to the liquid product crossover, the FEs of liquid products were calculated based on the total amount collected in the anode and cathode sides during the same period. The rest procedure was identical to that in a flow cell.
To fabricate the ZnO/RhCu cs-GDE, the uniform RhCu (0.35 mg) segment (1.8 (L) × 0.5 (W) cm2) was first coated onto the GDL, followed by spraying a ZnO (~0.04 mg) segment (0.2 (L) × 0.5 (W) cm2) at the inlet. The area and position of each segment were precisely controlled by a machined template43.
In situ Raman spectroscopy measurements
In situ Raman experiments were carried out using a modified flow cell developed by our group36. The Raman spectrum was recorded on a Renishaw inVia Raman microscope with a 785 nm laser. For each in situ Raman measurement, the acquisition time was 10 s, and the accumulation of scans was 5. Cathode potentials were applied in the potentiostatic mode and converted to the RHE scale accordingly.
Data availability
All the data that support the findings of this study are available in the main text and the Supplementary Information. The atomic coordinates of the optimized computational models are available in the Materials Cloud repository (https://doi.org/10.24435/materialscloud:4b-cf). Source data are provided with this paper.
References
Stephens, I. E. L. et al. 2022 roadmap on low temperature electrochemical CO2 reduction. J. Phys. Energy 4, 042003 (2022).
Nitopi, S. et al. Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte. Chem. Rev. 119, 7610–7672 (2019).
Xiang, K. et al. Boosting CO2 electroreduction towards C2+ products via CO* intermediate manipulation on copper-based catalysts. Environ. Sci. Nano 9, 911–953 (2022).
Murata, A. & Hori, Y. Product selectivity affected by cationic species in electrochemical reduction of CO2 and CO at a Cu electrode. Bull. Chem. Soc. Jpn 64, 123–127 (1991).
Lu, X., Shinagawa, T. & Takanabe, K. Product distribution control guided by a microkinetic analysis for CO reduction at high-flux electrocatalysis using gas-diffusion Cu electrodes. ACS Catal. 13, 1791–1803 (2023).
Xiao, H., Cheng, T. & Goddard, W. A. Atomistic mechanisms underlying selectivities in C1 and C2 products from electrochemical reduction of CO on Cu(111). J. Am. Chem. Soc. 139, 130–136 (2017).
Cheng, T., Xiao, H. & Goddard, W. A. Full atomistic reaction mechanism with kinetics for CO reduction on Cu(100) from ab initio molecular dynamics free-energy calculations at 298 K. Proc. Natl Acad. Sci. USA 114, 1795–1800 (2017).
Luo, W., Nie, X., Janik, M. J. & Asthagiri, A. Facet dependence of CO2 reduction paths on Cu electrodes. ACS Catal. 6, 219–229 (2016).
Calle‐Vallejo, F. & Koper, M. T. M. Theoretical considerations on the electroreduction of CO to C2 species on Cu(100) electrodes. Angew. Chem. Int. Ed. 52, 7282–7285 (2013).
Cheng, D. et al. The nature of active sites for carbon dioxide electroreduction over oxide-derived copper catalysts. Nat. Commun. 12, 395 (2021).
Shen, S. et al. AuCu alloy nanoparticle embedded Cu submicrocone arrays for selective conversion of CO2 to ethanol. Small 15, 1902229 (2019).
Li, J. et al. Constraining CO coverage on copper promotes high-efficiency ethylene electroproduction. Nat. Catal. 2, 1124–1131 (2019).
Zhuang, T.-T. et al. Steering post-C–C coupling selectivity enables high efficiency electroreduction of carbon dioxide to multi-carbon alcohols. Nat. Catal. 1, 421–428 (2018).
Gu, Z. et al. Efficient electrocatalytic CO2 reduction to C2+ alcohols at defect-site-rich Cu surface. Joule 5, 429–440 (2021).
Wang, X. et al. Efficient electrically powered CO2-to-ethanol via suppression of deoxygenation. Nat. Energy 5, 478–486 (2020).
Li, J. et al. Enhanced multi-carbon alcohol electroproduction from CO via modulated hydrogen adsorption. Nat. Commun. 11, 3685 (2020).
Li, Y. C. et al. Binding site diversity promotes CO2 electroreduction to ethanol. J. Am. Chem. Soc. 141, 8584–8591 (2019).
Kuhl, K. P. et al. Electrocatalytic conversion of carbon dioxide to methane and methanol on transition metal surfaces. J. Am. Chem. Soc. 136, 14107–14113 (2014).
Feaster, J. T. et al. Understanding selectivity for the electrochemical reduction of carbon dioxide to formic acid and carbon monoxide on metal electrodes. ACS Catal. 7, 4822–4827 (2017).
Katayama, Y. et al. An in situ surface-enhanced infrared absorption spectroscopy study of electrochemical CO2 reduction: selectivity dependence on surface C-bound and O-bound reaction intermediates. J. Phys. Chem. C 123, 5951–5963 (2019).
Zhi, X., Jiao, Y., Zheng, Y., Davey, K. & Qiao, S.-Z. Directing the selectivity of CO2 electroreduction to target C2 products via non-metal doping on Cu surfaces. J. Mater. Chem. A 9, 6345–6351 (2021).
Zhi, X., Vasileff, A., Zheng, Y., Jiao, Y. & Qiao, S.-Z. Role of oxygen-bound reaction intermediates in selective electrochemical CO2 reduction. Energy Environ. Sci. 14, 3912–3930 (2021).
Piqué, O., Low, Q. H., Handoko, A. D., Calle-Vallejo, F. & Yeo, B. S. Selectivity map for the late stages of CO and CO2 reduction to C2 species on Cu electrodes. Angew. Chem. Int. Ed. 60, 10784–10790 (2021).
Schouten, K. J. P., Pérez Gallent, E. & Koper, M. T. M. Structure sensitivity of the electrochemical reduction of carbon monoxide on copper single crystals. ACS Catal. 3, 1292–1295 (2013).
Piqué, O., Viñes, F., Illas, F. & Calle-Vallejo, F. Elucidating the structure of ethanol-producing active sites at oxide-derived Cu electrocatalysts. ACS Catal. 10, 10488–10494 (2020).
Seh, Z. W. et al. Combining theory and experiment in electrocatalysis: insights into materials design. Science 355, eaad4998 (2017).
Wu, F. et al. Modulating the oxophilic properties of inorganic nanomaterials for electrocatalysis of small carbonaceous molecules. Nano Today 29, 100802 (2019).
Wang, Y.-R. et al. Reduction-controlled atomic migration for single atom alloy library. Nano Lett. 22, 4232–4239 (2022).
Xu, H. et al. Cation exchange strategy to single-atom noble-metal doped CuO nanowire arrays with ultralow overpotential for H2O splitting. Nano Lett. 20, 5482–5489 (2020).
Kibis, L. S., Stadnichenko, A. I., Koscheev, S. V., Zaikovskii, V. I. & Boronin, A. I. XPS study of nanostructured rhodium oxide film comprising Rh4+ species. J. Phys. Chem. C 120, 19142–19150 (2016).
Lum, Y. & Ager, J. W. Stability of residual oxides in oxide-derived copper catalysts for electrochemical CO2 reduction investigated with 18O labeling. Angew. Chem. Int. Ed. 57, 551–554 (2018).
Liu, H. et al. Efficient electrochemical nitrate reduction to ammonia with copper-supported rhodium cluster and single-atom catalysts. Angew. Chem. Int. Ed. 61, e202202556 (2022).
Watkins, N. B. et al. Hydrodynamics change Tafel slopes in electrochemical CO2 reduction on copper. ACS Energy Lett. 8, 2185–2192 (2023).
Su, D.-J. et al. Kinetic understanding of catalytic selectivity and product distribution of electrochemical carbon dioxide reduction reaction. JACS Au 3, 905–918 (2023).
Huang, Y., Handoko, A. D., Hirunsit, P. & Yeo, B. S. Electrochemical reduction of CO2 using copper single-crystal surfaces: effects of CO* coverage on the selective formation of ethylene. ACS Catal. 7, 1749–1756 (2017).
Li, Z. et al. Planar defect-driven electrocatalysis of CO2-to-C2H4 conversion. J. Mater. Chem. A 9, 19932–19939 (2021).
Bohra, D. et al. Lateral adsorbate interactions inhibit HCOO− while promoting CO selectivity for CO2 electrocatalysis on silver. Angew. Chem. Int. Ed. 58, 1345–1349 (2019).
Firet, N. J. & Smith, W. A. Probing the reaction mechanism of CO2 electroreduction over Ag films via operando infrared spectroscopy. ACS Catal. 7, 606–612 (2017).
Shan, W. et al. In situ surface-enhanced Raman spectroscopic evidence on the origin of selectivity in CO2 electrocatalytic reduction. ACS Nano 14, 11363–11372 (2020).
Goodpaster, J. D., Bell, A. T. & Head-Gordon, M. Identification of possible pathways for C–C bond formation during electrochemical reduction of CO2: new theoretical insights from an improved electrochemical model. J. Phys. Chem. Lett. 7, 1471–1477 (2016).
Birdja, Y. Y. et al. Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nat. Energy 4, 732–745 (2019).
Schouten, K. J. P., Kwon, Y., Ham, C. J. M., van der, Qin, Z. & Koper, M. T. M. A new mechanism for the selectivity to C1 and C2 species in the electrochemical reduction of carbon dioxide on copper electrodes. Chem. Sci. 2, 1902–1909 (2011).
Zhang, T. et al. Highly selective and productive reduction of carbon dioxide to multicarbon products via in situ CO management using segmented tandem electrodes. Nat. Catal. 5, 202–211 (2022).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Computat. Mater. Sci. 6, 15–50 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Methfessel, M. & Paxton, A. T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 40, 3616–3621 (1989).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Henkelman, G. & Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 113, 9978–9985 (2000).
Nie, X., Luo, W., Janik, M. J. & Asthagiri, A. Reaction mechanisms of CO2 electrochemical reduction on Cu(111) determined with density functional theory. J. Catal. 312, 108–122 (2014).
Nie, X., Esopi, M. R., Janik, M. J. & Asthagiri, A. Selectivity of CO2 reduction on copper electrodes: the role of the kinetics of elementary steps. Angew. Chem. Int. Ed. 52, 2459–2462 (2013).
Rostamikia, G., Mendoza, A. J., Hickner, M. A. & Janik, M. J. First-principles based microkinetic modeling of borohydride oxidation on a Au(111) electrode. J. Power Sources 196, 9228–9237 (2011).
Maheshwari, S., Li, Y., Agrawal, N. & Janik, M. J. in Advances in Catalysis, Vol. 63 (ed. Song, C.) 117–167 (Academic Press, 2018); https://doi.org/10.1016/bs.acat.2018.10.003
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541 (2005).
Melhem, G. A. et al. Kinetics of the reactions of ethylene oxide with water and ethylene glycols. Process Saf. Prog. 20, 231–246 (2001).
Acknowledgements
This work is supported by the Department of Energy (DOE) Office of Energy Efficiency and Renewable Energy under IEDO contract DE-EE0010836 and partly supported by the National Science Foundation (NSF) grant CBET-2033343. P.W. and T.P.S. acknowledge funding support from a Rice University Interdisciplinary Excellence Award and NSF grant CBET-2143941. S.Y. acknowledges the use of facilities within the Eyring Materials Center at Arizona State University, supported in part by NNCI-ECCS-1542160. Z.L. acknowledges URC Graduate Student Stipend awarded by the Office of Research at University of Cincinnati. Z.L. thanks X. Shang for discussing catalyst synthesis and reaction mechanisms. V.S., V.K.R.K., and Y.F. acknowledge the funding from the UC CEAS Pilot Program NOEMA. A.I.F. and S.X. acknowledge support by the NSF grant CHE 2102299. S.D.S. is supported by a US DOE Early Career Award. J.D.J. is supported by the Brookhaven National Laboratory Goldhaber Distinguished Fellowship. The work carried out at Brookhaven National Laboratory was supported by the DOE under contract DE-SC0012704. The XAS measurements used resources 7-BM and 8-ID of the National Synchrotron Light Source II, a DOE Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under contract DE-SC0012704. The 7-BM beamline operations were supported in part by the Synchrotron Catalysis Consortium (DOE Office of Basic Energy Sciences grant DE-SC0012335). We would like to thank E. Stavitski, S. Ehrlich, and N. Marinkovic for help with XAFS data collection.
Author information
Authors and Affiliations
Contributions
Z.L. and P.W. conceptualized the project under the supervision of T.P.S. and J.W.; Z.L. synthesized catalysts, performed the electrochemical tests, and analyzed experimental data with the help of T.I. and T.Z.; P.W. performed DFT simulation; S.Y., X.L. and Z.L. conducted the catalyst characterization with the help of Y.B., J.L. and A.S.; Z.L. and V.K.R.K. carried out in situ Raman measurements with the assistance of Y.F. and V.S.; S.X., J.D.J., L.M., A.I.F. and S.D.S. carried out XAS measurements and analyses. Z.L., P.W., J.R., S.Y., T.P.S. and J.W. wrote the paper. All authors discussed the results and commented on the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Chemical Engineering thanks Fengwang Li and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–49, Tables 1–8, methods and Notes 1 and 2
Source data
Source Data Fig. 1
Numerical Source Data.
Source Data Fig. 2
Numerical Source Data.
Source Data Fig. 3
Numerical Source Data.
Source Data Fig. 4
Numerical Source Data.
Source Data Fig. 5
Numerical Source Data.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, Z., Wang, P., Lyu, X. et al. Directing CO2 electroreduction pathways for selective C2 product formation using single-site doped copper catalysts. Nat Chem Eng 1, 159–169 (2024). https://doi.org/10.1038/s44286-023-00018-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s44286-023-00018-w
This article is cited by
-
Beyond C–C coupling in CO2 reduction
Nature Chemical Engineering (2024)
